
Abstract
Circular RNAs (circRNAs) are a class of covalently closed, endogenous ncRNAs. Most circRNAs are derived from exonic or intronic sequences by precursor RNA back-splicing. Advanced high-throughput RNA sequencing and experimental technologies have enabled the extensive identification and characterization of circRNAs, such as novel types of biogenesis, tissue-specific and cell-specific expression patterns, epigenetic regulation, translation potential, localization and metabolism. Increasing evidence has revealed that circRNAs participate in diverse cellular processes, and their dysregulation is involved in the pathogenesis of various diseases, particularly cancer. In this review, we systematically discuss the characterization of circRNAs, databases, challenges for circRNA discovery, new insight into strategies used in circRNA studies and biomedical applications. Although recent studies have advanced the understanding of circRNAs, advanced knowledge and approaches for circRNA annotation, functional characterization and biomedical applications are continuously needed to provide new insights into circRNAs. The emergence of circRNA-based protein translation strategy will be a promising direction in the field of biomedicine.
Similar content being viewed by others
Background
CircRNA was originally regarded as incorrect RNA cleavage products in viroids [1]. With the development of high-throughput sequencing technologies, an increasing number of circRNAs have been discovered and have received much attention [2, 3]. Unlike other well-known classes of linear RNAs, such as messenger RNA (mRNA), long noncoding RNA (lncRNA), small nucleolar RNA (snoRNA), microRNA (miRNA), etc., circular RNAs are covalently closed single-stranded RNAs (ssRNAs) that have recently become a widespread class of RNA species [3,4,5,6,7,8]. Although there is still a challenge to identify and annotate novel emerging circRNAs, advances in bioinformatics algorithms, detection methods, and molecular biological techniques have provided new opportunities to accelerate the understanding of circRNAs.
In recent years, several key characterizations of circRNAs have been identified [5, 9]. Although a few circRNAs were first identified during intron self-splicing from ribosomal RNAs, mitochondrial RNAs, and tRNAs, most annotated circRNAs are generated from pre-mRNA back-splicing [4, 5, 10, 11]. In this uncommon pre-mRNA splicing, a downstream 5′ splice site is joined to an upstream 3′ splice site to form circular RNAs with a 3′,5′-phosphodiester bond at the back-splicing junction site (BSJ) [4]. Many regulators have been revealed to improve circRNA biogenesis, including intronic complementary sequences (ICSs) in flanking introns of circle-forming exons, Alu elements and RNA-binding proteins (RBPs) [4, 10, 12,13,14]. Due to the lower efficiency of back-splicing than that of canonical splicing, the examined cells and tissues usually showed a generally low abundance of circRNAs. Once produced, the unique covalently closed conformation of circRNAs endows them with considerable stability and more resistance to RNase R than linear RNAs [15], which enables them to regulate cellular processes with a small number of molecules. Interestingly, there are some insights into circRNA clearance, including circRNA degradation by RNase H1 in circRNA:DNA hybrids [16, 17], endonuclease RNase L during innate immune response activation [18], and the RNaseP/MRP complex in m6A modification [19]. circRNA levels can also be reduced in cancer cells with a rapid proliferation rate [9, 20].
In the past few years, circRNAs have been regarded as competing endogenous RNAs that sponge miRNAs that silence their target genes [4, 10, 21, 22]. Recent studies have revealed that circRNAs perform cellular functions via several novel regulatory mechanisms, including circRNA-RBP [23], circRNA:DNA hybrids [16, 17], m6A modification [19, 24,25,26], guiding A-to-I editing [27, 28], and translation potential [29,30,31,32]. These features illustrated that circRNAs may comprehensively play important roles in pathological and physiological processes. Increasing evidence indicates that circRNAs are closely associated with proliferation, metastasis, DNA damage, drug resistance and other life activities of cancer cells [20, 33,34,35].
Given that circRNAs have structural stability advantages and that the negative effect of intron-derived circRNAs on triggering the immune response is smaller than that of other RNAs, the development of RNA drugs based on circRNAs has important application prospects [5, 9, 36]. circRNAs can be relatively stable in biological fluids and may serve as good biomarkers for early diagnosis and prognosis [36, 37]. Several tissue-specific circRNAs have been suggested to be used as targets for cancer treatment, even in therapy resistance and targeted drug development [38,39,40,41,42,43]. Of note, RNA circle-based translation technologies have emerged as a promising strategy in biomedicine [9, 30, 44, 45]. For example, the circRNA-RBD-Delta vaccine was designed to resist the COVID-19 pandemic [44].
In this review, we collected the recent progress in the biogenesis, degradation and biology of circRNAs and describe novel technologies for the identification, accurate quantification, and functional characterization of circRNAs. Based upon our findings, we also discuss the current challenges of circRNA analysis and new insight into strategies to determine circRNA functions and the biomedical implications of circRNA.
Characterization of circRNAs
Biogenesis of circRNAs
In general, circular RNA is usually derived from back-splicing of pre-mRNA to form a closed RNA transcript [3, 5, 10, 11]. Additionally, circular RNA can intermediately originate from small nuclear RNAs (snRNAs), mitochondrial RNAs, ribosomal RNAs (rRNAs), and transfer RNAs (tRNAs) during intron self-splicing [5, 42, 46,47,48]. Advancing RNA sequencing (RNA-seq) technologies and computational pipelines for circular RNA annotation, recent studies have found that circRNAs can be derived from exons, introns, 5' untranslated regions (UTRs), 3' UTRs or antisense sequences and can be classified into four main categories, intronic circRNAs (ciRNAs), exon‒intron circRNAs (EIciRNAs), exonic circRNAs (ecircRNAs), and others, detected in a variety of organisms, including viruses, archaea, plants, parasites, and most mammals [4, 5, 10, 11, 49, 50] (Fig. 1a). Evidence has shown that back-splicing of pre-mRNA is the predominant process for circRNA generation [3, 50]. In this back-splicing process of pre-mRNA, a splice donor that is downstream of the 5’ splice site is joined to a splice acceptor that is upstream of the 3’ splice site, producing a circular format with a 3’-5’ phosphodiester bond at the back-splicing junction site (BSJ) [3]. In addition, RBPs, special sequences of introns, etc., may assist in the production of circRNA [3, 12, 15]. Circularized RBPs can shorten the distance between the upstream and downstream of the circular exon by connecting related intron sequences, promote splicing, and induce the formation of circular RNA [11, 23, 51]. If the intron has a unique inverted repeat sequence (such as Alu) [12, 52], after base pairing occurs, the splicing donor is brought close to the splicing acceptor, which promotes nucleophilic attack and splicing and can also promote the production of circRNA. However, the biochemical environment and regulatory factors required for the occurrence of circRNA are not yet clear. It is still worth noting that one gene can generate different circRNAs, which can be affected by the competition of RNA pairing across the flanking introns [3, 11].

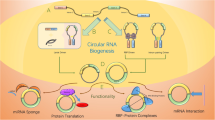
Characterization of circRNAs. a Biogenesis of circRNAs. circRNAs are produced from back-splicing or intron self-splicing of precursor RNAs. Exons, introns, 5′untranslated regions (UTR), 3′UTR or antisense sequences, can generate circRNAs, which include ciRNAs, EIciRNAs, ecircRNAs, and others. CircRNAs regulate biological process via various regulation mechanism, including circRNA-miRNA sponge (b), circRNA-protein interaction (c), circRNA immunity (d), circR-loop (e), guiding A-to-I editing (f), and translation (g). h, i Degradation mechanism of circRNA. h RNase H1 degrades a subgroup of ciRNAs in R-loops. i circRNAs are degraded by RNase L in PKR activated innate immunity. j Recruitment of endonucleases by m6A marks to degrade circRNAs
Function mechanisms of circRNAs
To date, studies using the application of emerging approaches have elucidated various regulatory mechanisms of circRNAs, which highlight many aspects of gene expression, DNA damage, RNA editing and immunity. We will focus on the representative epigenetic regulation of circRNAs (Fig. 1b–g), including circRNA-miRNA sponges, circRNA:DNA hybrids (circR-loops), guiding A-to-I editing, circRNA-protein interactions, and translation [16, 22, 27,28,29, 32].
A majority of studies have shown that circRNA can act as miRNA sponges in a manner similar to that of mRNA [22]. Circular RNA exists in the cytoplasm and has multiple miRNA binding sites. It can sponge miRNA to inhibit the regulatory function of miRNA. For example, miR-7 [53, 54] has been identified as a tumor-inducing factor or tumor suppressor in the process of tumorigenesis. Circular RNA (ciRS-7; also known as CDR1as) can specifically sponge miR-7, thereby inhibiting the function of miR-7 and upregulating the expression of IRS2, EGFR and other related genes [22, 55] (Fig. 1b). Another well-known epigenetic regulatory mechanism of circRNAs is their interaction with RNA-binding proteins [23, 56] (Fig. 1c). circRNA interactions with RBPs could function as protein antagonists or as inhibitors of protein activity [10, 57, 58]. For example, circ-Foxo3 interacts with cell cycle-related proteins (including p21 and p27), thereby blocking the role of these proteins in the cancer cell cycle [57]. CircPABPN1 binds to HuR, suppresses the interaction of HuR with PABPN1 mRNA and reduces its translation [58]. Besides, endogenous circRNAs tend to form 16–26 bp duplexes and interact with double-stranded RNA (dsRNA)-activated protein kinase (PKR), which blocks innate immunity [18, 40] (Fig. 1d). CircRNAs have an extensive ability to regulate cellular processes, which may explain the epigenetic differences between cells in the same organism.
In recent years, some emerging epigenetic regulatory mechanisms of circRNAs have been illuminated. DNA: RNA immunoprecipitation sequencing (DRIP-seq) data have also shown that circRNAs frequently form R-loop structures and tend to regulate DNA damage and genome instability [16, 59] (Fig. 1e). Some circRNAs can act as stable antisense RNAs to bind with RNAs to modulate RNA stability, structure, and activity [27, 60, 61] (Fig. 1f). For example, artificial antisense sequences in a circular RNA backbone can significantly reduce the proliferation of the SARS-CoV-2 virus [60]. Circular guide (g)RNAs were engineered to execute A-to-I editing on mRNAs by recruiting endogenous ADARs, which may realize the aim of treatment without disturbing genes [27].
Translation potential of circRNAs
As mentioned above, circRNAs are a class of noncoding RNAs, but recent scientific research has shown that some circRNAs also have certain coding capabilities [32]. The 5' cap and 3' poly(A) tail are necessary structures for the linear translation of mRNA [25]. Unlike ordinary mRNA, circRNA lacks a similar translational molecular structure, but it can utilize the N6-adenosine methylation (m6A) modification or internal ribosome entry site (IRES) translation to promote the direct binding of the initiation factors to the cyclic RNA [25, 32, 62,63,64] (Fig. 1g). The translation of linear mRNA is initiated by the elF4E complex [65, 66]. First, elF4F binds to the 5' cap end of the mRNA, and then elF4G serves as a protein binding scaffold to assemble the initiation complex [66]. Then, the combination of elF3 and elF4G recruits ribosomes to the mRNA and initiates translation [66]. For circRNA, a special eIF4G protein (eIF4G2) directly recognizes IRES and initiates eIF4 complex assembly without eIF4E in a 5' cap-independent manner, providing circRNA with translation ability [29, 67]. m6A modification can also regulate the protein-coding potential of circRNAs [25, 68, 69]. For example, a high m6A methylation level was found in circZNF609, which promotes internal ribosome entry site (IRES)-activated protein coding [25, 68]. Yang et al. also examined the coding landscape of the human transcriptome and found that many circRNAs contain m6A motifs with translational potential and that high m6A levels in circRNAs have the ability to improve the efficiency of translation [25]. Interestingly, according to mass spectrometry, 50% of translatable endogenous circRNAs undergo rolling ring translation [32, 63, 67]. Given that circRNA lacks the general translational elements, a large number of products translated from circular RNAs are short in length and lower efficiency than that from mRNAs. Moreover, there are still issues that need to be further answered, such as which factors regulate the translation of circRNA, and what is the relationship between the translation product of circRNA and that of its corresponding linear transcript?
CircRNA degradation
Due to the special structural characteristics of circRNA, it cannot be degraded by RNase H, which is conventionally used to eliminate linear RNA [15]. The specific degradation mechanism of circRNA is currently unclear. Several studies have found that miRNA can regulate the degradation of circRNA [22, 70]. For example, CDR1as can be degraded via sponging by miR-671 through Argonaute 2 (Ago2)-mediated degradation [55]. Circular intronic RNAs (ciRNAs) escape from DBR1 debranching of intron lariats and are cotranscriptionally produced from pre-mRNA splicing, but their turnover and mechanism of action have remained elusive [59]. Li et al. reported that RNase H1 degrades a subgroup of circular intronic RNAs (ciRNAs), which have high GC% and often form R-loops [16, 59] (Fig. 1h). For example, ci-ankrd52 facilitates R-loop formation, a process that allows the release of ankrd52 pre-mRNA from R-loops by ci-ankrd52 replacement and subsequent ciRNA removal via RNase H1-mediated degradation [59]. This RNase H1/R-loop-dependent ciRNA degradation likely limits ciRNA accumulation and resolves R-loops at some GC-rich ciRNA-producing loci. In the autoimmune disease systemic lupus erythematosus (SLE), endogenous circRNAs bind to PKR via forming 16–26 bp imperfect RNA duplexes [18]. Upon viral infection, PKR is activated by phosphorylation in early cellular innate immune responses, resulting in the release of circRNAs and global degradation by RNase L [18] (Fig. 1i). This study suggests that the structure of circRNAs is important in innate immunity and its degradation. Studies have also found that m6A RNA modification can promote the recruitment of endonucleases to degrade circRNA [9, 19] (Fig. 1j).
In addition to intracellular degradation, circRNA can also be transported out of the cell in the form of exosomes and into body fluids [36, 71, 72]. However, the reason why cells form exosomes is still unclear. Is it merely a tool for the exchange of information between cells? Alternatively, it may reduce the toxicity caused by excessive accumulation of circRNA in the cell and actively transport circRNA out of the cell. The degradation of exosomes may release the circRNA outside; but there is no conclusive mechanism yet [73]. Although there are some endeavors to understand the mechanism of circRNA decay in certain contexts, further studies are still needed to fully understand the common circRNA degradation mechanisms under different physiological conditions.
Principles and challenges for circRNA discovery and annotation
CircRNA constitutes a large amount of cell contents of unknown function [5, 9]. Accurate identification and annotation of novel emerging circRNAs are still urgently needed in this rapidly expanding research field. Recent advances in high-throughput RNA sequencing and related bioinformatics tools have accelerated research (Table 1). Since 2012, increasing numbers of bioinformatics tools have been developed to discover and annotate circRNAs. In 2013, find-circ became the first publicly available pipeline for identifying circRNAs from sequencing data [49]. Even today, many explorations of circRNAs still commence with RNA-seq data [74,75,76,77]. While RNase R-treated sequencing is considered easier and more accurate for circRNA detection, most circRNA detection tools can identify back-splice junction (BSJ) reads with high confidence from conventional RNA-seq datasets [2, 49, 78]. Nevertheless, achieving both sensitivity and specificity in circRNA discovery remains a challenge, particularly in the context of identifying and annotating novel emerging circRNAs.
Canonical BSJ-based circRNA identification
Many tools identify circRNAs by searching for specific BSJ sequences and performing different kinds of mapping (Fig. 2a). Most of the algorithms embedded in the tool are based on the segmentation of reads, while some other tools are based on predefined BSJ and circRNA flanking sequences. Examples include Find-circ [49], CIRI [79], CIRIexplorer [12, 13], Ularcirc [80], and circRNA-finder [81]. They all have their own merits or characteristics. Find_circ was the first circRNA prediction tool using the identification of back-spliced sequencing reads in RNA-Seq. CIRI, CIRI2 and CIRCexplorer2 [13, 79, 82] all scan through sequence data first to identify junction reads in backspliced exons, intron lariats, and alternative splicing sites and then implements multiple filtration strategies to remove false-positives. Other identification of BSJ reads is based on splicing, such as MapSplice [74] and segemehl [83]. MapSplice improves the quality and diversity of read alignments of a given splice to increase accuracy and can be used for both short (< 75 bp) and long reads (≥ 75 bp) to detect novel canonical as well as noncanonical splices [74].

Workflow of BSJ-based circRNA identification. a The canonical workflow of circRNA identification tools that search for the specific BSJ sequences in the sequencing data and map to genome. b circRNA enriched by rRNA deleted and RNase R treatment before library. c circRNAs fragment into short reads and BSJ detecting in RNA sequencing. d After enrichment, the circRNA pool was nicked to generate large fragments, and then the obtained circRNA pool aligns with the ONT long-read sequencing protocol. e Discovery tools identify the full-length circRNA isoforms using rolling circle amplification followed by nanopore long-read sequencing
Although circRNA library preparation of RNA-seq by rRNA deletion and RNase R treatment followed by many circRNA identification tools is a better method, there exist some RNase R-sensitive circRNAs, such as circ_CDR1as, which leads to the problem that these RNase R-sensitive circRNAs will be missing when only using RNase R-treated library preparation-based tools [15, 49, 82] (Fig. 2b, c). To improve circRNA identification efficiency and reduce the false-positive rate, some researchers integrate current prediction algorithms to make an ensemble tool (Table 1). For example, RAISE [84], CircRNAwrap [85], and PcircRNA_finder [75] that was used in the study of plants. Different integrated identification pipelines satisfy the different research purposes for users. Recently, Gaffo et al. developed CirComPara2 [86], which has been set to simultaneously use seven circRNA detection methods (integrated C2BW, C2SE, C2ST, C2TH, CIRI2 [82], DCC [78] and find_circ [49]) and identify the real circRNAs shared between at least two of these methods. The new trends of circRNA detection development are integrating variable tools because they can outperform single state-of-the-art circRNA identification tools and consistently achieve high recall rates without losing precision.
Fusion circRNA identification
Previous studies have shown that fusion genes can transcribe into not only linear but also chimeric fusion circular RNAs (f-circRNAs), which are functional in gene expression regulation and implicated in malignant transformation [87,88,89,90]. Currently, even though it remains a challenge to identify fusion circRNAs owing to their general sparsity, low abundance in cells, heavy background noise in RNA-seq and perhaps imperfect computational methods, researchers have endeavored to develop bioinformatics approaches to systematically identify fusion transcripts, specifically detecting f-circRNAs in cancer cells (Table 1). ACFS has the ability to detect fusion events and recognize f-circRNAs from RNA-Seq data accurately [91]. However, f-circRNA detectors may suffer from a high false-positive rate and a significant increase in the computational burden owing to the detection algorithm performance. Identification of f-circRNAs requires detection of the BSJ site within the gene fusion events. STAR Chimeric Post (STARChip) is an open-source software based on the STAR aligner that can simplify filter high-quality chimeric alignments and improve f-circRNA identification to annotate f-circRNA in a rapid, efficient and scalable manner [89]. Cai et al. developed a comprehensive Python-based workflow called “Fcirc” to identify linear and circular RNA transcripts from known fusion events in RNA-Seq datasets [92]. It requires already known gene fusions as a reference to build the bipartite graph of gene pairs, which is different from fusion detection tools such as ChimeraScan [88], FusionCatcher [93], JAFFA [94], TrinityFusion [95] and STAR-Fusion [95]. Therefore, Fcirc can detect f-circRNAs from known fusion events with higher specificity, a lower false-positive rate and shorter computing times [92]. Usefully, Fcirc is an open-friendly comprehensive pipeline that can allow users to add their own fusion gene pairs of interest at their convenience and regularly update newly emerging fusion genes from common multiple databases (COSMIC, FusionCancer, ChimerDB, FARE-CAFE, and TicDB) [93, 96,97,98,99].
circRNA identification using long-read sequencing data
The circRNA discovery tools above are mostly compatible with the reads of next-generation RNA-seq [2, 100]. Due to the short reads in RNA-seq, these alignment-based algorithms have difficulty distinguishing circular reads from the exonic regions that overlap the corresponding linear transcripts. In recent years, with emerging long-read sequencing technologies, including PacBio and Oxford Nanopore, reconstruction of transcript isoforms has become much easier [101,102,103,104]. Thus, the application of long-read sequencing technologies will lead to a novel generation of circRNA discovery tools that have the ability to achieve high-throughput detection of full-length circRNAs and improve sensitivity and specificity. circNick-LRS [103] (Fig. 2d) is the first reliable method to use long-read nanopore sequencing to detect circRNAs in both humans and mice.
Of note, due to circNick-LRS and circPanel-LRS eliminating the need for prior circRNA enrichment, a large number of nonconical splicing events in the global genome have been found to produce various types of circRNAs, including novel exons, intron retention and microexons. Both circFL-seq [105] and isoCirc [104] identify full-length circRNA isoforms using rolling circle amplification followed by nanopore long-read sequencing (Fig. 2e). Significantly, the low abundance circRNA reads could be enriched and identified using rolling circles and long-read sequencing. Zhao’s team developed an algorithm called the circRNA identifier using long-read sequencing data (CIRI-long) (Fig. 2e) to reconstruct the sequence of circRNAs [102, 106]. CIRI-long not only enables unbiased reconstruction of full-length circRNA sequences but also identifies mitochondria-derived circRNAs, transcriptional read-through circRNAs, and noncanonical AG/GT splicing circRNAs, which other methods to detect. Interestingly, CIRI-long identified a novel type of intronic self-ligated circRNA with a different incompletely characterized internal GT/AG splice signal rather than the flanking AG/GT signal in most exonic and intronic-exonic circRNAs [102]. With the development of sequencing technology, circRNA discovery tools provide insights into circRNA complexity that will further advance this rapidly expanding research field.
circRNA identification using machine learning
Because the above methods always require RNA-seq data as input, circRNA signals with low abundance are usually missed [78, 100]. It is necessary for us to develop a novel tool to identify circRNAs at low levels. Machine learning algorithms establish some mapping rules based on the knowledge and characteristics of the real known circRNAs (Table 1). For example, PredcircRNA [76] and StackCirRNAPred [107] predict whether an unknown RNA sequence possibly comes from circRNA by some common reliable features, such as ALU repeats, structural motifs and sequence motifs [15, 76]. Other machine learning circRNA prediction tools based on the characteristics of nucleotide sequences are PredicircRNATool [108], DeepCirCode [77], CirRNAPL [109], PCirc [110], circDeep [111], etc. CirRNAPL is a user-friendly web server that extracts the structural features and pseudo-ribonucleic acid composition of circRNA to optimize the extreme learning machine based on the particle swarm optimization algorithm, which achieves identification accuracy in three public datasets [109]. Further improvements in the sensitivity and specificity of classifying circRNA from other lncRNAs can be found in circDeep, which is an end-to-end deep learning framework [111]. Considering the growing number of circRNA sequences and their splicing complexity, advanced parallel technology is highly recommended in circRNA discovery.
Database for circRNA annotation and functional study
With the development of bioinformatic tools for circRNAs, an increasing number of public circRNA databases have emerged [20, 100, 112,113,114]. The most well-known and comprehensive database is circBase, which encompasses over 90,000 circRNAs along with their genomic coordinates, strands, annotations, and other relevant information [113]. These circRNA databases have become widely utilized in annotation pipelines, facilitating the research and analysis of circRNAs [100, 113]. Furthermore, several databases have been developed to gather diverse attributes of circRNAs beyond basic sequence information, offering unique features for research purposes [2, 64, 100, 115]. Notably, riboCIRC and TransCirc are comprehensive databases that specifically focus on potential translatable circRNAs [64, 116]. They provide predictions of circRNA-derived open reading frames (cORFs) and annotations of cORF-encoded peptides, supported by evidence of translation.
In recent years, the clinical significance of circRNAs has gained substantial attention, with increasing evidence showing their potential as clinical biomarkers and therapeutic targets [67, 114, 117, 118]. Specialized databases such as MiOncoCirc focus on providing information on the association between circRNAs and cancer [20]. Lnc2Cancer 3.0 has been updated to include circRNA-cancer associations and presents information on regulatory mechanisms, biological functions, and clinical applications of circRNAs in cancer [115]. Another comprehensive database, CircR2Disease v2.0 [119], provides experimentally validated relationships between circRNAs and various diseases. ExoRBase 2.0 concentrates on RNAs found in extracellular vesicles, encompassing circRNAs [120]. This database sheds light on the alterations of circRNAs in extracellular vesicles under both physiological and pathological conditions. At the same time, functional circRNA has emerged as a prominent research focus within the field of noncoding RNA. Several databases, including CircFunBase [112], deepBase [121], and circBank [122], provide valuable information on the interactions of circRNAs with various types of RNAs and proteins.
Despite progress in circRNA detection and annotation, the lack of standardized naming conventions remains a pressing issue in this field. The diverse naming methods used across different databases and articles have created a significant barrier for research, leading to information duplication and errors. Some databases use a 'circ_' prefix followed by a numeric ID or the parental gene symbol to name circRNAs [49, 113]. However, this inconsistent and arbitrary naming approach hampers the establishment of an integrated circRNA database. To address this issue, Chen et al. proposed a clear naming system for circRNAs. According to this system, a new circRNA can be named 'circ + ' followed by the parental gene name (separated by '::' in the case of fusion genes), the number of its exon, and 'RI' if it remains in an intron or 'S' if it exhibits different internal splicing patterns [50]. We strongly encourage researchers to embrace these clear naming rules to promote consistency and facilitate data integration.
New insight into strategies to determine circRNA functions
Several methods have been developed to study the functions of circRNAs [9, 46]. We systematically summarized current strategies used to explore circRNAs, including ceRNA prediction [22], knockdown or out of functional circRNAs, overexpression of functional circRNAs [123,124,125,126,127,128,129,130,131], and circRNA-RBP prediction [132]. The advantages and disadvantages of these methods have also been discussed. Some new insights may help improve the strategies of circRNA research and applications of therapeutic potential.
Strategies for circRNA detection
CircRNA sequencing of rRNA-depleted and RNase R-treated cells is the method used to discover novel circRNAs and was also used in all early circRNA profiling studies [20, 82, 133]. Based on the BSJ feature of circRNAs, candidate circRNAs were further identified and quantified. In recent years, many common detection techniques for various types of RNAs have also been applied in circRNA studies [78, 85, 105, 134]. Due to the lack of clarity regarding circRNA production or splicing, these detection methods have specific advantages and disadvantages (Fig. 3).

Strategies for circRNA detection. a Northern blot to identify and quantify circRNAs with a BSJ-spanning probe. b Quantification of circRNAs by reverse transcription (RT) and quantitative PCR (qPCR) assays. A pair of divergent primers are used. c Droplet digital PCR (DdPCR) is a novel technology determines the absolute quantification of a candidate circRNA using the ratio of positive to negative droplets. d NanoString Technology captured the BSJ flanking sequences by a biotinylated probe and a reporter probe loading fluorescent barcodes, followed by a high-resolution charge-coupled device (CCD) camera and digitization. e In situ hybridization used to visualize and quantify the interest circRNAs. There are two methods, which based on an oligonucleotide probe coupled to fluorescent dyes, and a sgRNA in a dCas13a-EGFP system
Northern blotting is the gold standard method for validating all kinds of RNAs, including circRNAs [9, 18, 123, 128]. Antisense probes are designed complementary to the sequences spanning the BSJ point in the circRNAs of interest, which are loaded on a denatured agarose gel containing formaldehyde, and hybridization is performed [18, 128] (Fig. 3a). This technique can precisely identify and quantify targeted circRNAs distinguished from linear RNAs transcribed from the same gene. However, the disadvantage of northern blotting is also obvious. This method requires a large amount of RNA, involves multiple steps, has a high background and often uses radioactively labeled probes [18]. This method generally requires many skills and is also time-consuming. Generally, candidate circRNAs are further validated and quantified by reverse transcription (RT) and quantitative PCR (qPCR) assays [2, 125, 135] (Fig. 3b). Although RT‒PCR is a timesaving and effective technique by means of a real-time PCR machine, the designed primer often cannot precisely distinguish the circular from the linear transcript during the fast PCR process with many copies of the amplified products [2]. The formation of concatemers by rolling circle amplification during the RT step is also a challenge that may hamper the accurate quantification of circRNAs.
Interestingly, droplet digital PCR (ddPCR) can overcome this shortcoming brought by RT‒qPCR [2, 33]. ddPCR is a novel technology that can determine the absolute quantification of a candidate circRNA using the ratio of positive to negative droplets, which exhibits a higher sensitivity even in plasma that has a very low amount of circRNA [33, 136] (Fig. 3c). However, the reagents for ddPCR assays are always expensive compared to other methods. If circRNAs can be quantified via high-throughput techniques, NanoString Technology is a good choice [4, 127] (Fig. 3d). The BSJ flanking sequences are captured by a biotinylated probe and a reporter probe loaded with fluorescent barcodes, and the circRNA-based barcodes on the reporter probes can finally be counted by a high-resolution charge-coupled device camera (CCD) and digitization. This enzyme-free technique also works well to detect paraffin-embedded RNA [4]. In situ hybridization (ISH) is another technique used to visualize and quantify circRNAs of interest [4, 125] (Fig. 3e). This technique designs an oligonucleotide probe, spanned to the BSJ site of circRNA, coupled to fluorescent dyes, to visualize a circRNA of interest in fixed and permeabilized cells using confocal microscopy. The value of fluorescent signals can reflect the quantity of circRNA to some extent. However, the ISH approach always requires the use of multiple probes covering the unique BSJ region, which may result in poor efficiency and a high false-positive rate. Interestingly, the dCas13a-EGFP system can be used to image and track specific circRNAs [137,138,139]. The special BSJ sequences could be a limit of guide RNA design in this approach.
New insight into the knockdown/out of functional circRNAs
Downregulating the expression of circRNAs is a popular strategy to explore their cellular functions [4, 5, 9]. Most circRNA knockdown methods are based on the complementary base pairing of seed sequences to BSJ junction sites, including siRNA, shRNA, ASO, or CRISPR/Cas series systems (Fig. 4a–c).

Strategies for knockdown/overexpression of circRNAs. a Lentivirus carrying shRNA according to the siRNA sequence to make the stable knockdown. sh/siRNA method executes the knockdown based on the complementary base pairing of seed sequences, which has 6–8 bases sponged to BSJ junction site. b CRISPR/Cas13d system degrades circRNAs that requires 28–30 nt long spacers and is intolerant to mismatches in spacers. c CRISPR/Cas9 system knocks out the special circRNA by deleting intronic complementary sequence neighboring the circularized exons. d Overexpression circRNA in a tRNA-derived intronic-circRNA system that followed a fluorescence-based RNA reporter allows to characterize the expression and localization visualization of circRNA. e Conformation of circRNA expression vector containing flanking introns from SUZ12 that splices to express circRNA without extra sequences. f In vitro synthesized RNA circles produced by T4 RNA ligase without extraneous fragments
Introducing siRNA corresponding to circRNA specifically targeting BSJ into transfected cells is a convenient and effective method to inhibit the expression of circRNA in cancer cells [46, 125]. The cells can also be transfected with lentivirus carrying shRNA according to the siRNA sequence to achieve stable knockdown [46, 125] (Fig. 4a). However, the siRNA method executes the knockdown based on the complementary base pairing of seed sequences, which only has 6–8 bases sponged to the BSJ junction site, which may produce an off-target effect on the linear lncRNA or mRNA. The CRISPR/Cas13d system is a useful tool for efficiently degrading circRNAs and reducing false targeting [124, 129] (Fig. 4b). Efficient Cas13d knockdown requires 28–30 nt long spacers and is intolerant to mismatches in spacers [129, 140, 141]. For example, Li et al. constructed a CRISPR–RfxCas13d system and found that gRNA spacers with the BSJ in the center (–7 to 7 nucleotides spanning the BSJ site) exhibited high knockdown efficiencies without affecting linear cognate RNAs [124]. Because circular and linear RNA have distinct biogenesis efficiencies, conformations and turnover rates, RfxCas13d-based RNA interference specifically suppresses circular but not linear RNA [124]. Another advantage is that CRISPR/Cas13-based gRNA, which carry a spacer sequence specifically targeting and spanning the BSJ site within a relatively long sequence, should have the capability to distinguish between circular and linear RNAs and thereby reduce off-target effects on linear lncRNA or mRNA. The combination of lentiviral vehicle and CRISPR/Cas13d can help in investigating the function of circRNA specificity in a xenotransplantation model and drug sensitivity screening.
In recent years, CRISPR/Cas9, which is a highly specific and efficient tool to edit the genome, has also been used in circRNA knockout [123, 142]. In general, the CRISPR/Cas9 system knocks out special circRNAs by deleting intronic complementary sequences neighboring circularized exons in circRNA biogenesis [5, 46, 143,144,145] (Fig. 4c). For example, sgRNA specifically targeting the inverted complementary sequence in the intron of GCN1L1 can knock circGCN1L1 out but not disturb the corresponding linear mRNA [145]. Similarly, CRISPR/Cas9 removal of the downstream inverted repeat ALU element can prevent circHIPK3 formation [144]. However, due to the complexity of circRNA biogenesis, it is difficult to determine which intronic sequences are targeted by sgRNAs in the CRISPR/Cas9 system. Apart from targeting intronic sequences, another challenge of circRNA knockout using the CRISPR/Cas9 system is that many circRNAs are produced from alternative splicing between exons and introns in the genome. Alternative splicing-based circRNA cannot directly target the sequence by sgRNAs, which may interfere with linear mRNA production [146].
Therefore, it is still necessary to gain insight into circRNA knockdown-based strategies, which should be considered with many different factors involved in circRNA production.
Overexpression of functional circRNAs
Several methods based on chemical synthesis and enzymatic ligation have been used to generate circRNAs in vitro; however, circRNA production in vivo has only recently been delineated [47, 128, 147, 148]. There is a circRNA-expressing vector that splices intron-containing tRNAs to produce circRNAs in cells [47, 148] (Fig. 4d). Construction of the tRNA-derived intronic-circRNA with a fluorescence-based RNA reporter allows us to characterize the expression of and visually localize circRNA. Because tRNA is constitutively expressed in all cells, tRNA-derived intronic circRNAs are theoretically expressed at high-copy and stable levels [47, 148]. Due to the feature of tRNA biogenesis by the processivity of pol III, this method have a circRNA size limitation (generally < 250 nt) [47]. Another in vivo circularized RNA was generated by the Group I intron of the phage T4 thymidylate synthase (td) gene transfected into cultured mammalian cells [62, 149]. However, both tRNA- and td gene-based RNA circles induced some extra sequences that tended to form 16–26 bp imperfect dsRNA regions, which generally activated remarkable immune responses via recognition by the pattern recognition receptor retinoic-acid-inducible gene I (RIG-I) or PKR [62, 128, 149]. We previously constructed a universal circRNA expression vector containing flanking introns from SUZ12 that ensured correct splicing to express circRNA without extra sequences [125] (Fig. 4e). We added a sequence that is the reverse complement repeat of the first 100 bp of the 5’ intron component into the vector following the 3’ intron to promote the interaction between the flanking introns, facilitating circRNA production. For example, the sequence of exons 8–9 of MYBL2 was inserted into the vector, and circMYBL2 was highly expressed, i.e., approximately 100-fold, in 293 T cells [125].
Considering the complexity of circRNA biogenesis, suitable strategies are needed for studying the different structural and functional features of circular RNA occurring in cells [5].
The replacement of stronger enhancers including ICSs, Alu elements, other RNA pairing structures and adding BSJ associated RBPs may be strategies to improve circRNA overexpression [5, 12, 150]. In contrast, Chen’s laboratory introduced in vitro synthesized RNA circles produced by T4 RNA ligase without extraneous fragments that present minimized immunogenicity, suggesting a useful method for the future synthesis of circular RNAs [128] (Fig. 4f).
ceRNA prediction
When circRNAs enter the cytoplasm, some of them become competitive endogenous RNAs (ceRNAs) [22, 100]. CircRNA can bind miRNA to prevent it from binding to target genes and changing the regulatory ability of target gene mRNA. Bioinformatics algorithms can be used to predict whether circRNAs have matching miRNAs [151,152,153] (Fig. 5a). The AGO2 protein was identified by analyzing the experimental data for CLIP-seq and functional genomic annotations, and the communication between miRNA and targeted circRNA was predicted after analysis and processing [151].

Methods to explore the possible mechanisms of circRNAs. a AGO2 CLIP-seq to predict the communication between miRNA and targeted circRNA. b, c circRNA-RBP interactions is mainly through RNA pull-down assay (b) or RNA immunoprecipitation (RIP) for experimental analysis (c). b In the RNA pull-down assay, a biotin-labeled probe recognized the BSJ of circRNA and then captured by biotin coupled magnetic beads. Finally, mass spectrum (MS) and western blot analysis to determinate the circRNA binding proteins. c In RIP-seq assay, the candidate RBP was first binds to the magnetic beads via antibody, and then the interacted circRNAs were analyzed by sequencing and RT-PCR. d circR-loops are identified by immunoprecipitation with the R loop-specific S9.6 antibody or catalytically inactive human RNase H1. Discovery the circR-loops in DRIP-seq data. circR-loops regulate diversity types of biological process, including transcriptional pausing, DNA damage, and double-strand DNA breaks (DSBs)
circRNA-RBP prediction
Although circRNA-miRNA sponging is the most well-known function, increasing evidence has also shown that circRNAs can interact with RBPs to exert widespread regulatory effects [56, 132]. For example, circPABPN1 can bind to HuR and prevent HuR from binding to PABPN1 mRNA, thereby reducing the translation of PABPN1 [154]. Some databases have summarized the interactions between circRNAs and RBPs. For example, CircInteractome provides miRNA and RBP binding sites on circRNA [132]. starBase also concentrated and systematically identified RNA‒RNA and protein‒RNA interaction networks [151].
To date, experimental research on circRNA-RBP interactions has mainly been conducted through RNA pulldown assays or RNA immunoprecipitation (RIP) for experimental analysis [56, 155] (Fig. 5b and c). Although these methods have been popularly used in many important discoveries, they still face many difficulties such as high costs, large tasks, and time consumption. Therefore, some programs that can predict the interaction of circRNA and RBP have been developed to compensate for the defects of classic experiments [56, 156]. Wang's team used matrix factorization and neural networks (MFNNs) to construct a prediction framework based only on interaction matrices, which has a high prediction accuracy and is an effective prediction method [156]. CirRBP, a stacked operation ensemble deep learning model, can fuse binding sites from multiple databases via a localization algorithm and compensates for the defect that most previous prediction methods only identify circRNA-RBP binding sites based on a single data resource [56]. However, CirRBP cannot provide accurate binding sites but only provides probability values of sequence fragments. Then, CirRBP was developed into an open-source web application called CRWS, which can allow users to change the codes in their own needs. CRWS is a useful online tool to use multi-source data to train models and predict precise binding sites [56]. Therefore, highly efficient and convenient circRNA-RBP prediction strategies will undoubtedly be useful for the study of circRNA functions.
circR-loops: circRNA:DNA hybrids
R-loops are widespread structures that are often formed co-transcriptionally [59, 157,158,159]. The genome-wide R-loop signature was generally identified by immunoprecipitation with the R loop-specific S9.6 antibody or catalytically inactive human RNase H1 (dRNH1) coupled with high-throughput sequencing of the resident DNA and RNA [59, 158, 160]. Apart from nascent mRNAs, DRIP-seq data have also shown that lncRNAs and circRNAs frequently form R-loop structures [17, 161] (Fig. 5d). These pervasive formations of circR-loops regulate diverse types of biological processes, including gene expression and DNA damage in cells [16, 17, 161,162,163]. For example, circSEP3 can form an R-loop by binding strongly to its cognate DNA locus, leading to SEPALLATA3 transcriptional pausing and coinciding with alternative splicing [163]. Overexpression of circSMARCA5 can generate a circR-loop at its parent gene locus, which results in transcriptional pausing at exon 15 of SMARCA5 and is sufficient to improve sensitivity to cytotoxic drugs in breast cancer [162]. Interestingly, a recent study showed that a set of circRNAs are enriched within the breakpoint cluster region (bcr) of MLL and can form circR-loops at their cognate loci [17]. These circR-loops promote transcriptional pausing, proteasome inhibition, chromatin reorganization, and double-strand DNA breaks (DSBs). Overexpressing circMLL (9,10) can trigger the de novo generation of clinically relevant chromosomal translocations mimicking the MLL recombinome in mouse leukemia xenograft models [17]. These studies suggest that nuclear circRNAs may form circR-loops and play both physiological and pathological roles in cells. Abnormalities in circRNA export from the nucleus can lead to diseases. Chen et al. identified that conserved exportin 4 (XPO4) can modulate circRNA nuclear export [16]. They observed that knockdown of XPO4 can improve circRNA nuclear retention, circR loop formation and DNA damage [16].
Recent studies may suggest that many circRNAs in circR-loops regulate the cognate DNA locus or mRNA transcription in a cis manner [16, 17]. It is still unclear whether these circRNAs in circR-loops can play roles in trans. There is still an interesting question that whether circR-loops interact with special RBPs to mediate chromatin marks, chromatin accessibility or active chromatin landscape.
New insights into biomedical application of cancer-related circRNA
Because circRNA has tissue- and cancer-specific expression and stability in body fluids, it can be used as a rapid, accurate, and noninvasive biomarker for early diagnosis and prognosis [20, 37, 114, 130, 164]. Several circRNAs are reported to play important roles in tumorigenesis and progression, as well as in chemotherapeutic resistance, and are potential promising targets in cancer treatment [66, 115, 130].
CircRNA is a promising biomarker in cancer
Cancer cells present aberrant expression of circRNAs, which are usually related to some clinical characteristics, such as tumor type, tumor size, histological grade, tumor invasion and metastasis (Table 2). For example, in non-small cell lung cancer, low expression of hsa_circ_0001073 may distinguish adenocarcinoma from squamous cell carcinoma [165]. In breast cancer, circRNA expression profiles may distinguish between estrogen receptor-positive, HER2-positive, and triple-negative breast cancer [166]. In tissue samples, the upregulation of hsa_circ_0003823, circPUM1, circCYP24A1, and circCNOT6L presented diagnostic performance with considerable sensitivity and specificity values, which exhibited relatively higher recurrence of esophageal squamous cell carcinoma (ESCC) [167,168,169,170]. In the plasma samples, Hu et al., found that highly concentration of plasma circGSK3β and CEA can indicate the recurrence/metastasis of ESCC [171]. CircRNA also showed the ability to distinguish different nontumor diseases [172]. The hsa_circRNA_0001599 was highly expressed in large-artery atherosclerosis (LAA)-stroke patients, revealing its potential as a biomarker of LAA-stroke diagnosis [172]. The plasma concentration of CircBRAP can be a predictor of preeclampsia [173]. CircRNA can be quite stable in biological fluids, and detection of circulating circRNA may be an excellent noninvasive biopsy that is likely to become a new method for cancer detection in the future.
CircRNAs can not only distinguish different tumor subtypes but also indicate different prognostic levels in the body [130, 174, 175]. For example, CIRS-7 is associated with poor prognosis in most cancers [174]; circUBAP2 has also been identified as an oncogenic factor associated with poor prognosis [174], while circLARP4 is a tumor suppressor associated with good prognosis in several cancers [176]. circRNA-CREIT was also recently found to be abnormally downregulated in doxorubicin-resistant triple-negative breast cancer (TNBC) cells and associated with poor prognosis [40].
CircRNAs are promising therapeutic targets
In recent years, numerous dysregulated circRNAs have been found to affect the proliferation, apoptosis, metastasis, DNA damage and other life activities of cancer cells [3, 10, 99, 130]. Therefore, similar to miRNAs and lncRNAs, circRNAs can also be used as therapeutic targets for cancer treatment [54, 130, 177, 178] (Table 2). For example, intratumoral injection of circNRIP1 siRNA could significantly inhibit the growth of gastric cancer in PDX mouse models, suggesting that oncogenic circNRIP1 may be a promising target for gastric cancer treatment [179]. Antisense oligonucleotides (ASOs) against circIPO11 combined with the TOP1 inhibitor camptothecin (CPT) exert synergistic effects and can significantly suppress liver cell self-renewal and HCC propagation [123]. The knockdown of circMYBL2 in vitro and in vivo by siRNA and shRNA significantly inhibited the FLT3-ITD protein level and inhibited the proliferation of FLT3-ITD AML cells but had no effect on normal cells [125]. circIPO11 knockout using CRISPR/Cas9 technology suppresses the progression of chemically induced liver cancer development [123]. Notably, several circRNAs act as suppressors in cancer progression, indicating their antitumor effects [154, 180,181,182,183,184]. circANAPC7, newly discovered tumor suppressors, can significantly inhibit tumor growth and muscle atrophy in pancreatic cancer [180]. In vivo delivery of these kinds of tumor suppressor circRNAs may be a promising approach for anticancer therapy.
CircRNA regulates therapy resistance and targeted drug development
In the current clinical treatment of cancer, various chemotherapeutic drugs have been developed to inhibit the growth of cancer cells and have achieved good clinical effects [185,186,187]. However, with the prolonged time of medication at any time, the drug resistance of cancer cells gradually increases, resulting in the gradual weakening of the therapeutic effect, which is a major problem that has to be solved in clinical treatment [187, 188]. Recent studies show that circRNAs play a role in the resistance of cancer cells to anticancer agents [33, 189, 190]. They found that circRNA-SORE (also known as circRNA_104,797 and circ_0087293) was upregulated in sorafenib-resistant HCC cells, acting as ceRNA to isolate miR-103a-2-5p and miR-660-3p and competitively activate the Wnt/β-catenin pathway to promote sorafenib resistance [191] (Fig. 6a). Interestingly, this team also reported that circRNA-SORE binds YBX1 and blocks PRP19-mediated YBX1 degradation. They found that silencing circRNA-SORE by injection of siRNA in vivo could substantially overcome sorafenib resistance [41] (Fig. 6a). CircVMP1 could upregulate the expression of methyltransferase 3, N6-adenosine-methyltransferase complex catalytic subunit (METTL3) and SOX2 by acting as a sponge of miR-524-5p, thereby promoting the progression of NSCLC and cisplatin (DDP) resistance [192]. These studies put forward a new idea for solving chemotherapeutic drug resistance by knocking down specific circRNAs to inhibit their function of promoting drug resistance.

CircRNA regulates therapy resistance. a circRNA-SORE acts as ceRNA to miR-103a-2-5p and miR-660-3p, and competitively activates Wnt2b translation to promote sorafenib resistance in HCC; it also binds to YBX1 and blocks PRP19-mediated YBX1 degradation to regulate sorafenib resistance. b circCDYL2 enhances the interaction between GRB7 and FAK, thereby activates AKT and ERK1/2 signaling pathways to promote trastuzumab resistance in breast cancer. Circ-HER2 can also encode a small protein HER2-103, which promotes homo/hetero dimerization of epidermal growth factor receptor (EGFR)/HER3, and actives AKT phosphorylation, which endows the sensitivity to Pertuzumab in triple-negative breast cancer. c circMYBL2 regulates FLT3-ITD translation by binding of PTBP1 to FLT3 messenger RNA in quizartinib-resistant FLT3-ITD AML. d circRNA-CREIT facilitates the interaction between PKR and the E3 ligase HACE1 and promoted proteasomal degradation of PKR protein, thereby attenuating the stress granules (SGs) assembly to activate the RACK1/MTK1 apoptosis signaling pathway and overcome doxorubicin resistance
CircRNAs can also interact with oncoproteins to help cancer cells establish drug resistance [33, 189, 193, 194]. For example, circCDYL2 enhances the interaction between GRB7 and FAK by inhibiting the ubiquitination degradation of GRB7, thereby maintaining the activation of downstream AKT and ERK1/2 signaling pathways and leading to trastuzumab resistance in breast cancer [193] (Fig. 6b). Circ-HER2 encodes the small protein HER2-103, which promotes homo/heterodimerization of epidermal growth factor receptor (EGFR)/HER3 and activates AKT phosphorylation and malignant phenotypes [194]. Pertuzumab inhibits the tumorigenicity of circ-HER2/HER2-103-expressing TNBC cells but not circ-HER2/HER2-103-negative TNBC cells in vivo [194]. These studies suggest that both knockdown of circCDYL2 and overexpression of circ-HER2/HER2-103 together can improve the outcome of drug therapy targeting HER2 signaling in TNBC. We previously also showed that circMYBL2 is more highly expressed in AML patients with FLT3-ITD mutations [125] (Fig. 6c). Relapse of FLT3_ITD AML has been observed due to acquired resistance with secondary mutations in FLT3. shRNA-mediated circMYBL2 knockdown specifically inhibited FLT3-ITD translation by preventing the binding of polypyrimidine tract-binding protein 1 (PTBP1) from FLT3 messenger RNA and impaired the cytoactivity of inhibitor-resistant FLT3-ITD AML, suggesting that circMYBL2 knockdown was effective against FLT3-ITD AML with quizartinib resistance [125]. Notably, circRNAs can regulate the assembly of membraneless organelles to overcome drug resistance [40, 189]. For example, circRNA-CREIT facilitates the interaction between PKR and the E3 ligase HACE1 to promote proteasomal degradation of PKR, which attenuates the assembly of stress granules (SGs) to activate the RACK1/MTK1 apoptosis signaling pathway and overcome doxorubicin resistance in TNBC [40] (Fig. 6d).
Drug resistance is an urgent problem to be solved in current tumor therapy treatments. Recent studies have shown that circRNAs can regulate drug tolerance pathways by interacting with miRNAs, proteins and translated proteins in tumor cells [33, 130, 189]. Targeting drug resistance-related circRNAs may improve the efficiency of chemotherapeutics in cancers.
Challenges of circRNAs as therapeutic targets
Although recent studies have suggested that circRNAs are promising therapeutic targets in many diseases, there are still some challenges [67, 99, 130]. Currently, two targeted therapies are commonly used: gene editing systems and RNAi [123, 141,142,143, 146]. The gene editing method uses the CRISPR‒Cas9 system to specifically delete the Alu sequence, which is important for circRNA formation [4, 10, 15, 143]. Such an operation does not affect the mRNA content of the corresponding linear product of the gene but only affects the formation of circRNA, thus regulating the life activities of the cell. However, this method often leads to the occurrence of unpredictable selective shearing events, and DNA editing is an irreversible operation with potential ethical problems. On the contrary, RNAi technology is relatively safe to change cellular RNA levels for it will not cause gene changes [67, 125, 141, 195,196,197]. It induces circRNA cleavage by delivering small interfering RNA or short hairpin RNA to cells and reduces the content of circRNA. In addition, the CRISPR‒Cas13 system is increasingly being utilized to effectively target circRNA without affecting mRNA and has been shown to have an overall advantage in the efficiency and specificity of circRNA knockdown [124, 126, 141]. However, the efficiency of introducing gRNA and Cas13 enzymes into target cells is not high, and there is a certain off-target effect. For CRISPR‒Cas13 technology to be truly applied to clinical practice, these problems still need to be further solved.
Therapeutic potential based on circular RNA translation
Recent studies have found that some circRNAs can also be directly translated into small peptides and play a role in cells [9, 65, 198]. Interestingly, a number of circRNAs can encode carcinogenic or cancer-inhibiting protein products [199,200,201] (Fig. 7). For example, circAKT3 has a predicted ORF and encodes a small 174-amino acid peptide, AKT3-174aa, which competitively binds p-PDK1 to inhibit downstream targets of p-PDK1, suppressing glioblastoma tumorigenicity [199] (Fig. 7a). MAPK1-109aa, encoded by circMAPK1, can inhibit the proliferation and migration of gastric cancer cells [200] (Fig. 7b). circPLCE1-411 promotes the ubiquitin-dependent degradation of the critical NF-κB regulator RPS3 by directly binding the HSP90α/RPS3 complex to inhibit the NF-κB signaling pathway in colorectal carcinoma (CRC) [201] (Fig. 7c). In vivo experiments showed that circular LINC-PINT and vSP27 could inhibit the growth of cancer and had no adverse effects on mice [202, 203] (Fig. 7d).

Therapeutic potential based on circRNA translation. a circAKT3 encodes a small 174-amino acid peptide, AKT3-174aa, which competitively binds p-PDK1 to inhibit downstream targets of AKT signaling pathway in glioblastoma. b MAPK1-109aa, encoded by circMAPK1, can inhibit the phosphorylation of MAPK1 by competitively binding to MEK1, thereby suppressing MAPK pathway and inhibits the proliferation and migration of gastric cancer cells. c circPLCE1 encodes circPLCE1-411 that promotes the ubiquitin-dependent degradation of the critical NF-κB regulator RPS3 via directly binding the HSP90α/RPS3 complex to facilitate the dissociation of RPS3 from the complex, thereby reducing NF-κB nuclear translocation in CRC cells. d circ-0082389 encodes a small peptide, LINC-PINT, interacts with polymerase associated factor complex (PAF1c) and inhibits the transcriptional elongation of multiple oncogenes in glioblastoma tumorigenesis. e A workflow of circRNA-RBD vaccine. SP, signal peptide sequence of human tPA. Foldon, the trimerization domain from bacteriophage T4 fibritin. The circRNARBD-Delta vaccine based on lipid nanoparticle (LNP), which is injected into mice and rhesus macaque, and elicits potent neutralizing antibodies and T cell responses, providing robust protection against SARS-CoV-2
Given that circRNAs have the perfect characteristics of stable conformation, high stability, and special immunogenicity, RNA circle-based technologies were developed [9, 18, 67]. Recently, circRNAs harboring the translational capability of SARS-CoV-2 receptors were used to generate mRNA vaccines, such as the circRNA-RBD-Delta vaccine, which was used to protect against the COVID-19 pandemic [44] (Fig. 7e). However, few studies have investigated circRNAs with mRNA-based therapeutics in cancer treatment. It is a promising strategy to synthesize translational circRNAs with antineoplastic genes in cancer therapy. Similar to small antisense oligonucleotides, efficient introduction of circRNA into target cells is key to clinical implementation. To improve the delivery efficiency of circRNA delivery boxes, vectors can be replaced with lentiviruses or adeno-associated viruses [28, 54, 190, 196]. circRNA expression boxes in target cells may produce a large number of linear products in addition to target circRNA, which may adversely affect cells. We may directly introduce circRNA, which has been synthesized in vitro, into the target cells and deliver it with nonviral nanoparticles [45, 130, 192, 204]. However, in vitro circularized RNAs generally induce extra coding genes or sequences and often activate remarkable immune responses and other unknown side effects. Therefore, future studies may develop specific and effective approaches to improve circular RNA-based therapeutics.
Conclusions and perspectives
With advances in bioinformatics and biotechnologies, circRNA research has become an increasingly popular and important field [2, 5, 9, 10, 50, 99, 130]. There are many new insights into aspects of circRNA studies, including biogenesis, epigenetic regulation and degradation [4, 5, 9, 10, 67]. Increasing evidence has revealed that circRNAs have dysregulated expression patterns and diverse regulatory mechanisms underlying cellular processes and are always related to the pathogenesis of various diseases, including cancer [20, 130]. However, the study of the regulation, functions and biomedical application of these molecules is still at an early stage, and the complexity of circRNA already appears. For example, diverse biogenesis mechanisms of circRNAs are still emerging. Most annotated circRNAs are produced by back-splicing of pre-mRNA or intron self-splicing of small RNAs [5, 13, 148, 149]. With advances in deep sequencing, especially the development of long-read sequencing, a majority of novel circRNAs are generated by unknown splicing and differential locations on chromatin, such as from incomplete introns or exons with splicing complexity [100, 102, 127]. Some circRNAs were derived from intergenic sequences [50, 205]. The factors regulating these unknown production mechanisms of circRNA should be further delineated. In addition, although many significant advances in identification tools of circRNAs have appeared, it is still difficult to precisely define their length, location, and expression, which are always different from those in experimental validations. This is an important and challenging task in this field, which requires scientists to work together. Advanced parallel technologies will be helpful for circRNA discovery. Some open friendly comprehensive pipelines, such as Fcirc, may offer platforms for users to optimize the discovery tools of circRNAs [64, 89, 92].
The sequence overlaps of circRNAs with their cognate linear RNA sequences usually restrict the determination of circRNA functions [5, 11]. Although recent progress in biotechnologies for knockdown and knockout has been made, uncertain efficiency and off-targeting in si/shRNA or CRISPR/Cas series systems always occur. A recent design based on CRISPR‒Cas13 systems can improve the specificity of targeting BSJ sites [124, 129, 140, 141]. However, the efficiency of expression of Cas13 and sgRNA together is low in cells, especially in cells in suspension, which may restrict their widespread application. Importing some extra sequences and immunogenicity are two difficulties in circRNA overexpression in cells, which affect the application of circRNAs in biomedicine [18, 149]. Novel strategies for circRNA overexpression are urgently needed. In vitro synthesized circRNAs via T4 RNA ligase without extraneous fragments that present minimized immunogenicity may be developed to be a useful method to meet the sufficient quantity of circRNAs in biomedical applications [128].Kindly check and confirm the section headings are correct.Yes, we check and confirm the section headings are correct.
Considering the structural stability advantages, cancer-specific expression, and drug resistance exhibited by circRNAs, they hold significant promise as noninvasive biomarkers for cancer and as targets in cancer treatment [20, 67, 99, 130]. Nonetheless, in clinical practice, the challenge lies in determining the extraction and processing methods for test substances, hindering the quest to establish circRNA as the quickest and most precise biopsy marker for clinical assessments. Additionally, achieving precise in vivo delivery of si/shRNA-based knockdown or tumor suppressor circRNAs in anticancer therapy should be continually optimized. We hope that these issues can be addressed in future research.
The discovery of circRNA translation not only brings exciting new perspectives for translation machines but also brings novel design concepts for the treatment of major diseases based on circRNA translation [32, 62, 206]. The considerable intra- and extracellular stability of circRNA seems to make it a more ideal tool than other ncRNAs in many aspects of biomedical applications [62, 67]. A novel SARS-CoV-2 vaccine based on circRNA-RBD translation was able to produce a higher and longer-lasting antigen and induce a higher proportion of neutralizing antibodies than an mRNA vaccine [44]. However, circRNA-based protein translation strategies are still in the exploratory stage. Many problems remain unresolved. The most important problem is that the translation efficiency of circRNA based on IRES is low. Therefore, the common translational elements of circRNA need to be further optimized. For example, a team found that five elements upstream of the IRES topology, the 5′ PABP spacer, the HBA1 3′ UTR and the HRV-B3 IRES with proximal loop Apt-eIF4G insertion, can considerably improve the translational efficiency of circRNA in vivo [30]. In addition, the search for candidate proteins suitable for circRNA translation strategies should also be continued. A precision medicine approach based on personalized circRNA construction-candidate target-host may be possible in the future. The emergence of circRNA-based protein translation strategies has brought new directions to the field of biomedicine.
Availability of data and materials
Not applicable.
Abbreviations
- circRNAs:
-
Circular RNAs
- ncRNAs:
-
Non-coding RNAs
- mRNA:
-
Messenger RNA
- lncRNA:
-
Long noncoding RNA
- snoRNA:
-
Small nucleolar RNA
- miRNA:
-
MicroRNA
- ssRNAs:
-
Single-stranded RNAs
- tRNAs:
-
Transfer RNAs
- pre-mRNA:
-
Precursor mRNA
- BSJ:
-
Back-splicing junction site
- ICSs:
-
Intronic complementary sequences
- RBPs:
-
RNA-binding proteins
- RNase R:
-
Ribonuclease R
- m6A:
-
N(6)-Methyladenosine
- COVID-19:
-
Corona Virus Disease 2019
- snRNAs:
-
Small nuclear RNAs
- rRNAs:
-
Ribosomal RNAs
- RNA-seq:
-
RNA sequencing
- UTRs:
-
Untranslated regions
- ciRNAs:
-
Intronic circRNAs
- EIciRNAs:
-
Exon‒intron circRNAs
- ecircRNAs:
-
Exonic circRNAs
- circR-loops:
-
CircRNA:DNA hybrids
- IRS2:
-
Insulin receptor substrate 2
- EGFR:
-
Epidermal growth factor receptor
- PABPN1:
-
Nuclear poly (A) binding protein 1
- HuR:
-
Human antigen R
- dsRNA:
-
Double-stranded RNA
- PKR:
-
Double-stranded RNA-activated protein kinase
- DRIP-seq:
-
DNA:RNA immunoprecipitation sequencing
- R-loop:
-
DNA:RNA hybrids
- SARS-CoV-2:
-
Severe Acute Respiratory Syndrome Coronavirus 2
- gRNAs:
-
Guide RNAs
- ADARs:
-
Adenosine Deaminase
- IRES:
-
Internal ribosome entry
- elF4:
-
Eukaryotic translation initiation factor 4
- elF4G:
-
Eukaryotic translation initiation factor 4G
- elF3:
-
Eukaryotic translation initiation factor 3
- elF4G38:
-
Eukaryotic translation initiation factor 4G38
- eIF4E:
-
Eukaryotic translation initiation factor 4E
- Ago2:
-
Argonaute 2
- DBR1:
-
Debranching RNA Lariats 1
- RNase H1:
-
Ribonuclease H1
- SLE:
-
Systemic lupus erythematosus
- RNase L:
-
Ribonuclease L
- f-circRNAs:
-
Fusion circular RNAs
- ceRNA:
-
Competing endogenous RNA
- RT:
-
Reverse transcription
- PCR:
-
Polymerase Chain Reaction
- qPCR:
-
Quantitative PCR
- ddPCR:
-
Droplet digital PCR
- CCD:
-
Charge-coupled device camera
- ISH:
-
In situ hybridization
- siRNA:
-
Small interfering RNA
- shRNA:
-
Short hairpin RNA
- ASO:
-
Antisense oligonucleotides
- CRISPR:
-
Clustered Regularly Interspaced Short Palindromic Repeats
- GCN1:
-
GCN1 Activator Of EIF2AK4
- sgRNAs:
-
Small guide RNAs
- td:
-
T4 thymidylate synthase
- RIG-I:
-
Retinoic-acid-inducible gene I
- SUZ12:
-
SUZ12 polycomb repressive complex 2 subunit
- MYBL2:
-
V-Myb avian myeloblastosis viral oncogene homolog-like 2
- CLIP-seq:
-
Crosslinking immunoprecipitation-high-throughput-sequencing
- RIP:
-
RNA immunoprecipitation
- MFNNs:
-
Matrix factorization and neural networks
- dRNH1:
-
Human RNase H1
- DRIP-seq:
-
DNA:RNA hybrid immunoprecipitation and sequencing
- SMARCA5:
-
SWI/SNF Related, Matrix Associated, Actin Dependent Regulator of Chromatin, Subfamily A, Member 5
- DSBs:
-
Double-strand DNA breaks
- XPO4:
-
Exportin 4
- AML:
-
Acute Myelocytic Leukemia
- ALL:
-
Acute Lymphocytic Leukemia
- CLL:
-
Chronic Lymphocytic Leukemia
- CML:
-
Chronic Myeloid Leukemia
- MM:
-
Multiple Myeloma
- CRC:
-
Colorectal Carcinoma
- HCC:
-
Hepatocellular Carcinoma
- GC:
-
Gastric Carcinoma
- BC:
-
Bladder Cancer
- PC:
-
Pancreatic Cancer
- OSCC:
-
Oral Squamous Cell Carcinoma
- ESCC:
-
Esophageal Squamous Cell Carcinoma
- EC:
-
Esophagus Cancer
- LC:
-
Lung Cancer
- RC:
-
Renal Carcinoma
- GM:
-
Glioma Malignancy
- OC:
-
Ovarian Cancer
- TC:
-
Thyroid Cancer
- CC:
-
Cervical Cancer
- CEA:
-
Carcinoembryonic antigen
- LAA:
-
Large-artery atherosclerosis
- TNBC:
-
Triple-negative breast cancer
- PDX:
-
Patient-Derived Xenograft
- ASOs:
-
Antisense oligonucleotides
- TOP1:
-
DNA topoisomerase I
- CPT:
-
Camptothecin
- FLT3:
-
FMS-like tyrosine kinase 3
- ITD:
-
Internal tandem duplication
- YBX1:
-
Y-box binding protein 1
- PRP19:
-
Pre-mRNA processing factor 19
- METTL3:
-
Methyltransferase Like 3
- NSCLC:
-
Non-Small Cell Lung Carcinoma
- DDP:
-
Cisplatin
- GRB7:
-
Growth factor receptor bound protein 7
- FAK:
-
Focal Adhesion Kinase
- AKT:
-
Protein Kinase B, PKB
- ERK:
-
Extracellular regulated protein kinases
- HER2:
-
Human epidermal growth factor receptor 2
- PTBP1:
-
Polypyrimidine tract-binding protein 1
- HACE1:
-
HECT domain and ankyrin-repeat-containing E3 ubiquitin-protein ligase 1
- SGs:
-
Stress granules
- RACK1:
-
Receptor for Activated C kinase1
- MTK1:
-
Mitogen-activated protein kinase kinase kinase 4
- RNAi:
-
RNA interference
- ORF:
-
Open reading frame
- PDK1:
-
3-Phosphoinositide-dependent protein kinase 1
- NF-κB:
-
Nuclear factor kappa-B
- RPS3:
-
Ribosomal protein S3
- HSP:
-
Heat Shock Proteins
- PABP:
-
Poly A binding protein
- HBA1:
-
Hemoglobin Subunit Alpha 1
References
Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A. 1976;73(11):3852–6.
Szabo L, Salzman J. Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet. 2016;17(11):679–92.
Chen LL. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17(4):205–11.
Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20(11):675–91.
Yang L, Wilusz JE, Chen L-L. Biogenesis and regulatory roles of circular RNAs. Annu Rev Cell Dev Biol. 2022;38(1):263–89.
Chen ZH, Chen TQ, Zeng ZC, Wang D, Han C, Sun YM, et al. Nuclear export of chimeric mRNAs depends on an lncRNA-triggered autoregulatory loop in blood malignancies. Cell Death Dis. 2020;11(7):566.
Huang W, Sun Y-M, Pan Q, Fang K, Chen X-T, Zeng Z-C, et al. The snoRNA-like lncRNA LNC-SNO49AB drives leukemia by activating the RNA-editing enzyme ADAR1. Cell Discov. 2022;8(1):117.
Sun YM, Chen YQ. Principles and innovative technologies for decrypting noncoding RNAs: from discovery and functional prediction to clinical application. J Hematol Oncol. 2020;13(1):109.
Liu CX, Chen LL. Circular RNAs: Characterization, cellular roles, and applications. Cell. 2022;185(12):2016–34.
Chen LL. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat Rev Mol Cell Biol. 2020;21(8):475–90.
Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, et al. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell. 2014;56(1):55–66.
Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence-mediated exon circularization. Cell. 2014;159(1):134–47.
Zhang XO, Dong R, Zhang Y, Zhang JL, Luo Z, Zhang J, et al. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016;26(9):1277–87.
Stagsted LVW, O’Leary ET, Ebbesen KK, Hansen TB. The RNA-binding protein SFPQ preserves long-intron splicing and regulates circRNA biogenesis in mammals. Life. 2021;10:e63088.
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19(2):141–57.
Chen L, Wang Y, Lin J, Song Z, Wang Q, Zhao W, et al. Exportin 4 depletion leads to nuclear accumulation of a subset of circular RNAs. Nat Commun. 2022;13(1):5769.
Conn VM, Gabryelska M, Toubia J, Kirk K, Gantley L, Powell JA, et al. Circular RNAs drive oncogenic chromosomal translocations within the MLL recombinome in leukemia. Cancer Cell. 2023;41(7):1309–26.
Liu C-X, Li X, Nan F, Jiang S, Gao X, Guo S-K, et al. Structure and Degradation of Circular RNAs Regulate PKR Activation in Innate Immunity. Cell. 2019;177(4):865–80.
Park OH, Ha H, Lee Y, Boo SH, Kwon DH, Song HK, et al. Endoribonucleolytic Cleavage of m(6)A-Containing RNAs by RNase P/MRP Complex. Mol Cell. 2019;74(3):494–507.
Vo JN, Cieslik M, Zhang Y, Shukla S, Xiao L, Zhang Y, et al. The landscape of circular RNA in Cancer. Cell. 2019;176(4):869–81.
Huang W, Fang K, Chen T-Q, Zeng Z-C, Sun Y-M, Han C, et al. circRNA circAF4 functions as an oncogene to regulate MLL-AF4 fusion protein expression and inhibit MLL leukemia progression. J Hematol Oncol. 2019;12(1):103.
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–8.
Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, et al. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015;160(6):1125–34.
Zhou C, Molinie B, Daneshvar K, Pondick JV, Wang J, Van Wittenberghe N, et al. Genome-Wide Maps of m6A circRNAs Identify Widespread and Cell-Type-Specific Methylation Patterns that Are Distinct from mRNAs. Cell Rep. 2017;20(9):2262–76.
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 2017;27(5):626–41.
Zeng ZC, Pan Q, Sun YM, Huang HJ, Chen XT, Chen TQ, et al. METTL3 protects METTL14 from STUB1-mediated degradation to maintain m(6) A homeostasis. EMBO Rep. 2023;24(3):e55762.
Yi Z, Qu L, Tang H, Liu Z, Liu Y, Tian F, et al. Engineered circular ADAR-recruiting RNAs increase the efficiency and fidelity of RNA editing in vitro and in vivo. Nat Biotechnol. 2022;40(6):946–55.
Katrekar D, Yen J, Xiang Y, Saha A, Meluzzi D, Savva Y, et al. Efficient in vitro and in vivo RNA editing via recruitment of endogenous ADARs using circular guide RNAs. Nat Biotechnol. 2022;40(6):938–45.
Shi Y, Jia X, Xu J. The new function of circRNA: translation. Clin Transl Oncol. 2020;22(12):2162–9.
Chen R, Wang SK, Belk JA, Amaya L, Li Z, Cardenas A, et al. Engineering circular RNA for enhanced protein production. Nat Biotechnol. 2022;41(2):262–72.
Kameda S, Ohno H, Saito H. Synthetic circular RNA switches and circuits that control protein expression in mammalian cells. Nucleic Acids Res. 2023;51(4):e24.
Fan X, Yang Y, Chen C, Wang Z. Pervasive translation of circular RNAs driven by short IRES-like elements. Nat Commun. 2022;13(1):3751.
Zhang F, Jiang J, Qian H, Yan Y, Xu W. Exosomal circRNA: emerging insights into cancer progression and clinical application potential. J Hematol Oncol. 2023;16(1):67.
Shen H, Liu B, Xu J, Zhang B, Wang Y, Shi L, et al. Circular RNAs: characteristics, biogenesis, mechanisms and functions in liver cancer. J Hematol Oncol. 2021;14(1):134.
Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015;22(3):256–64.
Kalluri R. The biology and function of exosomes in cancer. J Clin Invest. 2016;126(4):1208–15.
Zhou Q, Xie D, Wang R, Liu L, Yu Y, Tang X, et al. The emerging landscape of exosomal CircRNAs in solid cancers and hematological malignancies. Biomark Res. 2022;10(1):28.
Gao M, Li C, Xiao H, Dong H, Jiang S, Fu Y, et al. hsa_circ_0007841: a novel potential biomarker and drug resistance for multiple myeloma. Front Oncol. 2019;9:1261.
Sun Q, Wang L, Zhang C, Hong Z, Han Z. Cervical cancer heterogeneity: a constant battle against viruses and drugs. Biomark Res. 2022;10(1):85.
Wang X, Chen T, Li C, Li W, Zhou X, Li Y, et al. CircRNA-CREIT inhibits stress granule assembly and overcomes doxorubicin resistance in TNBC by destabilizing PKR. J Hematol Oncol. 2022;15(1):122.
Xu J, Ji L, Liang Y, Wan Z, Zheng W, Song X, et al. CircRNA-SORE mediates sorafenib resistance in hepatocellular carcinoma by stabilizing YBX1. Signal Transduct Target Ther. 2020;5(1):298.
Zhao Q, Liu J, Deng H, Ma R, Liao J-Y, Liang H, et al. Targeting Mitochondria-Located circRNA SCAR Alleviates NASH via Reducing mROS Output. Cell. 2020;183(1):76-93.e22.
Lyu L, Zhang S, Deng Y, Wang M, Deng X, Yang S, et al. Regulatory mechanisms, functions, and clinical significance of CircRNAs in triple-negative breast cancer. J Hematol Oncol. 2021;14(1):41.
Qu L, Yi Z, Shen Y, Lin L, Chen F, Xu Y, et al. Circular RNA vaccines against SARS-CoV-2 and emerging variants. Cell. 2022;185(10):1728–44.
Li H, Peng K, Yang K, Ma W, Qi S, Yu X, et al. Circular RNA cancer vaccines drive immunity in hard-to-treat malignancies. Theranostics. 2022;12(14):6422–36.
Tang X, Ren H, Guo M, Qian J, Yang Y, Gu C. Review on circular RNAs and new insights into their roles in cancer. Comput Struct Biotechnol J. 2021;19:910–28.
Schmidt CA, Giusto JD, Bao A, Hopper AK, Matera AG. Molecular determinants of metazoan tricRNA biogenesis. Nucleic Acids Res. 2019;47(12):6452–65.
Clark CG, Cross GA. Circular ribosomal RNA genes are a general feature of schizopyrenid amoebae. J Protozool. 1988;35(2):326–9.
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–8.
Chen L-L, Bindereif A, Bozzoni I, Chang HY, Matera AG, Gorospe M, et al. A guide to naming eukaryotic circular RNAs. Nat Cell Biol. 2023;25(1):1–5.
Aktaş T, Avşar Ilık İ, Maticzka D, Bhardwaj V, Pessoa Rodrigues C, Mittler G, et al. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature. 2017;544(7648):115–9.
Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015;10(2):170–7.
Zhao J, Tao Y, Zhou Y, Qin N, Chen C, Tian D, et al. MicroRNA-7: a promising new target in cancer therapy. Cancer Cell Int. 2015;15:103.
Li Z, Rana TM. Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov. 2014;13(8):622–38.
Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, et al. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J. 2011;30(21):4414–22.
Wang Z, Lei X. A web server for identifying circRNA-RBP variable-length binding sites based on stacked generalization ensemble deep learning network. Methods. 2022;205:179–90.
Du WW, Yang W, Liu E, Yang Z, Dhaliwal P, Yang BB. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016;44(6):2846–58.
Li XX, Xiao L, Chung HK, Ma XX, Liu X, Song JL, et al. Interaction between HuR and circPABPN1 Modulates Autophagy in the Intestinal Epithelium by Altering ATG16L1 Translation. Mol Cell Biol. 2020;40(6):e00492-e519.
Li X, Zhang JL, Lei YN, Liu XQ, Xue W, Zhang Y, et al. Linking circular intronic RNA degradation and function in transcription by RNase H1. Sci China Life Sci. 2021;64(11):1795–809.
Pfafenrot C, Schneider T, Muller C, Hung LH, Schreiner S, Ziebuhr J, et al. Inhibition of SARS-CoV-2 coronavirus proliferation by designer antisense-circRNAs. Nucleic Acids Res. 2021;49(21):12502–16.
Ma J, Du WW, Zeng K, Wu N, Fang L, Lyu J, et al. An antisense circular RNA circSCRIB enhances cancer progression by suppressing parental gene splicing and translation. Mol Ther. 2021;29(9):2754–68.
Wesselhoeft RA, Kowalski PS, Anderson DG. Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat Commun. 2018;9(1):2629.
Abe N, Matsumoto K, Nishihara M, Nakano Y, Shibata A, Maruyama H, et al. Rolling circle translation of circular RNA in living human cells. Sci Rep. 2015;5:16435.
Huang W, Ling Y, Zhang S, Xia Q, Cao R, Fan X, et al. TransCirc: an interactive database for translatable circular RNAs based on multi-omics evidence. Nucleic Acids Res. 2021;49(D1):D236–42.
Sun L, Wang W, Han C, Huang W, Sun Y, Fang K, et al. The oncomicropeptide APPLE promotes hematopoietic malignancy by enhancing translation initiation. Mol Cell. 2021;81(21):4493–508.
Fabbri L, Chakraborty A, Robert C, Vagner S. The plasticity of mRNA translation during cancer progression and therapy resistance. Nat Rev Cancer. 2021;21(9):558–77.
Wen SY, Qadir J, Yang BB. Circular RNA translation: novel protein isoforms and clinical significance. Trends Mol Med. 2022;28(5):405–20.
Ho-Xuan H, Glazar P, Latini C, Heizler K, Haase J, Hett R, et al. Comprehensive analysis of translation from overexpressed circular RNAs reveals pervasive translation from linear transcripts. Nucleic Acids Res. 2020;48(18):10368–82.
Huang W, Chen TQ, Fang K, Zeng ZC, Ye H, Chen YQ. N6-methyladenosine methyltransferases: functions, regulation, and clinical potential. J Hematol Oncol. 2021;14(1):117.
Lin J, Wang X, Zhai S, Shi M, Peng C, Deng X, et al. Hypoxia-induced exosomal circPDK1 promotes pancreatic cancer glycolysis via c-myc activation by modulating miR-628-3p/BPTF axis and degrading BIN1. J Hematol Oncol. 2022;15(1):128.
Zhu L, Sun HT, Wang S, Huang SL, Zheng Y, Wang CQ, et al. Isolation and characterization of exosomes for cancer research. J Hematol Oncol. 2020;13(1):152.
Paskeh MDA, Entezari M, Mirzaei S, Zabolian A, Saleki H, Naghdi MJ, et al. Emerging role of exosomes in cancer progression and tumor microenvironment remodeling. J Hematol Oncol. 2022;15(1):83.
Shi X, Wang B, Feng X, Xu Y, Lu K, Sun M. circRNAs and exosomes: a mysterious frontier for human cancer. Mol Ther Nucleic Acids. 2020;19:384–92.
Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38(18): e178.
Chen L, Yu Y, Zhang X, Liu C, Ye C, Fan L. PcircRNA_finder: a software for circRNA prediction in plants. Bioinformatics. 2016;32(22):3528–9.
Pan X, Xiong K. PredcircRNA: computational classification of circular RNA from other long non-coding RNA using hybrid features. Mol Biosyst. 2015;11(8):2219–26.
Wang J, Wang L. Deep learning of the back-splicing code for circular RNA formation. Bioinformatics. 2019;35(24):5235–42.
Cheng J, Metge F, Dieterich C. Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics. 2016;32(7):1094–6.
Gao Y, Wang J, Zhao F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015;16(1):4.
Humphreys DT, Fossat N, Demuth M, Tam PPL, Ho JWK. Ularcirc: visualization and enhanced analysis of circular RNAs via back and canonical forward splicing. Nucleic Acids Res. 2019;47(20): e123.
Westholm JO, Miura P, Olson S, Shenker S, Joseph B, Sanfilippo P, et al. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014;9(5):1966–80.
Gao Y, Zhang J, Zhao F. Circular RNA identification based on multiple seed matching. Brief Bioinform. 2018;19(5):803–10.
Hoffmann S, Otto C, Doose G, Tanzer A, Langenberger D, Christ S, et al. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing and fusion detection. Genome Biol. 2014;15(2):R34.
Li L, Zheng YC, Kayani MUR, Xu W, Wang GQ, Sun P, et al. Comprehensive analysis of circRNA expression profiles in humans by RAISE. Int J Oncol. 2017;51(6):1625–38.
Li L, Bu D, Zhao Y. CircRNAwrap—a flexible pipeline for circRNA identification, transcript prediction, and abundance estimation. FEBS Lett. 2019;593(11):1179–89.
Gaffo E, Buratin A, Dal Molin A, Bortoluzzi S. Sensitive, reliable and robust circRNA detection from RNA-seq with CirComPara2. Brief Bioinform. 2022;23(1):418.
Babin L, Andraos E, Fuchs S, Pyronnet S, Brunet E, Meggetto F. From circRNAs to fusion circRNAs in hematological malignancies. JCI Insight. 2021;6(21): e151513.
Iyer MK, Chinnaiyan AM, Maher CA. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics. 2011;27(20):2903–4.
Akers NK, Schadt EE, Losic B. STAR Chimeric Post for rapid detection of circular RNA and fusion transcripts. Bioinformatics. 2018;34(14):2364–70.
Guarnerio J, Bezzi M, Jeong JC, Paffenholz SV, Berry K, Naldini MM, et al. Oncogenic role of fusion-circRNAs derived from cancer-associated chromosomal translocations. Cell. 2016;165(2):289–302.
You X, Conrad TO. Acfs: accurate circRNA identification and quantification from RNA-Seq data. Sci Rep. 2016;6:38820.
Cai Z, Xue H, Xu Y, Köhler J, Cheng X, Dai Y, et al. Fcirc: A comprehensive pipeline for the exploration of fusion linear and circular RNAs. Gigascience. 2020;9(6):54.
LaHaye S, Fitch JR, Voytovich KJ, Herman AC, Kelly BJ, Lammi GE, et al. Discovery of clinically relevant fusions in pediatric cancer. BMC Genomics. 2021;22(1):872.
Davidson NM, Majewski IJ, Oshlack A. JAFFA: High sensitivity transcriptome-focused fusion gene detection. Genome Med. 2015;7(1):43.
Haas BJ, Dobin A, Li B, Stransky N, Pochet N, Regev A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019;20(1):213.
Kim N, Kim P, Nam S, Shin S, Lee S. ChimerDB–a knowledgebase for fusion sequences. Nucleic Acids Res. 2006;34:D21–4.
Novo FJ, de Mendibil IO, Vizmanos JL. TICdb: a collection of gene-mapped translocation breakpoints in cancer. BMC Genomics. 2007;8:33.
Korla PK, Cheng J, Huang CH, Tsai JJ, Liu YH, Kurubanjerdjit N, et al. FARE-CAFE: a database of functional and regulatory elements of cancer-associated fusion events. Database. 2015;2015:8.
Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, Forbes SA. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer. 2018;18(11):696–705.
Chen L, Wang C, Sun H, Wang J, Liang Y, Wang Y, et al. The bioinformatics toolbox for circRNA discovery and analysis. Brief Bioinform. 2021;22(2):1706–28.
Gao Y, Wang H, Zhang H, Wang Y, Chen J, Gu L. PRAPI: post-transcriptional regulation analysis pipeline for Iso-Seq. Bioinformatics. 2018;34(9):1580–2.
Zhang J, Hou L, Zuo Z, Ji P, Zhang X, Xue Y, et al. Comprehensive profiling of circular RNAs with nanopore sequencing and CIRI-long. Nat Biotechnol. 2021;39(7):836–45.
Rahimi K, Venø MT, Dupont DM, Kjems J. Nanopore sequencing of brain-derived full-length circRNAs reveals circRNA-specific exon usage, intron retention and microexons. Nat Commun. 2021;12(1):4825.
Xin R, Gao Y, Gao Y, Wang R, Kadash-Edmondson KE, Liu B, et al. isoCirc catalogs full-length circular RNA isoforms in human transcriptomes. Nat Commun. 2021;12(1):266.
Liu Z, Tao C, Li S, Du M, Bai Y, Hu X, et al. circFL-seq reveals full-length circular RNAs with rolling circular reverse transcription and nanopore sequencing. Elife. 2021;10: e69457.
Hou L, Zhang J, Zhao F. Full-length circular RNA profiling by nanopore sequencing with CIRI-long. Nat Protoc. 2023;18(6):1795–813.
Wang X, Liu Y, Li J, Wang G. StackCirRNAPred: computational classification of long circRNA from other lncRNA based on stacking strategy. BMC Bioinform. 2022;23(1):563.
Liu Z, Han J, Lv H, Liu J, Liu R. Computational identification of circular RNAs based on conformational and thermodynamic properties in the flanking introns. Comput Biol Chem. 2016;61:221–5.
Niu M, Zhang J, Li Y, Wang C, Liu Z, Ding H, et al. CirRNAPL: A web server for the identification of circRNA based on extreme learning machine. Comput Struct Biotechnol J. 2020;18:834–42.
Yin S, Tian X, Zhang J, Sun P, Li G. PCirc: random forest-based plant circRNA identification software. BMC Bioinform. 2021;22(1):10.
Chaabane M, Williams RM, Stephens AT, Park JW. circDeep: deep learning approach for circular RNA classification from other long non-coding RNA. Bioinformatics. 2019;36(1):73–80.
Meng X, Hu D, Zhang P, Chen Q, Chen M. CircFunBase: a database for functional circular RNAs. Database. 2019;2019:003.
Glazar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. RNA. 2014;20(11):1666–70.
Maass PG, Glazar P, Memczak S, Dittmar G, Hollfinger I, Schreyer L, et al. A map of human circular RNAs in clinically relevant tissues. J Mol Med (Berl). 2017;95(11):1179–89.
Gao Y, Shang S, Guo S, Li X, Zhou H, Liu H, et al. Lnc2Cancer 3.0: an updated resource for experimentally supported lncRNA/circRNA cancer associations and web tools based on RNA-seq and scRNA-seq data. Nucleic Acids Res. 2021;49(D1):D1251.
Li H, Xie M, Wang Y, Yang L, Xie Z, Wang H. riboCIRC: a comprehensive database of translatable circRNAs. Genome Biol. 2021;22(1):79.
Zhou X, Zhan L, Huang K, Wang X. The functions and clinical significance of circRNAs in hematological malignancies. J Hematol Oncol. 2020;13(1):138.
Deng F, Zhang C, Lu T, Liao EJ, Huang H, Wei S. Roles of circRNAs in hematological malignancies. Biomark Res. 2022;10(1):50.
Fan C, Lei X, Tie J, Zhang Y, Wu FX, Pan Y. CircR2Disease v2.0: An Updated Web Server for Experimentally Validated circRNA-disease Associations and Its Application. Genomics Proteom Bioinform. 2022;20(3):435–45.
Lai H, Li Y, Zhang H, Hu J, Liao J, Su Y, et al. exoRBase 20: an atlas of mRNA, lncRNA and circRNA in extracellular vesicles from human biofluids. Nucleic Acids Res. 2022;50(D1):D118–28.
Yang JH, Shao P, Zhou H, Chen YQ, Qu LH. deepBase: a database for deeply annotating and mining deep sequencing data. Nucleic Acids Res. 2010;38:D123–30.
Liu M, Wang Q, Shen J, Yang BB, Ding X. Circbank: a comprehensive database for circRNA with standard nomenclature. RNA Biol. 2019;16(7):899–905.
Gu Y, Wang Y, He L, Zhang J, Zhu X, Liu N, et al. Circular RNA circIPO11 drives self-renewal of liver cancer initiating cells via Hedgehog signaling. Mol Cancer. 2021;20(1):132.
Li S, Li X, Xue W, Zhang L, Yang LZ, Cao SM, et al. Screening for functional circular RNAs using the CRISPR-Cas13 system. Nat Methods. 2021;18(1):51–9.
Sun YM, Wang WT, Zeng ZC, Chen TQ, Han C, Pan Q, et al. circMYBL2, a circRNA from MYBL2, regulates FLT3 translation by recruiting PTBP1 to promote FLT3-ITD AML progression. Blood. 2019;134(18):1533–46.
Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, Hsu PD. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell. 2018;173(3):665–76.
Zhang J, Zhao F. Reconstruction of circular RNAs using Illumina and Nanopore RNA-seq datasets. Methods. 2021;196:17–22.
Liu CX, Guo SK, Nan F, Xu YF, Yang L, Chen LL. RNA circles with minimized immunogenicity as potent PKR inhibitors. Mol Cell. 2022;82(2):420–34.
Yan WX, Chong S, Zhang H, Makarova KS, Koonin EV, Cheng DR, et al. Cas13d Is a Compact RNA-Targeting Type VI CRISPR Effector Positively Modulated by a WYL-Domain-Containing Accessory Protein. Mol Cell. 2018;70(2):327–39.
Kristensen LS, Jakobsen T, Hager H, Kjems J. The emerging roles of circRNAs in cancer and oncology. Nat Rev Clin Oncol. 2022;19(3):188–206.
Zhang J, Zhao F. Characterizing circular RNAs using nanopore sequencing. Trends Biochem Sci. 2021;46(9):785–6.
Dudekula DB, Panda AC, Grammatikakis I, De S, Abdelmohsen K, Gorospe M. CircInteractome: A web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 2016;13(1):34–42.
Song X, Zhang N, Han P, Moon BS, Lai RK, Wang K, et al. Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic Acids Res. 2016;44(9): e87.
Zheng Y, Ji P, Chen S, Hou L, Zhao F. Reconstruction of full-length circular RNAs enables isoform-level quantification. Genome Med. 2019;11(1):2.
He J, Chu Z, Lai W, Lan Q, Zeng Y, Lu D, et al. Circular RNA circHERC4 as a novel oncogenic driver to promote tumor metastasis via the miR-556-5p/CTBP2/E-cadherin axis in colorectal cancer. J Hematol Oncol. 2021;14(1):194.
Das A, Das D, Panda AC. Quantification of Circular RNAs Using Digital Droplet PCR. J Vis Exp. 2022. https://doi.org/10.3791/64464.
Han C, Sun LY, Luo XQ, Pan Q, Sun YM, Zeng ZC, et al. Chromatin-associated orphan snoRNA regulates DNA damage-mediated differentiation via a non-canonical complex. Cell Rep. 2022;38(13): 110421.
Chen B, Shi H, Zhang J, Zhou C, Han M, Jiang W, et al. CRISPR-based RNA-binding protein mapping in live cells. Biochem Biophys Res Commun. 2021;583:79–85.
Chen M, Sui T, Yang L, Qian Y, Liu Z, Liu Y, et al. Live imaging of RNA and RNA splicing in mammalian cells via the dcas13a-SunTag-BiFC system. Biosens Bioelectron. 2022;204: 114074.
Shi P, Murphy MR, Aparicio AO, Kesner JS, Fang Z, Chen Z, et al. Collateral activity of the CRISPR/RfxCas13d system in human cells. Commun Biol. 2023;6(1):334.
Zhang Y, Nguyen TM, Zhang XO, Wang L, Phan T, Clohessy JG, et al. Optimized RNA-targeting CRISPR/Cas13d technology outperforms shRNA in identifying functional circRNAs. Genome Biol. 2021;22(1):41.
Huang W, Zeng ZC, Wang WT, Sun YM, Chen YQ, Luo XQ, et al. A CRISPR/CAS9-based strategy targets the personalized chimeric neosequence in fusion-driven cancer genome for precision medicine. Clin Transl Med. 2021;11(3): e355.
Yang J, Meng X, Pan J, Jiang N, Zhou C, Wu Z, et al. CRISPR/Cas9-mediated noncoding RNA editing in human cancers. RNA Biol. 2018;15(1):35–43.
Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7:11215.
Zhu H, Hu Y, Wang C, Zhang X, He D. CircGCN1L1 promotes synoviocyte proliferation and chondrocyte apoptosis by targeting miR-330-3p and TNF-alpha in TMJ osteoarthritis. Cell Death Dis. 2020;11(4):284.
Gao X, Ma XK, Li X, Li GW, Liu CX, Zhang J, et al. Knockout of circRNAs by base editing back-splice sites of circularized exons. Genome Biol. 2022;23(1):16.
Petkovic S, Muller S. RNA circularization strategies in vivo and in vitro. Nucleic Acids Res. 2015;43(4):2454–65.
Schmidt CA, Matera AG. tRNA introns: Presence, processing, and purpose. Wiley Interdiscip Rev RNA. 2020;11(3): e1583.
Chen YG, Kim MV, Chen X, Batista PJ, Aoyama S, Wilusz JE, et al. Sensing Self and Foreign Circular RNAs by Intron Identity. Mol Cell. 2017;67(2):228–38.
Li X, Yang L, Chen LL. The Biogenesis, Functions, and Challenges of Circular RNAs. Mol Cell. 2018;71(3):428–42.
Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v20: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–7.
Li Y, Huo C, Lin X, Xu J. Computational Identification of Cross-Talking ceRNAs. Adv Exp Med Biol. 2018;1094:97–108.
Digby B, Finn SP. nf-core/circrna: a portable workflow for the quantification, miRNA target prediction and differential expression analysis of circular RNAs. BMC Bioinformatics. 2023;24(1):27.
Abdelmohsen K, Panda AC, Munk R, Grammatikakis I, Dudekula DB, De S, et al. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol. 2017;14(3):361–9.
Dong X, Chen K, Chen W, Wang J, Chang L, Deng J, et al. circRIP: an accurate tool for identifying circRNA-RBP interactions. Brief Bioinform. 2022;23(4):186.
Wang Z, Lei X. Matrix factorization with neural network for predicting circRNA-RBP interactions. BMC Bioinform. 2020;21(1):229.
Brickner JR, Garzon JL, Cimprich KA. Walking a tightrope: The complex balancing act of R-loops in genome stability. Mol Cell. 2022;82(12):2267–97.
Garcia-Muse T, Aguilera A. R Loops: from physiological to pathological roles. Cell. 2019;179(3):604–18.
Niehrs C, Luke B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat Rev Mol Cell Biol. 2020;21(3):167–78.
Crossley MP, Brickner JR, Song C, Zar SMT, Maw SS, Chedin F, et al. Catalytically inactive, purified RNase H1: A specific and sensitive probe for RNA-DNA hybrid imaging. J Cell Biol. 2021;220(9): e202101092.
Sanz LA, Hartono SR, Lim YW, Steyaert S, Rajpurkar A, Ginno PA, et al. Prevalent, Dynamic, and Conserved R-loop structures associate with specific epigenomic signatures in mammals. Mol Cell. 2016;63(1):167–78.
Xu X, Zhang J, Tian Y, Gao Y, Dong X, Chen W, et al. CircRNA inhibits DNA damage repair by interacting with host gene. Mol Cancer. 2020;19(1):128.
Conn VM, Hugouvieux V, Nayak A, Conos SA, Capovilla G, Cildir G, et al. A circRNA from SEPALLATA3 regulates splicing of its cognate mRNA through R-loop formation. Nat Plants. 2017;3:17053.
Reese M, Dhayat SA. Small extracellular vesicle non-coding RNAs in pancreatic cancer: molecular mechanisms and clinical implications. J Hematol Oncol. 2021;14(1):141.
Wang C, Tan S, Liu W-R, Lei Q, Qiao W, Wu Y, et al. RNA-Seq profiling of circular RNA in human lung adenocarcinoma and squamous cell carcinoma. Mol Cancer. 2019;18(1):134.
Nair AA, Niu N, Tang X, Thompson KJ, Wang L, Kocher JP, et al. Circular RNAs and their associations with breast cancer subtypes. Oncotarget. 2016;7(49):80967–79.
Wang YM, Zhao QW, Sun ZY, Lin HP, Xu X, Cao M, et al. Circular RNA hsa_circ_0003823 promotes the tumor progression, metastasis and apatinib resistance of esophageal squamous cell carcinoma by miR-607/CRISP3 Axis. Int J Biol Sci. 2022;18(15):5787–808.
Gong W, Xu J, Wang Y, Min Q, Chen X, Zhang W, et al. Nuclear genome-derived circular RNA circPUM1 localizes in mitochondria and regulates oxidative phosphorylation in esophageal squamous cell carcinoma. Signal Transduct Target Ther. 2022;7(1):40.
Gu L, Sang Y, Nan X, Zheng Y, Liu F, Meng L, et al. circCYP24A1 facilitates esophageal squamous cell carcinoma progression through binding PKM2 to regulate NF-kappaB-induced CCL5 secretion. Mol Cancer. 2022;21(1):217.
Zhang Y, Tang Q, Huang XM, Liao DZ. Circular RNA circCNOT6L regulates cell development through modulating miR-384/FN1 axis in esophageal squamous cell carcinoma. Eur Rev Med Pharmacol Sci. 2020;24(7):3674–85.
Hu X, Wu D, He X, Zhao H, He Z, Lin J, et al. circGSK3beta promotes metastasis in esophageal squamous cell carcinoma by augmenting beta-catenin signaling. Mol Cancer. 2019;18(1):160.
Li S, Hu W, Deng F, Chen S, Zhu P, Wang M, et al. Identification of Circular RNA hsa_circ_0001599 as a novel biomarker for large-artery atherosclerotic stroke. DNA Cell Biol. 2021;40(3):457–68.
Zhang Y, Yang H, Zhang Y, Shi J, Long Y. A Novel Circular RNA CircBRAP may be used as an early predictor of preeclampsia and its potential mechanism. Reprod Sci. 2022;29(9):2565–79.
Zou Y, Zheng S, Deng X, Yang A, Kong Y, Kohansal M, et al. Diagnostic and prognostic value of circular RNA CDR1as/ciRS-7 for solid tumours: A systematic review and meta-analysis. J Cell Mol Med. 2020;24(17):9507–17.
Zou T, Wang PL, Gao Y, Liang WT. Circular RNA_LARP4 is lower expressed and serves as a potential biomarker of ovarian cancer prognosis. Eur Rev Med Pharmacol Sci. 2018;22(21):7178–82.
Zhang X, Su X, Guo Z, Jiang X, Li X. Circular RNA La-related RNA-binding protein 4 correlates with reduced tumor stage, as well as better prognosis, and promotes chemosensitivity to doxorubicin in breast cancer. J Clin Lab Anal. 2020;34(7): e23272.
Wang WT, Chen TQ, Zeng ZC, Pan Q, Huang W, Han C, et al. The lncRNA LAMP5-AS1 drives leukemia cell stemness by directly modulating DOT1L methyltransferase activity in MLL leukemia. J Hematol Oncol. 2020;13(1):78.
Zhao R, Fu J, Zhu L, Chen Y, Liu B. Designing strategies of small-molecule compounds for modulating non-coding RNAs in cancer therapy. J Hematol Oncol. 2022;15(1):14.
Zhang X, Wang S, Wang H, Cao J, Huang X, Chen Z, et al. Circular RNA circNRIP1 acts as a microRNA-149-5p sponge to promote gastric cancer progression via the AKT1/mTOR pathway. Mol Cancer. 2019;18(1):20.
Shi X, Yang J, Liu M, Zhang Y, Zhou Z, Luo W, et al. Circular RNA ANAPC7 Inhibits Tumor Growth and Muscle Wasting via PHLPP2-AKT-TGF-beta Signaling Axis in Pancreatic Cancer. Gastroenterology. 2022;162(7):2004–17.
Zhou X, Liu K, Cui J, Xiong J, Wu H, Peng T, et al. Circ-MBOAT2 knockdown represses tumor progression and glutamine catabolism by miR-433-3p/GOT1 axis in pancreatic cancer. J Exp Clin Cancer Res. 2021;40(1):124.
Gao C, Xu YJ, Qi L, Bao YF, Zhang L, Zheng L. CircRNA VIM silence synergizes with sevoflurane to inhibit immune escape and multiple oncogenic activities of esophageal cancer by simultaneously regulating miR-124/PD-L1 axis. Cell Biol Toxicol. 2022;38(5):825–45.
Liu Y, Chen X, Liu J, Jin Y, Wang W. Circular RNA circ_0004277 Inhibits Acute Myeloid Leukemia Progression Through MicroRNA-134-5p / Single stranded DNA binding protein 2. Bioengineered. 2022;13(4):9662–73.
Wang N, Yang B, Jin J, He Y, Wu X, Yang Y, et al. Circular RNA circ_0040823 inhibits the proliferation of acute myeloid leukemia cells and induces apoptosis by regulating miR-516b/PTEN. J Gene Med. 2022;24(3): e3404.
Pomeroy AE, Schmidt EV, Sorger PK, Palmer AC. Drug independence and the curability of cancer by combination chemotherapy. Trends Cancer. 2022;8(11):915–29.
Parikh K, Banna G, Liu SV, Friedlaender A, Desai A, Subbiah V, et al. Drugging KRAS: current perspectives and state-of-art review. J Hematol Oncol. 2022;15(1):152.
Jin H, Wang L, Bernards R. Rational combinations of targeted cancer therapies: background, advances and challenges. Nat Rev Drug Discov. 2023;22(3):213–34.
Jin P, Jiang J, Zhou L, Huang Z, Nice EC, Huang C, et al. Mitochondrial adaptation in cancer drug resistance: prevalence, mechanisms, and management. J Hematol Oncol. 2022;15(1):97.
Liu XY, Zhang Q, Guo J, Zhang P, Liu H, Tian ZB, et al. The Role of Circular RNAs in the drug resistance of cancers. Front Oncol. 2021;11: 790589.
Wang WT, Han C, Sun YM, Chen TQ, Chen YQ. Noncoding RNAs in cancer therapy resistance and targeted drug development. J Hematol Oncol. 2019;12(1):55.
Xu J, Wan Z, Tang M, Lin Z, Jiang S, Ji L, et al. N(6)-methyladenosine-modified CircRNA-SORE sustains sorafenib resistance in hepatocellular carcinoma by regulating β-catenin signaling. Mol Cancer. 2020;19(1):163.
Xie H, Yao J, Wang Y, Ni B. Exosome-transmitted circVMP1 facilitates the progression and cisplatin resistance of non-small cell lung cancer by targeting miR-524-5p-METTL3/SOX2 axis. Drug Deliv. 2022;29(1):1257–71.
Ling Y, Liang G, Lin Q, Fang X, Luo Q, Cen Y, et al. circCDYL2 promotes trastuzumab resistance via sustaining HER2 downstream signaling in breast cancer. Mol Cancer. 2022;21(1):8.
Li J, Ma M, Yang X, Zhang M, Luo J, Zhou H, et al. Circular HER2 RNA positive triple negative breast cancer is sensitive to Pertuzumab. Mol Cancer. 2020;19(1):142.
Sullenger BA, Nair S. From the RNA world to the clinic. Science. 2016;352(6292):1417–20.
Saw PE, Song EW. siRNA therapeutics: a clinical reality. Sci China Life Sci. 2020;63(4):485–500.
Ruger J, Ioannou S, Castanotto D, Stein CA. Oligonucleotides to the (Gene) Rescue: FDA Approvals 2017–2019. Trends Pharmacol Sci. 2020;41(1):27–41.
Meng X, Chen Q, Zhang P, Chen M. CircPro: an integrated tool for the identification of circRNAs with protein-coding potential. Bioinformatics. 2017;33(20):3314–6.
Xia X, Li X, Li F, Wu X, Zhang M, Zhou H, et al. A novel tumor suppressor protein encoded by circular AKT3 RNA inhibits glioblastoma tumorigenicity by competing with active phosphoinositide-dependent Kinase-1. Mol Cancer. 2019;18(1):131.
Jiang T, Xia Y, Lv J, Li B, Li Y, Wang S, et al. A novel protein encoded by circMAPK1 inhibits progression of gastric cancer by suppressing activation of MAPK signaling. Mol Cancer. 2021;20(1):66.
Liang ZX, Liu HS, Xiong L, Yang X, Wang FW, Zeng ZW, et al. A novel NF-kappaB regulator encoded by circPLCE1 inhibits colorectal carcinoma progression by promoting RPS3 ubiquitin-dependent degradation. Mol Cancer. 2021;20(1):103.
Zhang M, Zhao K, Xu X, Yang Y, Yan S, Wei P, et al. A peptide encoded by circular form of LINC-PINT suppresses oncogenic transcriptional elongation in glioblastoma. Nat Commun. 2018;9(1):4475.
Zhang Y, Zhang X, Dai K, Zhu M, Liang Z, Pan J, et al. Bombyx mori Akirin hijacks a viral peptide vSP27 encoded by BmCPV circRNA and activates the ROS-NF-kappaB pathway against viral infection. Int J Biol Macromol. 2022;194:223–32.
Ju M, Kim D, Son G, Han J. Circular RNAs in and out of Cells: Therapeutic Usages of Circular RNAs. Mol Cells. 2023;46(1):33–40.
Chuang TJ, Wu CS, Chen CY, Hung LY, Chiang TW, Yang MY. NCLscan: accurate identification of non-co-linear transcripts (fusion, trans-splicing and circular RNA) with a good balance between sensitivity and precision. Nucleic Acids Res. 2016;44(3): e29.
Chen CK, Cheng R, Demeter J, Chen J, Weingarten-Gabbay S, Jiang L, et al. Structured elements drive extensive circular RNA translation. Mol Cell. 2021;81(20):4300–18.
Ye CY, Zhang X, Chu Q, Liu C, Yu Y, Jiang W, et al. Full-length sequence assembly reveals circular RNAs with diverse non-GT/AG splicing signals in rice. RNA Biol. 2017;14(8):1055–63.
Szabo L, Morey R, Palpant NJ, Wang PL, Afari N, Jiang C, et al. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015;16(1):126.
Wan C, Gao J, Zhang H, Jiang X, Zang Q, Ban R, et al. CPSS 2.0: a computational platform update for the analysis of small RNA sequencing data. Bioinformatics. 2017;33(20):3289–91.
Andres-Leon E, Nunez-Torres R, Rojas AM. miARma-Seq: a comprehensive tool for miRNA, mRNA and circRNA analysis. Sci Rep. 2016;6:25749.
Gao Y, Wang J, Zheng Y, Zhang J, Chen S, Zhao F. Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat Commun. 2016;7:12060.
Wang D. hppRNA-a Snakemake-based handy parameter-free pipeline for RNA-Seq analysis of numerous samples. Brief Bioinform. 2018;19(4):622–6.
Pan X, Xiong K, Anthon C, Hyttel P, Freude KK, Jensen LJ, et al. WebCircRNA: Classifying the Circular RNA Potential of Coding and Noncoding RNA. Genes (Basel). 2018;9(11):89.
Jia GY, Wang DL, Xue MZ, Liu YW, Pei YC, Yang YQ, et al. CircRNAFisher: a systematic computational approach for de novo circular RNA identification. Acta Pharmacol Sin. 2019;40(1):55–63.
Mangul S, Yang HT, Strauli N, Gruhl F, Porath HT, Hsieh K, et al. ROP: dumpster diving in RNA-sequencing to find the source of 1 trillion reads across diverse adult human tissues. Genome Biol. 2018;19(1):36.
Sekar S, Geiger P, Adkins J, Tassone E, Serrano G, Beach TG, et al. ACValidator: A novel assembly-based approach for in silico verification of circular RNAs. Biol Methods Protoc. 2020;5(1):010.
Li X, Wu Y. Detecting circular RNA from high-throughput sequence data with de Bruijn graph. BMC Genomics. 2020;1:749.
Li X, Chu C, Pei J, Mandoiu I, Wu Y. CircMarker: a fast and accurate algorithm for circular RNA detection. BMC Genomics. 2018;6:572.
Ma XK, Wang MR, Liu CX, Dong R, Carmichael GG, Chen LL, et al. CIRCexplorer3: A CLEAR Pipeline for Direct Comparison of Circular and Linear RNA Expression. Genomics Proteomics Bioinform. 2019;17(5):511–21.
Chen CY, Chuang TJ. NCLcomparator: systematically post-screening non-co-linear transcripts (circular, trans-spliced, or fusion RNAs) identified from various detectors. BMC Bioinformatics. 2019;20(1):3.
Zhang J, Chen S, Yang J, Zhao F. Accurate quantification of circular RNAs identifies extensive circular isoform switching events. Nat Commun. 2020;11(1):90.
Wu J, Li Y, Wang C, Cui Y, Xu T, Wang C, et al. CircAST: full-length assembly and quantification of alternatively spliced isoforms in circular RNAs. Genomics Proteomics Bioinformatics. 2019;17(5):522–34.
Nguyen DT, Trac QT, Nguyen TH, Nguyen HN, Ohad N, Pawitan Y, et al. Circall: fast and accurate methodology for discovery of circular RNAs from paired-end RNA-sequencing data. BMC Bioinform. 2021;22(1):495.
Stefanov SR, Meyer IM. CYCLeR—a novel tool for the full isoform assembly and quantification of circRNAs. Nucleic Acids Res. 2023;51(2):e10.
Jakobi T, Uvarovskii A, Dieterich C. circtools-a one-stop software solution for circular RNA research. Bioinformatics. 2019;35(13):2326–8.
Wu Y, Zhao B, Chen X, Geng X, Zhang Z. Circ_0009910 sponges miR-491-5p to promote acute myeloid leukemia progression through modulating B4GALT5 expression and PI3K/AKT signaling pathway. Int J Lab Hematol. 2022;44(2):320–32.
Chang W, Shang Z, Ming X, Wu J, Xiao Y. Circ-SFMBT2 facilitates the malignant growth of acute myeloid leukemia cells by modulating miR-582-3p/ZBTB20 pathway. Histol Histopathol. 2022;37(2):137–49.
Guo L, Kou R, Song Y, Li G, Jia X, Li Z, et al. Serum hsa_circ_0079480 is a novel prognostic marker for acute myeloid leukemia. J Clin Lab Anal. 2022;36(4): e24337.
Yang X, Li Y, Zhang Y, Liu J. Circ_0000745 promotes acute lymphoblastic leukemia progression through mediating miR-494-3p/NET1 axis. Hematology. 2022;27(1):11–22.
Tang YL, Su JY, Luo JS, Zhang LD, Zheng LM, Liang C, et al. Gene Expression Network and Circ_0008012 Promote Progression in MLL/AF4 Positive Acute Lymphoblastic Leukemia. Recent Pat Anticancer Drug Discov. 2023;18(4):538–48.
Xia L, Wu L, Bao J, Li Q, Chen X, Xia H, et al. Circular RNA circ-CBFB promotes proliferation and inhibits apoptosis in chronic lymphocytic leukemia through regulating miR-607/FZD3/Wnt/β-catenin pathway. Biochem Biophys Res Commun. 2018;503(1):385–90.
Yang F, Jin H, Que B, Chao Y, Zhang H, Ying X, et al. Dynamic m(6)A mRNA methylation reveals the role of METTL3-m(6)A-CDCP1 signaling axis in chemical carcinogenesis. Oncogene. 2019;38(24):4755–72.
Wu Z, Sun H, Liu W, Zhu H, Fu J, Yang C, et al. Circ-RPL15: a plasma circular RNA as novel oncogenic driver to promote progression of chronic lymphocytic leukemia. Leukemia. 2020;34(3):919–23.
Li S, Chen J, Fan Y, Xu X, Xiong M, Qi Y, et al. circZNF91 promotes the malignant phenotype of chronic lymphocytic leukemia cells by targeting the miR-1283/WEE1 Axis. Biomed Res Int. 2022;2022:2855394.
Zhong AN, Yin Y, Tang BJ, Chen L, Shen HW, Tan ZP, et al. CircRNA Microarray Profiling Reveals hsa_circ_0058493 as a novel biomarker for imatinib-resistant CML. Front Pharmacol. 2021;12: 728916.
Lu YH, Huang ZY. Global identification of circular RNAs in imatinib (IM) resistance of chronic myeloid leukemia (CML) by modulating signaling pathways of circ_0080145/miR-203/ABL1 and circ 0051886/miR-637/ABL1. Mol Med. 2021;27(1):148.
Feng Y, Zhang L, Wu J, Khadka B, Fang Z, Gu J, et al. CircRNA circ_0000190 inhibits the progression of multiple myeloma through modulating miR-767-5p/MAPK4 pathway. J Exp Clin Cancer Res. 2019;38(1):54.
Liu J, Du F, Chen C, Li D, Chen Y, Xiao X, et al. CircRNA ITCH increases bortezomib sensitivity through regulating the miR-615-3p/PRKCD axis in multiple myeloma. Life Sci. 2020;262: 118506.
Ye DX, Wang SS, Huang Y, Chi P. A 3-circular RNA signature as a noninvasive biomarker for diagnosis of colorectal cancer. Cancer Cell Int. 2019;19:276.
Tian J, Xi X, Wang J, Yu J, Huang Q, Ma R, et al. CircRNA hsa_circ_0004585 as a potential biomarker for colorectal cancer. Cancer Manag Res. 2019;11:5413–23.
Wang J, Li X, Lu L, He L, Hu H, Xu Z. Circular RNA hsa_circ_0000567 can be used as a promising diagnostic biomarker for human colorectal cancer. J Clin Lab Anal. 2018;32(5): e22379.
Pan B, Qin J, Liu X, He B, Wang X, Pan Y, et al. Identification of Serum Exosomal hsa-circ-0004771 as a novel diagnostic biomarker of colorectal cancer. Front Genet. 2019;10:1096.
Lei B, Zhou J, Xuan X, Tian Z, Zhang M, Gao W, et al. Circular RNA expression profiles of peripheral blood mononuclear cells in hepatocellular carcinoma patients by sequence analysis. Cancer Med. 2019;8(4):1423–33.
Zhu K, Zhan H, Peng Y, Yang L, Gao Q, Jia H, et al. Plasma hsa_circ_0027089 is a diagnostic biomarker for hepatitis B virus-related hepatocellular carcinoma. Carcinogenesis. 2020;41(3):296–302.
Yang C, Dong Z, Hong H, Dai B, Song F, Geng L, et al. circFN1 mediates sorafenib resistance of hepatocellular carcinoma cells by sponging miR-1205 and regulating E2F1 expression. Mol Ther Nucleic Acids. 2020;22:421–33.
Li Z, Zhou Y, Yang G, He S, Qiu X, Zhang L, et al. Using circular RNA SMARCA5 as a potential novel biomarker for hepatocellular carcinoma. Clin Chim Acta. 2019;492:37–44.
Yao T, Chen Q, Shao Z, Song Z, Fu L, Xiao B. Circular RNA 0068669 as a new biomarker for hepatocellular carcinoma metastasis. J Clin Lab Anal. 2018;32(8): e22572.
Jiang Z, Shen L, Wang S, Wu S, Hu Y, Guo J, et al. Hsa_circ_0028502 and hsa_circ_0076251 are potential novel biomarkers for hepatocellular carcinoma. Cancer Med. 2019;8(17):7278–87.
Liu B, Tian Y, Chen M, Shen H, Xia J, Nan J, et al. CircUBAP2 promotes MMP9-mediated oncogenic effect via sponging miR-194-3p in hepatocellular carcinoma. Front Cell Dev Biol. 2021;9: 675043.
Tian M, Chen R, Li T, Xiao B. Reduced expression of circRNA hsa_circ_0003159 in gastric cancer and its clinical significance. J Clin Lab Anal. 2018;32(3): e22281.
Li P, Chen H, Chen S, Mo X, Li T, Xiao B, et al. Circular RNA 0000096 affects cell growth and migration in gastric cancer. Br J Cancer. 2017;116(5):626–33.
Li P, Chen S, Chen H, Mo X, Li T, Shao Y, et al. Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin Chim Acta. 2015;444:132–6.
Chen S, Li T, Zhao Q, Xiao B, Guo J. Using circular RNA hsa_circ_0000190 as a new biomarker in the diagnosis of gastric cancer. Clin Chim Acta. 2017;466:167–71.
Zhao Q, Chen S, Li T, Xiao B, Zhang X. Clinical values of circular RNA 0000181 in the screening of gastric cancer. J Clin Lab Anal. 2018;32(4): e22333.
Lu J, Zhang PY, Xie JW, Wang JB, Lin JX, Chen QY, et al. Hsa_circ_0000467 promotes cancer progression and serves as a diagnostic and prognostic biomarker for gastric cancer. J Clin Lab Anal. 2019;33(3): e22726.
Shao Y, Chen L, Lu R, Zhang X, Xiao B, Ye G, et al. Decreased expression of hsa_circ_0001895 in human gastric cancer and its clinical significances. Tumour Biol. 2017;39(4):1010428317699125.
Yang C, Deng S. Hsa_circ_0017728 as an oncogene in gastric cancer by sponging miR-149 and modulating the IL-6/STAT3 pathway. Arch Med Sci. 2022;18(6):1558–71.
Shen Y, Zhang N, Chai J, Wang T, Ma C, Han L, et al. CircPDIA4 induces gastric cancer progression by promoting ERK1/2 activation and enhancing biogenesis of oncogenic circRNAs. Cancer Res. 2023;83(4):538–52.
Tang G, Xie W, Qin C, Zhen Y, Wang Y, Chen F, et al. Expression of circular RNA circASXL1 correlates with TNM classification and predicts overall survival in bladder cancer. Int J Clin Exp Pathol. 2017;10(8):8495–502.
Song Z, Zhang Q, Zhu J, Yin G, Lin L, Liang C. Identification of urinary hsa_circ _0137439 as potential biomarker and tumor regulator of bladder cancer. Neoplasma. 2020;67(1):137–46.
Liu F, Zhang H, Xie F, Tao D, Xiao X, Huang C, et al. Hsa_circ_0001361 promotes bladder cancer invasion and metastasis through miR-491-5p/MMP9 axis. Oncogene. 2020;39(8):1696–709.
Lu Q, Liu T, Feng H, Yang R, Zhao X, Chen W, et al. Circular RNA circSLC8A1 acts as a sponge of miR-130b/miR-494 in suppressing bladder cancer progression via regulating PTEN. Mol Cancer. 2019;18(1):111.
Li J, Li Z, Jiang P, Peng M, Zhang X, Chen K, et al. Circular RNA IARS (circ-IARS) secreted by pancreatic cancer cells and located within exosomes regulates endothelial monolayer permeability to promote tumor metastasis. J Exp Clin Cancer Res. 2018;37(1):177.
Li L, Wang N, Wang J, Li J. Hsa_circRNA_001859 regulates pancreatic cancer progression and epithelial-mesenchymal transition through the miR-21-5p/SLC38A2 pathway. Cancer Biomark. 2023;37(1):39–52.
Zhao SY, Wang J, Ouyang SB, Huang ZK, Liao L. Salivary Circular RNAs Hsa_Circ_0001874 and Hsa_Circ_0001971 as novel biomarkers for the diagnosis of oral squamous cell carcinoma. Cell Physiol Biochem. 2018;47(6):2511–21.
Zhang H, Shen Y, Zhang B, Qian M, Zhang Y, Yang H. Hsa_circ_0003829 serves as a potential diagnostic predictor for oral squamous cell carcinoma. J Int Med Res. 2020;48(9):300060520936880.
Gao F, Han J, Wang Y, Jia L, Luo W, Zeng Y. Circ_0109291 promotes cisplatin resistance of oral squamous cell carcinoma by sponging miR-188-3p to Increase ABCB1 Expression. Cancer Biother Radiopharm. 2022;37(4):233–45.
Lu GJ, Cui J, Qian Q, Hou ZB, Xie HY, Hu W, et al. Overexpression of hsa_circ_0001715 is a potential diagnostic and prognostic biomarker in lung adenocarcinoma. Onco Targets Ther. 2020;13:10775–83.
Liu XX, Yang YE, Liu X, Zhang MY, Li R, Yin YH, et al. A two-circular RNA signature as a noninvasive diagnostic biomarker for lung adenocarcinoma. J Transl Med. 2019;17(1):50.
Wang J, Zhao X, Wang Y, Ren F, Sun D, Yan Y, et al. circRNA-002178 act as a ceRNA to promote PDL1/PD1 expression in lung adenocarcinoma. Cell Death Dis. 2020;11(1):32.
Zhao D, Liu H, Liu H, Zhang X, Zhang M, Kolluri VK, et al. Downregulated expression of hsa_circ_0037515 and hsa_circ_0037516 as novel biomarkers for non-small cell lung cancer. Am J Transl Res. 2020;12(1):162–70.
Wang K, Sun Y, Tao W, Fei X, Chang C. Androgen receptor (AR) promotes clear cell renal cell carcinoma (ccRCC) migration and invasion via altering the circHIAT1/miR-195-5p/29a-3p/29c-3p/CDC42 signals. Cancer Lett. 2017;394:1–12.
Chen Z, Xiao K, Chen S, Huang Z, Ye Y, Chen T. Circular RNA hsa_circ_001895 serves as a sponge of microRNA-296-5p to promote clear cell renal cell carcinoma progression by regulating SOX12. Cancer Sci. 2020;111(2):713–26.
Yan Y, Wang H, Hu J, Guo T, Dong Q, Yin H, et al. CircRNA-104718 promotes glioma malignancy through regulation of miR-218-5p/HMGB1 signalling pathway. Metab Brain Dis. 2023;38(5):1531–42.
Li G, Lan Q. Exosome-mediated transfer of circ-GLIS3 enhances temozolomide resistance in glioma cells through the miR-548m/MED31 Axis. Cancer Biother Radiopharm. 2023;38(1):62–73.
Li Y, Liu Y, Zhang J, Li J, Shu Y. Propofol suppresses glioma tumorigenesis by regulating circ_0047688/miR-516b-5p/IFI30 Axis. Biochem Genet. 2023;61(1):151–69.
Ma Z, Ma J, Lang B, Xu F, Zhang B, Wang X. Circ_0001982 Up-regulates the expression of E2F1 by adsorbing miR-1205 to facilitate the progression of glioma. Mol Biotechnol. 2023;65(3):466–76.
Zhao S, Li B, Zhao R, Pan Z, Zhang S, Qiu W, et al. Hypoxia-induced circADAMTS6 in a TDP43-dependent manner accelerates glioblastoma progression via ANXA2/ NF-κB pathway. Oncogene. 2023;42(2):138–53.
Hua X, Zhang C, Ba Y, Zhao S, Fan K, Wang B. CircRNA circ_POSTN promotes the malignancy of glioma by regulating the miR-433-3p/SPARC axis. Metab Brain Dis. 2023;38(2):543–55.
Hu Y, Song Q, Zhao J, Ruan J, He F, Yang X, et al. Identification of plasma hsa_circ_0008673 expression as a potential biomarker and tumor regulator of breast cancer. J Clin Lab Anal. 2020;34(9): e23393.
Hu Y, Zhu Y, Zhang W, Lang J, Ning L. Utility of plasma circBNC2 as a diagnostic biomarker in epithelial ovarian cancer. Onco Targets Ther. 2019;12:9715–23.
Lan X, Cao J, Xu J, Chen C, Zheng C, Wang J, et al. Decreased expression of hsa_circ_0137287 predicts aggressive clinicopathologic characteristics in papillary thyroid carcinoma. J Clin Lab Anal. 2018;32(8): e22573.
Cui X, Chen J, Zheng Y, Shen H. Circ_0000745 promotes the progression of cervical cancer by regulating miR-409-3p/ATF1 Axis. Cancer Biother Radiopharm. 2022;37(9):766–78.
Acknowledgements
Not applicable.
Funding
This research was supported by National Key R&D Program of China (Nos. 2021YFA1300502, 2022YFA1300020), National Natural Science Foundation of China (No. 32170570) and Guangdong Province (Nos. 2021B1515020002, 2019JC05N394, 2022A1515140018), and Fundamental Research Funds for the Central Universities, Sun Yat-sen University (No. 231gbj010).
Author information
Authors and Affiliations
Contributions
WTW and YQC designed the study and drafted the manuscript. XYF and SXZ prepared the tables and figures and drafted the manuscript. KJP and HJH provided some important guidance on the revised manuscript. All authors participated in the process of drafting and revising the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Feng, XY., Zhu, SX., Pu, KJ. et al. New insight into circRNAs: characterization, strategies, and biomedical applications. Exp Hematol Oncol 12, 91 (2023). https://doi.org/10.1186/s40164-023-00451-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-023-00451-w