
Abstract
Background
Numerous studies have shown that bacteria form stable associations with host corals and have focused on identifying conserved “core microbiomes” of bacterial associates inferred to be serving key roles in the coral holobiont. Because studies tend to focus on only stony corals (order Scleractinia) or soft corals (order Alcyonacea), it is currently unknown if there are conserved bacteria that are shared by both. A meta-analysis was done of 16S rRNA amplicon data from multiple studies generated via identical methodology to allow direct comparisons of bacterial associates across seven deep-sea corals, including both stony and soft species: Anthothela grandiflora, Anthothela sp., Lateothela grandiflora, Lophelia pertusa, Paramuricea placomus, Primnoa pacifica, and Primnoa resedaeformis.
Results
Twenty-three operational taxonomic units (OTUs) were consistently present in greater than 50% of the coral samples. Seven amplicon sequence variants (ASVs), five of which corresponded to a conserved OTU, were consistently present in greater than 30% of the coral samples including five or greater coral species. A majority of the conserved sequences had close matches with previously identified coral-associated bacteria. While known to dominate tropical and temperate coral microbiomes, Endozoicomonas were extremely rare or absent from these deep-sea corals. An Endozoicomonas OTU associated with Lo. pertusa in this study was most similar to those from shallow-water stony corals, while an OTU associated with Anthothela spp. was most similar to those from shallow-water gorgonians.
Conclusions
Bacterial sequences have been identified that are conserved at the level of class Anthozoa (i.e., found in both stony and soft corals, shallow and deep). These bacterial associates are therefore hypothesized to play important symbiotic roles and are highlighted for targeted future study. These conserved bacterial associates include taxa with the potential for nitrogen and sulfur cycling, detoxification, and hydrocarbon degradation. There is also some overlap with kit contaminants that need to be resolved. Rarely detected Endozoicomonas sequences are partitioned by whether the host is a stony coral or a soft coral, and the finer clustering pattern reflects the hosts’ phylogeny.
Similar content being viewed by others
Background
Numerous shallow-water studies of stony and soft corals, both tropical and temperate, have shown that bacteria form stable associations with host coral species [1,2,3,4,5,6]. Studies of cold-water corals from the deep ocean (also referred to as deep-sea corals) have also shown conserved bacterial communities that differ between coral species [7,8,9,10,11]. Within those coral-associated bacterial communities, individual conserved bacterial associates began to be identified in clone library studies [2, 12, 13]. With the increase in sequencing depth afforded by second-generation sequencing, studies began focusing on identifying “core microbiomes” of bacterial associates consistently found in some percentage, preferably 100%, of samples of a particular coral [14,15,16]. These conserved bacterial associates are inferred to be serving key roles in the coral holobiont, and therefore, identifying and studying them should yield insights into coral biology and microbial symbiosis.
The conserved core microbiome in corals has frequently been found to include Endozoicomonas [17,18,19,20]. Further, bacteria from the genus Endozoicomonas, or within the same family, have often been found to dominate shallow-water coral microbiomes both tropical and temperate [17, 18, 20,21,22,23,24,25,26]. However, recent studies of deep-sea corals, both stony and soft, have found Endozoicomonas to be rare or undetected in their microbiomes [10, 11, 27, 28]. This raises questions about the important role this bacterial group is hypothesized to play in corals and why this group is largely absent in deep-sea corals.
Ainsworth et al. recently identified rare but consistent core bacterial associates shared across multiple stony coral genera in tropical and mesophotic habitats, although at a level of 30–50% of the samples rather than 100% [29]. However, because studies tend to focus on only stony corals (order Scleractinia) or soft corals (order Alcyonacea), it is currently unknown if there are conserved bacteria that are shared by both. Tackling the question is further impeded by the differences in extraction method, primer choice, and sequencing platform between studies on different corals, limiting our ability to compare across hosts.
In this study, I tested the hypothesis that there are conserved bacterial associates present across both stony and soft deep-sea corals by reanalyzing aggregated data from previous deep-sea coral investigations [11, 27, 28, 30]. Further, I investigated rare Endozoicomonas sequences present in some of these corals to determine how similar they are to the sequences derived from tropical and temperate coral microbiomes. Pyrosequencing of 16S ribosomal rRNA amplicons using identical methodology from extraction to sequencing in all of the aggregated studies allows for the first time a direct comparison of bacterial associates across seven species of deep-sea corals: Anthothela grandiflora, Anthothela sp., Lateothela grandiflora, Lophelia pertusa, Paramuricea placomus, Primnoa pacifica, and Primnoa resedaeformis.
Methods
Sample data
Datasets of 16S rRNA amplicons and associated environmental data from four prior publications covering seven species of deep-sea corals were combined and reanalyzed in aggregate (Additional file 1; [11, 27, 28, 30]). These raw sequence data are available from the NCBI Sequence Read Archive under BioProjects PRJNA296835, PRJNA297333, PRJNA305617, and PRJNA348705 as well as USGS data releases for each of the original papers [31,32,33,34].
DNA extraction and 16S rRNA gene amplicon sequencing
Uniform methods were used across these four studies to preserve, extract, and sequence bacterial amplicons associated with the corals. Briefly, coral samples were preserved in the field using RNAlater and stored at − 80 °C until processed. DNA was extracted using the MO BIO Powerplant DNA Isolation Kit with the modifications suggested by Sunagawa et al. [4, 35]. DNA samples were amplified with primers 563F (5′-AYTGGGYDTAAAGNG) and 926R (5′-CCGT CAATTYYTTTRAGTTT) which target the V4-V5 hypervariable region of the 16S rRNA gene [36]. Samples were pyrosequenced using Roche 454 GS FLX Titanium chemistry.
Sequence analysis of amplicon datasets
Bioinformatic analysis was conducted using QIIME 1.9.1 [37] and DADA2 1.9.2 [38] following the workflow of specific scripts and parameters for each step listed in Additional file 2. In brief, for QIIME, individual datasets had been previously denoised and trimmed of primers, and the resulting files were combined into a single fasta file for an open-reference operational taxonomic unit (OTU) picking [39]. Greengenes release 13_8 was used as the reference database [40, 41], and chimeras were removed by usearch61 [42]. Non-bacterial sequences and singletons were filtered out. A non-rarefied OTU table (Additional file 3) was used to determine the core OTUs shared across multiple coral species. Samples were then randomly rarefied to the size of the smallest library (4287 sequences) before the diversity metrics were calculated [43]. In brief, for DADA2 (run in RStudio using R version 3.5.1 [44]), the Roche 454 ssf files were converted to individual fastq files using QIIME scripts, and primers were removed using Biostrings [45] and Cutadapt [46]. Reads were trimmed to 325 bp based on quality profiles. Sequences were processed through the filter and inference modules of DADA2 in groups corresponding to original 454 runs (6 separate runs total) in order to more accurately estimate the error rates. The data were then merged for the chimera removal and taxonomy assignment. Greengenes release 13_8 was used as the reference database to keep the consistency with the OTU taxonomy. Amplicon sequence variants (ASVs) that were unclassified below the domain Bacteria level were removed. The resulting ASV table (Additional file 4) contains 4299 ASVs.
Sequence analysis of Endozoicomonas sequences
Operational taxonomic units that were classified by Greengenes as belonging to Endozoicomonadaceae were identified from the OTU table (Additional file 3). The Endozoicomonadaceae OTU sequences and those of reference clone library sequences previously derived from corals were aligned using Clustal X [47]. Stony corals that had comparable reference Endozoicomonas sequences included Acropora humilis (Genbank Accession KC668469.1) [22], Montipora aequituberculata (FJ347758.1) [48], Pocillopora damicornis (KC668770.1) [22], and Stylophora pistillata (KC669131.1) [22]. Soft corals that had comparable reference Endozoicomonas sequences included Eunicella cavolini (JQ691583.1) [21] and Gorgonia ventalina (GU118516.1) [4]. An online phylogenetic tree viewer was used to visualize the relationships revealed by the alignment [49].
Statistical analyses
Alpha and beta diversity metrics and relative abundance summaries were calculated within QIIME 1.9.1 [37]. Community similarity was assessed by principal coordinate analysis (PCoA) using weighted and unweighted Unifrac, Bray-Curtis, and Binary Sorenson Dice to determine the importance of taxonomic and phylogenetic relationships and sequence abundance. PERMANOVA analyses were conducted using PRIMER v7 software [50] on the Binary Sorenson Dice distance matrix from QIIME. A one-factor test design was based on the partial sum of squares type III, 9999 permutations of residuals under an unrestricted permutation of raw data. A two-factor test was based on the same parameters but with the factor “species” nested under the factor “genus.” Core microbiomes were identified based on the presence of an OTU in > 50% of the coral samples (n = 51) or the presence of an ASV in > 30% of the coral samples (n = 51) with the additional caveat of being present in > 5 coral hosts. This additional caveat was necessary to avoid detection of sequences that were conserved only at the level of species or genus.
Results
A total of 2,323,795 amplicon sequences were processed from 66 samples across 7 species of deep-sea corals (Additional file 1). After filtering and removal of low-read samples, this was reduced to 2,205,336 sequences across 51 samples: 12 samples of A. grandiflora, 4 samples of Anthothela sp., 3 samples of Anthothela ND (species not determined by genetics—either A. grandiflora or Anthothela sp.), 1 sample of L. grandiflora, 12 samples of Lo. pertusa, 3 samples of P. placomus, 6 samples of Pr. pacifica, and 10 samples of Pr. resedaeformis (Additional file 5).
Core bacterial associates
The final OTU table, generated after filtering to remove non-bacterial sequences but prior to rarefaction (3204 OTUs; Additional file 3), was searched for conserved OTUs across all 51 samples of the 7 deep-sea corals. While there were no OTUs that were present in 100% of the samples, there were 23 OTUs present in more than 50% of the coral samples (Table 1). The majority of these 23 OTUs could only be identified to the order or family level, and one was unassignable even to a phylum. The final ASV table, generated after filtering to remove sequences unclassified beyond the domain level (4299 ASVs; Additional file 4) was also searched for conserved ASVs, uncovering 7 ASVs that were present in more than 30% of coral samples, across at least 5 of the 7 species (Table 2). Five of these ASVs were the same sequences as conserved OTUs identified in Table 1. A majority (80%) of these conserved sequences had close matches (≥ 96% identity) with previously identified coral-associated bacteria (Table 3).
A Propionibacterium sequence was present in all 7 coral hosts, across 92% of the samples based on an OTU and 63% of the samples based on an ASV (Tables 1 and 2). This finding required some sleuthing, since the sequence appears under two different OTU numbers. In Lo. pertusa, P. placomus, and Anthothela sp., this sequence appears as OTU 4447394, whereas in the 2 Primnoa species, it shows up as OTU 4462014; however, the only difference between the OTUs is two additional bases (GG) at the beginning of OTU 4462014. There is a 100% sequence identity between OTU 4462014 and ASV_50. The Propionibacterium sequences from Primnoa, Paramuricea, and Anthothela have 100% identity across 331 base pairs (bp). The sequence from Lo. pertusa is 99% similar, sharing 217/220 bp with the other coral sequences, but containing 3 single bp insertions.
The second most commonly conserved OTU (245163, Pirellulaceae, found in 84% of samples and identical to a conserved ASV found in 53% of samples) was present in 6 coral species. A Bradyrhizobiaceae sequence (OTU4475561 and ASV_53) was found in 61% and 53% of the samples depending on the pipeline. The other broadly conserved sequences include a number of Pirellulaceae (as OTUs or ASVs), plus orders Campylobacterales, Kiloniellales, and Phycisphaerales; families Enterobacteriaceae, Methylobacteriaceae, and Vibrionaceae; and the genera Acidovorax, Acinetobacter, Bacillus, Lysobacter, Pseudomonas, and Sphingobium (Tables 1 and 2).
Endozoicomonas
There were 7 OTUs and 6 ASVs that were classified as belonging to the family Endozoicomonaceae (Additional files 3 and 4). These rare sequences were typically found in 1–7 samples, with low numbers of reads. The exceptions were OTU 743665 (ASV_56) which was found in 6 Lo. pertusa samples with over 100 reads each for a total of 2574 sequences, and New.ReferenceOTU28 (ASV_45) which was found in 7 Anthothela spp. samples with a total of 3446 sequences. When aligned with known coral-associated Endozoicomonas sequences derived from shallow-water stony and soft corals, Endozoicomonaceae OTU 743665 (Lo. pertusa) grouped with other stony corals and New.ReferenceOTU28 (Anthothela spp.) grouped closely with other soft corals (Fig. 1).

Newick phylogenetic tree visualization of the alignment of Endozoicomonas-like sequences. Sequences are listed by the name of the coral host. Operational taxonomic units from this study are in bold; reference sequences include NCBI accession numbers
Bacterial community composition
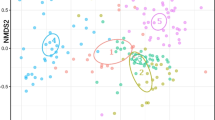
For comparisons of the entire bacterial community composition between the 7 corals, the most informative principal coordinate analysis was that based on the Binary Sorenson Dice distance matrix (Fig. 2). The samples appear to cluster at the level of host genus, with the two Anthothela spp. grouping together and the two Primnoa spp. grouping together. To test the robustness of this pattern, I performed a PERMANOVA across corals using genus as a factor. There were significant differences between the coral genera (PPERMANOVA = 0.0001, pseudo-F = 8.066, 9787 unique permutations). Subsequent pair-wise tests showed all coral genera were significantly different from each other with the exception of pairs that included L. grandiflora, since there is only one replicate of that coral genus and therefore the test did not have sufficient statistical power. Nesting the factor of “species” within “genus,” there were significant differences between Pr. pacifica and Pr. resedaeformis (PPERMANOVA = 0.0005, t = 1.9316, 5645 unique permutations) in spite of their close clustering. Conversely, the Anthothela spp. group was not significantly different: A. grandiflora vs. Anthothela sp. (PPERMANOVA = 0.3426, t = 1.0376, 1808 unique permutations), Anthothela sp. vs. Anthothela ND (PPERMANOVA = 0.3186, t = 1.0464, 35 unique permutations), and A. grandiflora vs. Anthothela ND (PPERMANOVA = 0.0238, t = 1.3249, 455 unique permutations).

Principal coordinate analysis of coral microbiomes based on Binary Sorensen Dice distance matrix
Discussion
Using the most stringent criteria (presence in 100% of samples), previous studies of each of these 7 deep-sea coral species identified from 1 to 48 bacterial taxa that constituted a conserved core [11, 27, 28, 30]. However, in most cases, this was done using rarefied datasets, which could lead to an underestimation if in fact the OTUs were present in the randomly discarded sequences. This meta-analysis across all 7 corals identified 23 highly conserved OTUs based on an unrarefied OTU table and an additional 2 conserved ASVs in addition to ASVs that matched the conserved OTUs. Eighty percent of these sequences (20/25) were highly similar (≥ 96% identity) to sequences previously identified as coral-associated (Table 3).
Conserved vs. contaminants
It is problematic that acquisition of these sequence datasets predated the 2014 publication by Salter et al. [51] that triggered widespread recognition of the need to run kit blank controls to detect possible contamination from DNA extraction kits. Such controls are becoming a common practice and are particularly critical for Illumina datasets which are an order of magnitude deeper than 454 datasets such as the ones compiled in this study. Not having kit controls for these datasets means having to interpret the findings with caution; however, it would be an oversimplification to automatically assume that the presence of certain bacterial groups that have been detected as contaminants in some kits indicates contamination [51, 52]. Even studies that have identified kit contaminants acknowledge that many of the contaminants detected can be indistinguishable from bacteria genuinely present in samples and that arbitrarily discarding low prevalence microbes to “correct” for contamination could hinder the identification of relevant minor components of microbiomes [52]. Bearing this in mind, I have marked the 8 OTUs in Table 1 and 4 ASVs in Table 2 that represent bacterial families/genera also commonly found as kit contaminants. All of these bacterial groups (Acidovorax, Acinetobacter, Bacillus, Bradyrhizobiaceae, Enterobacteriaceae, Propionibacterium, Pseudomonas, Methylobacteriaceae, Sphingobium) have commonly been found in coral microbiomes by recent high-throughput sequencing studies [10, 21, 24, 29, 53,54,55,56,57,58,59,60,61,62]; however, those studies also did not run kit blanks to assess the possible contamination. More convincing are studies that have cultured these groups from corals [23, 63,64,65,66,67,68,69,70,71,72] (e.g., see matching sequences listed in Table 3). The fact that so much of the coral microbiome literature is overshadowed by the uncertainty of whether these associates are real or contaminants makes it imperative to include kit blanks in future sequence surveys, as well as to focus future work on cultivation and confirmative microscopy of these bacterial taxa.
Core bacterial associates
The Propionibacterium OTU was found in 92% of the 51 coral samples and was present across all 7 coral hosts (Table 1). This OTU was included in the 100% conserved core of individual species Anthothela sp., Lo. pertusa, P. placomus, Pr. resedaformis, and Pr. pacifica. Of the 9 Propionibacterium ASVs, the most common was ASV_50 which was 100% identical to the OTU sequence and was found in 63% of the samples (Table 2). The presence of Propionibacterium has previously been noted in the conserved cores (90–100% sample inclusion) of a number of stony corals: Acropora hycinthus, Acropora muricata, Acropora rosaria, Coelastrea aspera, and Porites lutea [16, 54, 56]. Using less stringent requirements to define the conserved core, Propionibacterium has also been described as a rare but consistent endosymbiont in stony corals Acropora granulosa, Leptosiris spp., and Montipora capitata and has been detected at low relative abundance in a number of other stony coral species [29]. Similar sequences were found from a number of coral studies, including one cultured isolate (Table 3). The fact that this Propionibacterium sequence was found in both stony corals (order Scleratinia) and soft corals (order Alcyonacea) raised the question of whether this putative symbiont might also be found in other members of class Anthozoa–like anemones (order Actiniaria) or zoanthids (order Zoantharia). Propionibacterium was a minor member of the core microbiome of the anemone Aiptasia [73] and was present in anemones Actinia equina, Anemonia viridis [74], and Edwardsiella andrillae [75]. Propionibacterium sequences were also detected in the microbiomes of zoanthids Palythoa caribaeorum [55] and Palythoa australiae [76]. Based on the literature, Propionibacterium appears to be conserved not just in corals but across class Anthozoa. However, given the recent finding that Propionibacterium is also a dominant contaminant in extraction kits [52] and most of these studies did not include kit controls, this finding should be confirmed by non-sequence-based methods, including cultivation and microscopy.
A single Betaproteobacteria OTU from the family Comamonadaceae was conserved in almost 70% of the samples. Greengenes [40, 41] identified the sequence as Limnohabitans, a relatively new genus [77], but RDP Classifier [78] identified the OTU as Acidovorax. Aligning the OTU sequence against representatives from both genera revealed that for the 328-bp length of the OTU, there were only 3 bp differences between Limnohabitans and Acidovorax; however, in all cases, OTU 4483490 shared the same base as Acidovorax. Further, OTU 4483490 shared 99% identity over 306 bp with an Acidovorax clone from coral Stephanocoenia intersepta (Table 3; [79]). Acidovorax has been detected in association with tropical stony corals, often as the most abundant OTU [60, 79,80,81] and as a rare associate of a temperate soft coral [21]. Some strains of Acidovorax are capable of heterotrophic denitrification of nitrate, so this bacterium may play a role in coral nitrogen cycling [82].
The possibility of complete nitrogen cycling within corals by bacterial associates has been previously hypothesized based on individual coral core microbiome compositions [11, 28]. A similar hypothesis could be sketched based on several of the conserved OTUs identified in this study. The Bradyrhizobiaceae OTU had over 100 identical matches to nitrogen-fixing Bradyrhizobium strains from root nodules. Methylobacterium sequences have been noted as abundant or core members of other tropical stony coral microbiomes [56, 60] and similar to Bradyrhizobiaceae; some Methylobacteriaceae have been found to form root nodules and act as nitrogen-fixing symbionts [83]. Fixed nitrogen in the form of ammonia could then be converted to nitrate by the Pirellulaceae [84]. From there, nitrate could undergo ammonification by the Campylobacterales [85] or alternately undergo denitrification by Kiloniellales [86] or Acidovorax [82]. This combination of conserved bacterial OTUs could indicate the importance of nitrogen cycling to deep-sea corals.
Gammaproteobacteria are common members of coral microbiomes [2, 87,88,89], and all 7 conserved gammaproteobacterial OTUs were similar to the previously identified coral associates (Table 3). Acinetobacter, Pseudomonas, and Vibro have been hypothesized to play a role in oil degradation to benefit the holobiont [10, 90]. Supporting this idea is the finding that flocculent material coating deep-sea soft corals impacted by an oil spill was dominated by Acinetobacter and Pseudomonas [91]. Acinetobacter, Pseudomonas, and Vibrio have also been implicated in the degradation of dimethylsulfoniopropionate (DMSP)/dimethylsulfide (DMS) [92], suggesting a role in sulfur cycling. Previously, zooxanthellae were credited with the production of DMSP in corals; however, recent research has shown that aposymbiotic corals also are capable of making DMSP, indicating relevance to all corals not just those in the photic zone [93]. Enterobacteriaceae are found in healthy tropical corals [24, 94], and their conserved presence in deep-sea corals which are not directly impacted by sewage suggests that they have a role other than as an indicator of poor water quality [95]. The conservation of specific OTUs from gammaproteobacterial families Enterobacteriaceae, Moraxellaceae, Pseudomonadaceae, Vibrionaceae, and Xanthomonadaceae and their common occurrence in shallow-water corals (Table 3) implies functional importance to the coral holobiont that requires further study.
Three OTUs belong to the order Phycisphaerales (phylum Planctomycetes) and had 99–100% identity with uncultured bacteria collected from deep ocean waters [96, 97]. The family Phycisphaeraceae within this same order was found to be associated with tropical stony corals and more abundant under highly variable environmental conditions [98]. However, no similar sequences to these Phycisphaerales OTUs appear to be associated with tropical corals, possibly suggesting a role specific to cold-water corals.
Endozoicomonas
The rare Endozoicomonaceae sequences (more properly, Hahellaceae [99]) found associated with Lo. pertusa and Anthothela spp. are very similar to the sequences previously found associated with shallow-water corals and are clearly partitioned by whether the host is a stony coral (order Scleractinia) or a soft coral (order Alcyonacea) (Fig. 1). This is supported by a recent study on five Mediterranean soft corals that also found Endozoicomonas-affiliated sequences formed a phylogenetic relationship that mirrored that of the hosts’ systematic classification [5]. A recent genomic comparison of functional capabilities of seven strains of coral-associated Endozoicomonas indicated that stains had differing abilities, and therefore, divergent genotypes are expected to have different specializations [100]. However, the enrichment of particular functional genes across all strains suggested potential roles centering on carbohydrate cycling and amino acid synthesis, with some strains also contributing vitamins, cofactors, and pigments [100].
Endozoicomonas have been found to dominate coral microbiomes in shallow waters, regardless of the host being tropical or temperate, stony or soft [18, 21,22,23, 25, 101]. Is the rarity in deep-sea corals an artifact of methodology or a reflection of environmental restriction? Acropora millepora nearer to natural CO2 seeps (pH range 7.28–8.01) had 50% less Endozoicomonas than corals at a control site (pH range 7.91–8.09) [102]. The Lo. pertusa sites had pH values 7.79–7.86 [13], and the deep-sea soft corals came from sites with pH ranges of 7.94–8.15 [103]. Three of the four cultured type strains of Endozoicomonas that have been isolated from shallow corals (E. montiporae, E. euniceicola, and E. gorgoniicola) all have an optimal growth pH of 8.0 [48, 104], while the fourth, E. acroporae, has an optimum of pH 7.0 [105]. A second environmental factor could be temperature: the cultured type strains have a minimum growth temperature of 15–20 °C and an optimal range of 22–30 °C; the deep-sea coral habitats sampled in this study had temperatures ranging from 5 to 11 °C (Additional file 1). It is possible that separately or in combination, the lower pH and lower temperatures found in deep-sea coral environments limit the growth of coral-associated Endozoicomonas.
Bacterial community composition
The Binary Sorenson Dice index is unweighted and does not take phylogenetic relationships into account, so separation of coral microbiomes into groups based on host genus is driven by the presence/absence of OTUs (Fig. 2). This makes sense since the presence/absence analyses better reflect the importance of rare OTUs, which make up the majority of these coral-associated bacterial communities. The previous finding by Lawler et al. [11] that the two Anthothela spp. microbiomes were not significantly different suggested that conservation of bacterial communities at the host genus rather than species level might be a trend for deep-sea corals. However, in spite of the two Primnoa species clustering together, there were significant differences between their microbiomes (PPERMANOVA = 0.0005, t = 1.9316, 5645 unique permutations). When examined using a weighted Unifrac dissimilarity matrix which takes into account both the abundance of taxa and phylogenetic relationships, Pr. pacifica and Pr. resedaeformis microbiomes separate by species [30]. The two Anthothela species had a very low sequence divergence from each other [11], suggesting the similarity in their microbiomes could be due to how recently the host species diverged, in contrast to the two Primnoa species that inhabit the Atlantic and Pacific Oceans respectively [106]. Alternately, it may be that limitations of molecular markers for discriminating soft coral taxonomy may be complicating the host resolution in Anthothela spp. [107]. Regardless, the host is consistently the dominant driver of deep-sea coral microbiome structure rather than the environment.
Conclusions
-
This meta-analysis of bacterial microbiome datasets from 7 deep-sea corals revealed 23 highly conserved OTUs and an additional 2 unique ASVs of which 80% were highly similar to the sequences previously identified as coral-associated.
-
Many of these sequences are conserved across two orders of corals, suggesting conservation across class Anthozoa and highlighting the bacterial groups for future targeted study.
-
The conserved OTUs included a combination of Bradyrhizobiaceae, Methylobacteriaceae, Pirellulaceae, Campylobacteriales, Kiloniellales, and Acidovorax, which have the functional possibility of a complete nitrogen cycle. This may indicate the importance of bacterial symbionts to nitrogen cycling in deep-sea corals.
-
Unlike the other conserved OTUs, Phycisphaerales have not been detected in tropical corals, possibly suggesting a role specific to cold-water corals.
-
Rare Endozoicomonaceae/Hahellaceae sequences are clearly segregated by whether the host is a stony coral (order Scleractinia) or a soft coral (order Alcyonacea), and the finer clustering pattern reflects the hosts’ phylogeny.
-
It is possible that the rarity of Endozoicomonaceae/Hahellaceae sequences in deep-sea corals is due to the lower pH and lower temperatures found in deep-sea coral environments limiting the growth of coral-associated Endozoicomonas.
-
Deep-sea coral microbiomes are not consistently organized at the level of host genus, but the host is consistently the dominant driver of deep-sea coral microbiome structure rather than the environment.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the NCBI Sequence Read Archive under BioProjects PRJNA296835, PRJNA297333, PRJNA305617, and PRJNA348705.
References
Rohwer F, Breitbart M, Jara J, Azam F, Knowlton N. Diversity of bacteria associated with the Caribbean coral Montastraea franksi. Coral Reefs. 2001;20:85–91.
Rohwer F, Seguritan V, Azam F, Knowlton N. Diversity and distribution of coral-associated bacteria. Mar Ecol Prog Ser. 2002;243:1–10.
Holm JB, Heidelberg KB. Microbiomes of Muricea californica and M. fruticosa: comparative analyses of two co-occurring eastern Pacific octocorals. Front Microbiol. 2016;7:917.
Sunagawa S, Woodley CM, Medina M. Threatened corals provide underexplored microbial habitats. PLoS One. 2010;5(3):e9554.
La Rivière M, Garrabou J, Bally M. Evidence for host specificity among dominant bacterial symbionts in temperate gorgonian corals. Coral Reefs. 2015;34(4):1087–98.
Sharp KH, Pratte ZA, Kerwin AH, Rotjan RD, Stewart FJ. Season, but not symbiont state, drives microbiome structure in the temperate coral Astrangia poculata. Microbiome. 2017;5:120.
Hansson L, Agis M, Maier C, Weinbauer MG. Community composition of bacteria associated with cold-water coral Madrepora oculata: within and between colony variability. Mar Ecol Prog Ser. 2009;397:89–102.
Meistertzheim A-L, Lartaud F, Arnaud-Haond S, Kalenitchenko D, Bessalam M, Le Bris N, Galand PE. Patterns of bacteria-host associations suggest different ecological strategies between two reef building cold-water coral species. Deep-Sea Research Part I-Oceanographic Research Papers. 2016;114:12–22.
Penn K, Wu D, Eisen JA, Ward N. Characterization of bacterial communities associated with deep-sea corals on Gulf of Alaska seamounts. Appl Environ Microbiol. 2006;72(2):1680–3.
Röthig T, Yum LK, Kremb SG, Roik A, Voolstra CR. Microbial community composition of deep-sea corals from Red Sea provides insight into functional adaptation to a unique environment. Sci Rep. 2017;7:44714.
Lawler SN, Kellogg CA, France SC, Clostio RW, Brooke SD, Ross SW. Coral-associated bacterial diversity is conserved across two deep-sea Anthothela species. Front Microbiol. 2016;7:458.
Neulinger SC, Järnegren J, Ludvigsen M, Lochte K, Dullo W-C. Phenotype-specific bacterial communities in the cold-water coral Lophelia pertusa (Scleractinia) and their implications for the coral’s nutrition, health, and distribution. Appl Environ Microbiol. 2008;74(23):7272–85.
Kellogg CA, Lisle JT, Galkiewicz JP. Culture-independent characterization of bacterial communities associated with the cold-water coral Lophelia pertusa in the northeastern Gulf of Mexico. Appl Environ Microbiol. 2009;75(8):2294–303.
Shade A, Handelsman J. Beyond the Venn diagram: the hunt for a core microbiome. Environ Microbiol. 2012;14(1):4–12.
Hernandez-Agreda A, Gates RD, Ainsworth TD. Defining the core microbiome in corals’ microbial soup. Trends Microbiol. 2017;25(2):125–40.
Sweet MJ, Bulling MT. On the importance of the microbiome and pathobiome in coral health and disease. Front Mar Sci. 2017;4:9.
Brener-Raffalli K, Clerissi C, Vidal-Dupiol J, Adjeroud M, Bonhomme F, Pratlong M, Aurelle D, Mitta G, Toulza E. Thermal regime and host clade, rather than geography, drive Symbiodinium and bacterial assemblages in the scleractinian coral Pocillopora damicornis sensu lato. Microbiome. 2018;6:39.
Neave MJ, Rachmawati R, Xun L, Michell CT, Bourne DG, Apprill A, Voolstra CR. Differential specificity between closely related corals and abundant Endozoicomonas endosymbionts across global scales. ISME J. 2017;11:186–200.
van de Water JAJM, Melkonian R, Junca H, Voolstra CR, Reynaud S, Allemand D, Ferrier-Pagès C. Spirochaetes dominate the microbial community associated with the red coral Corallium rubrum on a broad geographic scale. Sci Rep. 2016;6:27277.
van de Water JAJM, Melkonian R, Voolstra CR, Junca H, Beraud E, Allemand D, Ferrier-Pagès C. Comparative assessment of Mediterranean gorgonian-associated microbial communities reveals conserved core and locally variant bacteria. Microb Ecol. 2016;73(2):446–78.
Bayer T, Arif C, Ferrier-Pagès C, Zoccola D, Aranda M, Voolstra CR. Bacteria of the genus Endozoicomonas dominate the microbiome of the Mediterranean gorgonian coral Eunicella cavolini. Mar Ecol Prog Ser. 2013;479:75–84.
Bayer T, Neave MJ, Alsheikh-Hussain A, Aranda M, Yum LK, Mincer T, Hughen K, Apprill A, Voolstra CR. The microbiome of the Red Sea coral Stylophora pistillata is dominated by tissue-associated Endozoicomonas bacteria. Appl Environ Microbiol. 2013;79(15):4759–62.
Correa H, Haltli B, Duque C, Kerr R. Bacterial communities of the gorgonian octocoral Pseudopterogorgia elisabethae. Microb Ecol. 2013;66:972–85.
Morrow KM, Moss AG, Chadwick NE, Liles MR. Bacterial associates of two Caribbean coral species reveal species-specific distribution and geographic variability. Appl Environ Microbiol. 2012;78(18):6438–49.
Speck MD, Donachie SP. Widespread Oceanospirillaceae bacteria in Porites spp. Journal of Marine Biology. 2012;2012:1–7.
Apprill A, Weber LG, Santoro AE. Distinguishing between microbial habitats unravels ecological complexity in coral microbiomes. mSystems. 2016;1(5):e00143–16.
Kellogg CA, Goldsmith DB, Gray MA. Biogeographic comparison of Lophelia-associated bacterial communities in the western Atlantic reveals conserved core microbiome. Front Microbiol. 2017;8:796.
Kellogg CA, Ross SW, Brooke SD. Bacterial community diversity of the deep-sea octocoral Paramuricea placomus. PeerJ. 2016;4:e2529.
Ainsworth TD, Krause L, Bridge T, Torda G, Raina J-B, Zakrzewski M, Gates RD, Padilla-Gamiño JL, Spalding HL, Smith C, et al. The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 2015;9:2261–74.
Goldsmith DB, Kellogg CA, Morrison CL, Gray MA, Stone RP, Waller RG, Brooke SD, Ross SW. Comparison of microbiomes of cold-water corals Primnoa pacifica and Primnoa resedaformis, with possible link between microbiome composition and host genotype. Sci Rep. 2018;8:12383.
Kellogg CA, Lawler SN: Cold-water coral microbiomes (Anthothela spp.) from Baltimore and Norfolk canyons: raw and processed data. In.: U.S. Geologial Survey data release; 2015. https://doi.org/10.5066/F7CZ356K
Kellogg CA. Cold-water coral microbiomes (Paramuricea placomus) from Baltimore Canyon: raw and processed data. In: US Geological Survey data release. St. Petersburg: USGS; 2015. https://doi.org/10.5066/F7HQ3WZZ
Kellogg CA, Goldsmith DB: Cold-water coral microbiomes (Lophelia pertusa) from Gulf of Mexico and Atlantic Ocean: raw data. In.: U.S. Geological Survey data release; 2017. https://doi.org/10.5066/F7M32SXM
Kellogg CA, Goldsmith DB: Cold-water coral microbiomes (Primnoa spp.) from Gulf of Alaska, Baltimore Canyon, and Norfolk Canyon: raw data. In.: U.S. Geological Survey data release; 2017. https://doi.org/10.5066/F7P55KMJ
Baker EJ, Kellogg CA: Comparison of three DNA extraction kits to establish maximum yield and quality of coral-associated microbial DNA. USGS Open-File Report 2014-1066, 14p. http://pubs.usgs.gov/of/2014/1066/
Claesson MJ, Wang Q, O’Sullivan O, Greene-Diniz R, Cole JR, Ross RP, O’Toole PW. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucl Acids Res. 2010;38(22):e200.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Gonzalez Peña A, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–3.
Rideout JR, He Y, Navas-Molina JA, Walters WA, Ursell LK, Gibbons SM, Chase J, McDonald D, Gonzalez A, Robbins-Pianka A, et al. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ. 2014;2:e545.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–72.
McDonald D, Price MN, Goodrich J, Nawrock EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–8.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–1.
Gihring TM, Green SJ, Schadt CW. Massively parallel rRNA gene sequencing exacerbates the potential for biased community diversity comparisons due to variable library sizes. Environ Microbiol. 2012;14(2):285–90.
R Core Team. R: a language and environment for statistical computing. R Foundation for statistical computing. Vienna; 2018.
Pagès H, Aboyoun P, Gentleman R, DebRoy S. Biostrings: efficient manipulation of biological strings. R package version 2480; 2018.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet Journal. 2011;17(1):10–2.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res. 1997;24:4876–82.
Yang CS, Chen MH, Arun AB, Chen CA, Wang JT, Chen WM. Endozoicomonas montiporae sp. nov., isolated from the encrusting pore coral Montipora aequituberculata. Int J Syst Evol Microbiol. 2010;60:1158–62.
Huerta-Cepas J, Serra F, Bork P. ETE 3: reconstruction, analysis and visualization of phylogenomic data. Mol Biol Evol. 2016;33(6):1635–8.
Clarke KR, Gorley RN: PRIMER v7: User manual/tutorial. PRIMER-E 2015, Plymouth:296pp.
Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman NJ, Walker AW. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12(1):87.
Glassing A, Dowd SE, Galandiuk S, Davis B, Chiodini RJ. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathogens. 2016;8:24.
De Castro AP, Dias Araújo S Jr, Reis AMM, Moura RL, Francini-Filho RB, Pappas G Jr, Rodrigues TB, Thompson FL, Krüger RH. Bacterial community associated with healthy and diseased reef coral Mussismilia hispida from Eastern Brazil. Microb Ecol. 2010;59:658–67.
Hester ER, Barott KL, Nulton J, Vermeij MJA, Rohwer FL. Stable and sporadic symbiont communities of coral and algal holobionts. ISME J. 2016;10:1157–69.
Paulino GVB, Broetto L, Pylro VS, Landell MF. Compositional shifts in bacterial communities associated with the coral Palythoa caribaeorum due to anthropogenic effects. Mar Poll Bull. 2017;114(2):1024–30.
Sweet MJ, Brown BE, Dunne RP, Singleton I, Bulling M. Evidence for rapid tide-related shifts in the microbiome of the coral Coelastrea aspera. Coral Reefs. 2017;36(3):815–28.
Williams AD, Brown BE, Putchim L, Sweet MJ. Age-related shifts in bacterial diversity in a reef coral. PLoS One. 2016;10(12):e0144902.
Lee OO, Yang J, Bougouffa S, Wang Y, Batang Z, Tian R, Al-Suwailem A, Qian P-Y. Spatial and species variations in bacterial communities associated with corals from the Red Sea as revealed by pyrosequencing. Appl Environ Microbiol. 2012;78(20):7173–84.
van Bleijswijk JDL, Whalen C, Duineveld GCA, Lavaleye MSS, Witte HJ, Mienis F. Microbial assemblages on a cold-water coral mound at the SE Rockall Bank (NE Atlantic): interactions with hydrography and topography. Biogeosciences. 2015;12(14):4483–96.
Barott KL, Rodriguez-Brito B, Janouškovec J, Marhaver K, Smith JE, Keeling P, Rohwer FL. Microbial diversity associated with four functional groups of benthic reef algae and the reef-building coral Montastraea annularis. Environ Microbiol. 2011;13(5):1192–204.
Glasl B, Bongaerts P, Elisabeth NH, Hoegh-Guldberg O, Herndl GJ, Frade PR. Microbiome variation in corals with distinct depth distribution ranges across a shallow-mesophotic gradient (15-85 m). Coral Reefs. 2017;36(2):447–52.
La Rivière M, Roumagnac M, Garrabou J, Bally M. Transient shifts in bacterial communities associated with the temperate gorgonian Paramuricea clavata in the northwestern Mediterranean Sea. PLoS One. 2013;8(2):e57385.
Brück TB, Brück WM, Santiago-Vázquez LZ, McCarthy PJ, Kerr RG. Diversity of the bacterial communities associated with the azooxanthellate deep water octocorals Leptogorgia minimata, Iciligorgia schrammi, and Swiftia exertia. Mar Biotechnol. 2007;9(5):561–76.
Beleneva IA, Dautova TI, Zhukova NV. Characterization of communities of heterotrophic bacteria associated with healthy and diseased corals in Nha Trang Bay (Vietnam). Microbiol. 2005;74(5):579–87.
Ben-Dov E, Kramarsky-Winter E, Kushmaro A. An in situ method for cultivating microorganisms using a double encapsulation technique. FEMS Microbiol Ecol. 2009;68:363–71.
Chiu JMY, Li S, Li A, Po B, Zhang R, Shin PKS, Qiu J-W. Bacteria associated with skeletal tissue growth anomalies in the coral Platygyra carnosus. FEMS Microbiol Ecol. 2012;79(2):380–91.
Nithyanand P, Manju S, Pandian SK. Phylogenetic characterization of culturable actinomycetes associated with the mucus of the coral Acropora digitifera from Gulf of Mannar. FEMS Microbiol Let. 2011;314:112–8.
Nithyanand P, Pandian SK. Phylogenetic characterization of culturable bacterial diversity associated with the mucus and tissue of the coral Acropora digitifera from the Gulf of Mannar. FEMS Microbiol Ecol. 2009;69:384–94.
Santiago-Vázquez LZ, Brück TB, Brück W, Duque-Alarcón AP, McCarthy PJ, Kerr RG. The diversity of the bacterial communities associated with the azooxanthellate hexacoral Cirrhipathes lutkeni. ISME J. 2007;1:654–9.
Shnit-Orland M, Kushmaro A. Coral mucus-associated bacteria: a possible first line of defense. FEMS Microbiol Ecol. 2009;67:371–80.
Pereira LB, Palermo BRZ, Carlos C, Ottoboni LMM. Diversity and antimicrobial activity of bacteria isolated from different Brazilian coral species. FEMS Microbiol Let. 2017;364(16):fnx164.
Nithyanand P, Thenmozhi R, Rathna J, Pandian SK. Inhibition of Streptococcus pyogenes biofilm formation by coral-associated actinomycetes. Curr Microbiol. 2010;60:454–60.
Röthig T, Costa RM, Simona F, Baumgarten S, Torres AF, Radhakrishnan A, Aranda M, Voolstra CR. Distinct bacterial communities associated with the coral model Aiptasia in aposymbiotic and symbiotic states with Symbiodinium. Front Mar Sci. 2016;3:234.
Muller EM, Fine M, Ritchie KB. The stable microbiome of inter and sub-tidal anemone species under increasing pCO2. Sci Rep. 2016;6:37387.
Murray AE, Rack FR, Zook R, Williams MJM, Higham ML, Broe M, Kaufmann RS, Daly M. Microbiome composition and diversity of the ice-dwelling sea anemone, Edwardsiella andrillae. Integr Comp Biol. 2016;56(4):542–55.
Sun W, Zhang F, He L, Li Z. Pyrosequencing reveals diverse microbial community associated with the zoanthid Palythoa australiae from the South China Sea. Invertebrate Microbiology. 2014;67:942–50.
Hahn MW, Kasalický V, Jezbera J, Brandt U, Jezberová J, Šimek K. Limnohabitans curvus gen. nov., sp. nov., a planktonic bacterium isolated from a freshwater lake. Int J Syst Evol Microbiol. 2010;60:1358–65.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–7.
Sweet M, Burn D, Croquer A, Leary P. Characterization of the bacterial and fungal communities associated with different lesion sizes of dark spot syndrome occurring in the coral Stephanocoenia intersepta. PLoS One. 2013;8(4):e62580.
Carlos C, Torres TT, Ottoboni LMM. Bacterial communities and species-specific associations with the mucus of Brazilian coral species. Sci Rep. 2013;3:1624.
Lins-de-Barros MM, Vieira RP, Cardoso AM, Monteiro VA, Turque AS, Silveira CB, Albano RM, Clementino MM, Martins OB. Archaea, bacteria, and algal plastids associated with the reef-buliding corals Siderastrea stellata and Mussismilia hispida from Búzios, South Atlantic Ocean, Brazil. Microb Ecol. 2010;59:523–32.
Willems A, Gillis M: Genus II: Acidovorax. Bergey’s Manual of Systematic Bacteriology 2005, 2(Part C (one)):696–703.
Jourand P, Renier A, Rapior S, Miana de Faria S, Prin Y, Galiana A, Giraud E, Dreyfus B. Role of methylotrophy during symbiosis between Methylobacterium nodulans and Crotalaria podocarpa. MPMI. 2005;18(10):1061–8.
Mohamed NM, Saito K, Tal Y, Hill RT. Diversity of aerobic and anaerobic ammonia-oxidizing bacteria in marine sponges. ISME J. 2010;4:38–48.
Tiedje JM. Ecology of denitrification and dissimilatory nitrate reduction to ammonium. In: Zehnder AJB, editor. Environmental microbiology of anaerobes. New York: Wiley; 1988. p. 179–244.
Imhoff JF, Wiese J. The order Kiloniellales. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson CC, editors. The prokaryotes. Berlin: Springer; 2014. p. 301–6.
Wegley L, Edwards R, Rodriguez-Brito B, Liu H, Rohwer F. Metagenomic analysis of the microbial community associated with the coral Porites astreoides. Environ Microbiol. 2007;9(11):2707–19.
Bourne DG, Dennis PG, Uthicke S, Soo RM, Tyson GW, Webster N. Coral reef invertebrate microbiomes correlate with the presence of photosymbionts. ISME J. 2013;7:1452–8.
Ransome E, Rowley SJ, Thomas S, Tait K, Munn CB. Disturbance to conserved bacterial communities in the cold-water gorgonian coral Eunicella verrucosa. FEMS Microbiol Ecol. 2014;90:404–16.
Al-Dahash LM, Mahmoud HM. Harboring oil-degrading bacteria: a potential mechanism of adaptation and survival in corals inhabiting oil-contaminated reefs. Mar Poll Bull. 2013;72:364–74.
Simister RL, Antzis EW, White HK. Examining the diversity of microbes in a deep-sea coral community impacted by the Deepwater Horizon oil spill. Deep-Sea Res II. 2015; https://doi.org/10.1016/j.dsr2.2015.01.010i.
Raina J-B, Dinsdale EA, Willis BL, Bourne DG. Do the organic sulfur compounds DMSP and DMS drive coral microbial associations? Trends Microbiol. 2010;18(3):101–8.
Raina J-B, Tapiolas DM, Forêt S, Lutz A, Abrego D, Ceh J, Seneca FO, Clode PL, Bourne DG, Willis BL, et al. DMSP biosynthesis by an animal and its role in coral thermal stress response. Nature. 2013;502:677–80.
Roder C, Arif C, Bayer T, Aranda M, Daniels C, Shibl A, Chavanich S, Voolstra CR. Bacterial profiling of white plague disease in a comparative coral species framework. ISME J. 2014;8:31–9.
Haas AF, Fairoz MFM, Kelly LW, Nelson CE, Dinsdale EA, Edwards RA, Giles S, Hatay M, Hisakawa N, Knowles B, et al. Global microbialization of coral reefs. Nat Microbiol. 2016;1:16042.
Eloe E, Shulse CN, Fadrosh DW, Williamson SJ, Allen EE, Bartlett DH. Compositional differences in particle-associated and free-living microbial assemblages from an extreme deep-ocean environment. Environ Microbiol Rep. 2011;3(4):449–58.
Allers E, Wright JJ, Konwar KM, Howes CG, Beneze E, Hallam SJ, Sullivan MB. Diversity and population structure of marine group a bacteria in the northeast subarctic Pacific Ocean. ISME J. 2013;7(2):256–68.
Ziegler M, Seneca FO, Yum LK, Palumbi SR, Voolstra CR. Bacterial community dynamics are linked to patterns of coral heat tolerance. Nat Commun. 2017;8:14213.
Neave MJ, Apprill A, Ferrier-Pagès C, Voolstra CR. Diversity and function of prevalent symbiotic marine bacteria in the genus Endozoicomonas. Appl Microbiol Biotechnol. 2016;100(19):8315–24.
Neave MJ, Michell CT, Apprill A, Voolstra CR. Endozoicomonas genomes reveal functional adaptation and plasticity in bacterial strains symbiotically associated with diverse marine hosts. Sci Rep. 2017;7:40579.
Vezzulli L, Pezzati E, Huete-Stauffer C, Pruzzo C, Cerrano C. 16SrDNA pyrosequencing of the Mediterranean gorgonian Paramuricea clavata reveals a link among alterations in bacterial holobiont members, anthropogenic influence and disease outbreaks. PLoS One. 2013;8(6):e67745.
Morrow KM, Bourne DG, Humphrey C, Botté ES, Laffy P, Zaneveld J, Uthicke S, Fabricius KE, Webster NS. Natural volcanic CO2 seeps reveal future trajectories for host-microbial associations in corals and sponges. ISME J. 2015;9:894–908.
Brooke SD, Watts MW, Heil AD, Rhode M, Mienis F, Duineveld GCA, Davies AJ, Ross SW. Distributions and habitat associations of deep-water corals in Norfolk and Baltimore canyons, Mid-Atlantic Bight, USA. Deep-Sea Res II. 2017;137:131–47.
Pike RE, Haltli B, Kerr RG. Description of Endozoicomonas euniceicola sp. nov. and Endozoicomonas gorgoniicola sp. nov., bacteria isolated from the octocorals Eunicea fusca and Plexaura sp., and an emended description of the genus Endozoicomonas. Int J Syst Evol Microbiol. 2013;63:4294–302.
Sheu S-Y, Lin K-R, M-y H, Sheu D-S, Tang S-L, Chen W-M. Endozoicomonas acroporae sp. nov., isolated from Acropora coral. Int J Syst Evol Microbiol. 2017;67:3791–7.
Cairns SD, Bayer FM. A review of the genus Primnoa (Octocorallia: Gorgonacea: Primnoidae), with the description of two new species. Bull Mar Sci. 2005;77(2):225–56.
McFadden CS, Benayahu Y, Pante E, Thoma JN, Nevarez PA, France SC. Limitations of mitochondrial gene barcoding in Octocorallia. Mol Ecol Resour. 2011;11:19–31.
Acknowledgements
Any use of trade names is for descriptive purposes only and does not imply endorsement by the US Government.
Funding
Funding for this project was provided by the US Geological Survey’s Ecosystems Mission Area, Environments Program, through the Outer Continental Shelf study in support of Bureau of Ocean Energy Management research needs.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declares that she has no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional Files
Additional file 1:
Coral samples and corresponding environmental data. Table listing the 66 coral samples, their full sample ID, collection location, depth, latitude, longitude, water temperature, and salinity. (PDF 106 kb)
Additional file 2:
Workflow with commented scripts and parameters. System information, scripts, and comments for each step in the QIIME analysis to produce OTUs and the DADA2 analysis to produce ASVs. (TXT 52 kb)
Additional file 3:
Non-rarefied OTU table. List of OTUs organized by sample and coral host. Includes OTU abundance per sample, reference sequence for each OTU, and the taxonomy. (XLSX 985 kb)
Additional file 4:
Filtered ASV table. List of ASVs organized by sample and coral host, after the removal of unclassified sequences. Includes ASV abundance per sample, reference sequence for each ASV, and the taxonomy. (XLSX 1270 kb)
Additional file 5:
Summary statistics for sequencing 16S rRNA genes from 51 coral samples. Numbers of sequences per sample, and post-rarefaction to 4287; the calculated number of OTUs per sample; and diversity metrics Chao1, Shannon, Simpson Evenness, and Inverse Simpson. (PDF 95 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kellogg, C.A. Microbiomes of stony and soft deep-sea corals share rare core bacteria. Microbiome 7, 90 (2019). https://doi.org/10.1186/s40168-019-0697-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-019-0697-3