
Abstract
Background
Long-term outcome is unfavourable for relapsed/refractory (r/r) lymphoma patients who are resistant to salvage chemotherapy, even after subsequent autologous stem-cell transplantation (ASCT). Although anti-CD30 chimeric antigen receptor (CAR30) T-cell therapy induces high response rates in these patients, the duration of response is relatively limited.
Methods
This open-label, single-center and single-arm pilot study investigated the safety and efficacy of ASCT in tandem with CAR30 T-cell infusion in r/r CD30+ lymphoma. The primary endpoint was safety and key secondary endpoint was overall response rate, overall survival, progression-free survival, and duration of response.
Results
Five classical Hodgkin lymphoma (cHL) patients and 1 anaplastic lymphoma kinase (ALK)-negative anaplastic large cell lymphoma (ALCL) patient were enrolled. The median age was 24 years. No patient had prior ASCT. Three patients (50.0%) relapsed for ≥ 2 times and 3 patients (50.0%) had primary refractory diseases. All had a Deauville score of 4 or 5, and 5 patients (83.3%) had a stable or progressive disease (SD/PD) at enrollment. All patients received myeloablative chemotherapy and infused CD34-positive hematopoietic stem cells (HSCs) and CAR30 T cells in tandem, with a median dose of 3.9 × 106/kg and 7.6 × 106/kg, respectively. Five paitents presented with cytokine release syndrome (CRS), all of which were grade 1. No neurotoxicity was observed. All patients had successful HSCs engraftment and reached an objective response, including 5 (4 cHL and 1 ALCL, 83.3%) with a complete response (CR) and 1 with a partial response (PR). With a median follow-up of 20.4 (range, 12.1–34.4) months, all remained alive and maintained their responses.
Conclusion
Our work demonstrates the combined administration of ASCT and CAR30 T-cell therapy is well-tolerate and highly effective in r/r cHL and ALCL, even in PET-positive or chemorefractory patients who are expected to have inferior outcome after ASCT, although further large-scaled validation in prospective clinical trial is warranted.
Trial registration The trial was registered with the Chinese Clinical Trial Registry (ChiCTR, number ChiCTR2100053662).
Similar content being viewed by others


Background
Approximately 80% of patients with classical Hodgkin lymphoma (cHL) are treated with first-line therapy [1]. Second-line high-dose chemotherapy (HDT) or targeted chemotherapy followed by autologous stem cell transplantation (ASCT) is recommended in patients with relapsed/refractory (r/r) cHL [2]. However, durable remission after HDT/ASCT was achieved in only approximately half of the patients. Long-term survival is most likely achieved in patients who are sensitive to salvage therapy and have a complete response (CR) before ASCT, whereas outcomes are unfavorable in patients with residual disease. Currently, positron emission tomography (PET)-adapted approaches are widely used to treat cHL. Patients with a negative [18F] fluorodeoxyglucose (FDG) activity determined by PET (Deauville 1–3) before ASCT had an event-free survival (EFS) of 80.0% versus 28.6% for patients with a positive result (Deauville 4–5) [3]. With the goal of achieving a PET-negative CR, patients with active disease are candidates for alternative salvage chemotherapy, radiotherapy, CD30-directed therapy, checkpoint inhibitors, or participation in clinical trials.
The limited expression of CD30 in normal tissues and its consistent overexpression in cHL and anaplastic large cell lymphoma (ALCL) have encouraged the development of CD30-directed therapy [4]. Brentuximab vedotin (BV), a CD30-directed antibody–drug conjugate, has been approved for the treatment of cHL and peripheral T-cell lymphoma and was commercially available in China in 2020. Currently, clinical trials of anti-CD30 chimeric antigen receptor (CAR30) T-cell therapy are actively conducted in r/r cHL and ALCL, with an overall response rate (ORR) of 53.0–78.0% and a limited median progression-free survival (PFS) of approximately 6.0 months [5,6,7], indicating the need for novel strategies to further improve its long-term disease control. In our previous study on CAR30 T-cell therapy followed by anti-PD-1 antibody maintenance in r/r cHL and ALCL, the best CR rate was 77.8% with a median PFS of 13.0 months [5]. Owing to the potential synergistic effects of the combined administration of myeloablative ASCT and adoptive T-cell immunotherapy [8], we conducted an open-label, single-center, single-arm pilot study to explore the safety and efficacy of ASCT in tandem with CAR30 T-cell infusion in r/r CD30+ lymphoma.
Methods
Study design and enrollment
We carried out an open-label, single-center and single-arm pilot study to evaluate the safety and efficacy of tandem ASCT and CAR30 T cell infusion in r/r CD30 + lymphoma. This study was conducted in compliance with the principles of the Declaration of Helsinki. Approval from the Institutional Review Board of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, was obtained before the start of the study. Written informed consent was obtained from all participants. The primary endpoint was safety and the key secondary endpoint were ORR, overall survival (OS), PFS, and duration of response (DOR). This trial was registered in the Chinese Clinical Trial Registry (ChiCTR, number ChiCTR2100053662).
Eligible patients relapsed or refractory to at least two lines of salvage therapy. The diagnosis was verified according to the World Health Organization (WHO) classification of hematopoietic and lymphoid tissue tumors. CD30 expression was determined by immunohistochemical staining of malignant cells. The inclusion and exclusion criteria are outlined in the Supplemental Methods.
Clinical procedures
Eligible patients received hematopoietic stem cells (HSCs) mobilization and apheresis for ASCT and lymphocyte apheresis to obtain adequate lymphocytes from peripheral blood for chimeric antigen receptor (CAR) T cell manufacturing. The CAR used in this trial is a third-generation CAR composed of a single-chain variable fragment (scFv) targeting CD30 (patent number: CN106589139B), two costimulatory domains from CD28 and 4-1BB, and a CD3ζ chain as the activation domain [5]. Validation of the CAR constructs and procedures for cell production and quality control assays have been described previously [5]. Assessment of T-cell subsets of CAR30 T-cell products was performed as previously described [9] before infusion. Patients will receive a standard dose of BEAM (300 mg/m2 bis-carmusitine, − 7 days; 200 mg/m2 etoposide, − 6 to − 3 days; 400 mg/m2 cytarabine, − 6 to − 3 days; and 140 mg/m2 melphalan, − 2 days) with or without fludarabine (25 mg/m2 for− 6 day) as preconditioning, which was determined at the discretion of the investigators based on the clinical conditions of patients. HSCs were infused on day zero, followed by infusion of CAR30 T cells on day 2 to day 6. Cryopreserved HSCs collected before enrollment were considered acceptable for infusion. The response was assessed by imaging evaluations every 3 months in the 1st year and every 6–12 months thereafter. All patients were followed-up until death, loss to follow-up, or withdrawal of consent. The clinical procedure is illustrated in Fig. 1A.

Flow chart, immunophenotype of CAR30 T-cell products and copies of CAR30 transgenes. A. Flow chart of study procedures. B. CD8/CD4 ratio of CAR30 T-cell products. C. The proportion of naïve T cells (TN, CD45RA + CCR7 +), effector (TEFF, CD45RA + CCR7-), central memory T cells (TCM, CD45RA-CCR7 +) and effector memory T cells (TEM, CD45RA-CCR7-) in the CAR30 T-cell products. D. The copies of CAR30 transgenes in the peripheral blood detected by ddPCR. The lower limit of quantitation was 50 copies/μg (the horizontal red line)
Laboratory assessments and follow-up
The in vivo expansion of CAR30 T cells was detected using droplet digital polymerase chain reaction (ddPCR) [5]. The cytokine levels were measured according to the manufacturer’s instructions. The cytokine release syndrome (CRS) was evaluated according to the scale proposed by Lee et al. [10]. Immune effector cell-associated neurotoxicity syndrome (ICANS) and other adverse events (AEs) were graded according to the American Society for Transplantation and Cellular Therapy Consensus [10] and the National Cancer Institute Common Terminology Criteria for Adverse Events V5.0. Staging and responses were assessed according to the US National Comprehensive Cancer Network and Lugano Treatment Response Criteria. Disease stage and response to treatment were assessed using PET/CT or CT, and defined according to the National Comprehensive Cancer Network Guidelines and the Lugano 2014 Guidelines.
Statistical analysis
PFS and OS were defined as the time from the date of the infusion of CAR30 T cell to the date of the first relapse or death due to any reason, respectively. DOR was estimated as the time from the first response to first progression or death. The Kaplan–Meier method was used to analyze the survival rates. Statistical significance was set at a two-sided P-value of < 0.05 (and two-sided) was assumed to be statistically significant. All statistical analyses were performed using GraphPad Prism version 8.
Results
Patients and baseline features
Between June 1, 2019, and May 1, 2021, six patients were enrolled in this study, including five with cHL and one with anaplastic lymphoma kinase (ALK)-negative ALCL. The median age of the patients was 24 years. None of the patients had a history of undergoing ASCT. Three patients (50.0%) relapsed ≥ 2 times, and three patients (50.0%) had primary refractory diseases. All had a Deauville score of 4 or 5, and 5 patients (83.3%) had a stable or progressive disease (SD/PD) to previous treatments at enrollment. None of the patients had previously received ASCT. At enrollment, 5 patients had SD/PD. The baseline characteristics of the enrolled patients are summarized in Table 1.
Infusion and kinetics of CAR30 T-cells and ASCT
The clinical procedure is shown in the flowchart in Fig. 1A. All patients underwent HSCs mobilization, apheresis, and lymphocyte apheresis (Fig. 1A). The median manufacturing time of CAR30 T-cell was 14.0 (interquartile range [IQR], 13.3–14.0) days. The patients were administered BEAM for preconditioning, including two patients who received additional fludarabine treatment (Table 1). CD34+ cells were infused at day zero with a median dosage of 3.9 (IQR, 3.2–6.1) × 106/kg and followed by the infusion of CAR30 T cells with a median dosage of 7.6 (IQR, 5.5–9.7) × 106/kg at day 2 to day 6. The phenotype of CAR30 T cell products was detected before infusion, as shown in Fig. 1B, C. The CAR30 T-cell products of patient 2 showed a high proportion of CD4 and a low proportion of TN, which may suggest less persistence and efficacy [11]. The median copies of peak expansion of CAR30 T cells was 4.2 (IQR, 2.6–4.4) × 104/ug DNA (Fig. 1D). Persistent CAR30 transgenes were detected in the peripheral blood with a median duration time of 3.8 (IQR, 3.5–4.0) months (Fig. 1D).
Safety
AEs are listed in Table 2. Cytopenia was the most common severe AEs (grade ≥ 3) within the 1st month. All the patients had grade 3 neutropenia and thrombocytopenia. The engraftments of neutrophil or platelet occured at a median time of 13.5 (IQR,12.3–14.0) days or 11.5 (IQR, 11.0–12.8) days, respectively, suggesting rapid multilineage engraftments post ASCT (Fig. 2A). CRS developed in five (83.3%) patients at a median time of 3.0 (IQR, 3.0–4.0) days after chimeric antigen receptor (CAR) T-cell infusion (Additional file 1: Table S1), lagging behind the rise in interleukin-6 (Fig. 2B). All CRS were of grade 1 and mostly resolved within 7.0 (IQR, 2.0–7.0) days after supportive care. ICANS was not observed. Although preconditioned with BEAM, none of the patients experienced a severe infection within the 1st month. 8 months later, one (16.7%) of them developed mild pneumonia due to infection with pneumocystis jirovecii and streptococcus after withdrawal of co-trimoxazole prophylaxis, and recovered rapidly after antibiotic treatment. Infection is one of the most common AEs following CD19-directed CAR T-cell therapy, with incidence ranging from 17.0% to 42.0% in the 1st month and 14.0–31.0% in the 1st year [12], suggesting the need for standardized infection prophylaxis [13].

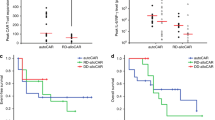
Multilineage engraftments and level of inflammatory cytokines of patients. A. Engraftment times of neutrophil or platelet of each patient after HSCs infusion. The red column represents the engraftment of neutrophil and the blue column represents the engraftment of platelet. B. The serum interleukin-6 (IL-6) level of each patient was assessed before and at serial time points after CAR T-cell infusion. C. The serum CRP level of each patient was assessed before and at serial time points after CAR T-cell infusion. D. The serum ferritin level of each patient was assessed before and at serial time points after CAR T-cell infusion. The horizontal red line denotes the normal limit of quantitation
Efficacy and long-term follow-up
At month 3 post-HSCs infusion, all patients achieved objective responses, including five (83.3%) with a CR and one (16.7%) with a patial response (PR) (Fig. 3A). With a median follow-up of 20.4 (range, 12.1–34.4) months, the median PFS and OS were not reached (Fig. 3B, C). On January 31, 2022, the data cutoff date, all patients maintained their responses and remained alive without disease relapse or progression (Fig. 3A), indicating that superior response and durable remission could be achieved when ASCT and CAR T-cell infusion were sequentially administered. Of note, CR was sustained in all five (83.3%) patients who had an SD/PD at enrollment, which highlighted that even in PET-positive or chemorefractory patients who were expected to have inferior outcomes after ASCT, long-term disease control could be realized when ASCT followed by CAR T-cell infusion.

Long-term outcome of patients. A. Duration time of remission after ASCT following CAR30 T-Cell infusion. The red column represents complete response (CR) and the blue column represents partial response (PR). Arrow indicates ongoing response. B, C. Propability of PFS (B) or OS (C) of enrolled patients after ASCT following CAR30 T-Cell infusion. Tick marks represent censored data. P patient
Discussion
Previous studies have demonstrated up to 30% of patients with r/r CD30+ lymphoma will not be ineligible for ASCT because of the lack of chemosensitivity, which have an extremely poor prognosis [14]. Even in approximately 50% of patients eligible for ASCT, recurrence occurs post ASCT, which partially due to less than a CR to second-line therapy before ASCT [15]. For ASCT, a remission status of PR and above is necessary. Patients with FDG-PET positivity after HDT showed poor outcomes [3]. Therefore, new treatment options are urgently needed. CD30 is often expressed in cHL and ALCL and has proven to be an excellent target for immune-based therapies [16]. In CD30+ lymphoma, the antibody–drug conjugate against CD30, BV has shown impressive activity, but the remissions are sustained only in less than 25% of patients with r/r lymphoma [17]. Although previous clinical trials of CAR30 T-cell therapy provided preliminary efficacy in r/r CD30+ lymphoma, a considerable number of patients failed to achieve durable CR. Carlos et al. reported the outcomes of 41 patients who received CAR30 T-cell. The complete remission rate for all evaluable patients was 60.0%, and 1-year PFS is 36.0% [7]. Wang et al. reported that out of 18 patients who received CAR30 T-cells, the remission rate for PR and above was 67.0%, with a median PFS of 6.0 months [18]. Timothy J et al. showed that the CR rate of patients receiving CAR30 T-cells was 67.0%, with a median PFS of 11 months [6]. Furthermore, our previous study have shown that the CR rate of patients received CAR30 T-cells followed by anti-PD-1 antibody maintenance was 77.8% in total of nine patients [5], with updated 2-year PFS of 44.4% with a median follow-up of 38.3 months (range, 0–63 months) (Additional file 1: Figure S1), indicating novel strategies to improve the efficacy of CAR30 T-cells were needed.
Accumulating evidence suggests that the efficacy of adoptive T-cell therapy can be enhanced by myeloablative stem cell transplantation for the treatment of B-cell lymphoma [8, 19]. Myeloablative preconditioning administered before adoptive T-cell infusion eliminates tumor cells and immunosuppressive elements in the microenvironment and induces a favorable cytokine profile to recruit and motivate immune cells, which may improve the efficacy of CAR T-cell therapy [20]. In addition, reduced tumor burden and suppressed monocytes and macrophages in the tumor microenvironment may alleviate the severity of CRS and ICANS [21]. Thus, this combination may result in enhanced synergistic response, mild toxicity, reduced relapse, and durable remission. In our study, when treated with ASCT in tandem with CAR30 T-cell infusion, superior response and survival can be obtained, even in 5 patients with a SD/PD (83.3%) before transplantation, for whom ASCT alone can hardly provide durable disease. We report our results in six r/r CD30+ lymphoma patients (five with cHL and one with ALCL), with ORR and CR rates of 100% and 83.3%, respectively. The median OS and PFS of the patients were not reached with a median follow-up of 20.4 months, which was far superior to that of the same CAR30 T cell infusion without ASCT [5] (Additional file 1: Figure S1). The therapy was well-tolerated, with only grade 1 CRS and no CRES. In contrast to our previous study of CAR30 T-cell therapy [5], the peak of IL-6 and serum ferritin levels during CRS were slightly lower in patients treated with ASCT in tandem with CAR30 T-cell infusion, while the expansion of CAR-T cells was no significant difference, although further large-scale validation in double-arm clinical trials is warranted. Tolerable CRS and excellent PFS suggest that potential synergistic effects may be generated when ASCT and CAR T cell therapies are combined. In addition, a second infusion of CAR19 T-cells has been considered a strategy to overcome CAR19 T-cell therapy failure in r/r B-cell malignancies [22]. In our study, patient 3 received mouse-derived CAR30 T-cell treatment prior to ASCT in tandem with CAR30 T-cell infusion, and ddPCR showed durable persistence of CAR30 T-cells, revealing that ASCT in tandem with CAR30 T-cell infusion may improve the efficacy of repeated CAR-T cell infusions.
The limitations of this study include the fact that only one patient received BV treatment in this study because of the approval of BV from the National Medical Products Administration in May 2020, later than the beginning of this study. However, all other patients received anti-PD-1 antibody treatment, which showed better clinical efficacy than that of BV [23].
Conclusions
Taken together, tandam administration in ASCT and CAR T-cell therapy is well tolerated and highly active in r/r cHL and ALCL, although further large-scale validation in prospective clinical trials is warranted.
Availability of data and materials
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AEs:
-
Adverse events
- ALCL:
-
Anaplastic large cell lymphoma
- ALK:
-
Anaplastic lymphoma kinase
- ASCT:
-
Autologous stem-cell transplantation
- BV:
-
Brentuximab vedotin
- CAR:
-
Chimeric antigen receptor
- CAR30:
-
Anti-CD30 chimeric antigen receptor
- ChiCTR:
-
Chinese clinical trial registry
- cHL:
-
Classical Hodgkin lymphoma
- CR:
-
Complete response
- CRS:
-
Cytokine release syndrome
- ddPCR:
-
Droplet digital polymerase chain reaction
- DOR:
-
Duration of response
- EFS:
-
Event-free survival
- FDG:
-
Fluorodeoxyglucose
- HDT:
-
High-dose chemotherapy
- HSCs:
-
Hematopoietic stem cells
- HSCs:
-
Hematopoietic stem cells
- ICANS:
-
Immune effector cell-associated neurotoxicity syndrome
- IQR:
-
Interquartile range
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- PET:
-
Positron emission tomography
- PFS:
-
Progression-free survival
- PR:
-
Partial response
- r/r:
-
Relapsed/refractory
- scFv:
-
Single-chain variable fragment
- SD/PD:
-
Stable or progressive disease
- WHO:
-
World Health Organization
References
Glimelius I, Ekberg S, Jerkeman M, Chang ET, Björkholm M, Andersson TM, et al. Long-term survival in young and middle-aged Hodgkin lymphoma patients in Sweden 1992–2009-trends in cure proportions by clinical characteristics. Am J Hematol. 2015;90(12):1128–34.
Ansell SM. Hodgkin lymphoma: a 2020 update on diagnosis, risk-stratification, and management. Am J Hematol. 2020;95(8):978–89.
Moskowitz CH, Matasar MJ, Zelenetz AD, Nimer SD, Gerecitano J, Hamlin P, et al. Normalization of pre-ASCT, FDG-PET imaging with second-line, non-cross-resistant, chemotherapy programs improves event-free survival in patients with Hodgkin lymphoma. Blood. 2012;119(7):1665–70.
Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J Clin Invest. 2017;127(9):3462–71.
Wang D, Zeng C, Xu B, Xu JH, Wang J, Jiang LJ, et al. Anti-CD30 chimeric antigen receptor T cell therapy for relapsed/refractory CD30(+) lymphoma patients. Blood Cancer J. 2020. https://doi.org/10.1038/s41408-020-0274-9.
Voorhees TJ, Zhao B, Oldan J, Hucks GE, Khandani A, Dittus C, et al. Pretherapy metabolic tumor volume associates with response to CD30 CAR T cells in Hodgkin lymphoma. Blood Adv. 2021. https://doi.org/10.1182/bloodadvances.2021005385.
Ramos CA, Grover NS, Beaven AW, Lulla PD, Wu MF, Ivanova A, et al. Anti-CD30 CAR-T cell therapy in relapsed and refractory Hodgkin lymphoma. J Clin Oncol. 2020;38(32):3794–804.
Sauter CS, Senechal B, Rivière I, Ni A, Bernal Y, Wang X, et al. CD19 CAR T cells following autologous transplantation in poor-risk relapsed and refractory B-cell non-Hodgkin lymphoma. Blood. 2019;134(7):626–35.
Rossi J, Paczkowski P, Shen YW, Morse K, Flynn B, Kaiser A, et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood. 2018;132(8):804–14.
Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transpl. 2019;25(4):625–38.
Bao F, Wan W, He T, Qi F, Liu G, Hu K, et al. Autologous CD19-directed chimeric antigen receptor-T cell is an effective and safe treatment to refractory or relapsed diffuse large B-cell lymphoma. Cancer Gene Ther. 2019;26(7–8):248–55.
Wudhikarn K, Palomba ML, Pennisi M, Garcia-Recio M, Flynn JR, Devlin SM, et al. Infection during the first year in patients treated with CD19 CAR T cells for diffuse large B cell lymphoma. Blood Cancer J. 2020;10(8):79.
Hill JA, Li D, Hay KA, Green ML, Cherian S, Chen X, et al. Infectious complications of CD19-targeted chimeric antigen receptor-modified T-cell immunotherapy. Blood. 2018;131(1):121–30.
Vassilakopoulos TP, Asimakopoulos JV, Konstantopoulos K, Angelopoulou MK. Optimizing outcomes in relapsed/refractory Hodgkin lymphoma: a review of current and forthcoming therapeutic strategies. Therapeutic Adv Hematol. 2020;11:2040620720902911.
Bentolila G, Pavlovsky A. Relapse or refractory Hodgkin lymphoma: determining risk of relapse or progression after autologous stem-cell transplantation. Leuk Lymphoma. 2020;61(7):1548–54.
Grover NS, Savoldo B. Challenges of driving CD30-directed CAR-T cells to the clinic. BMC Cancer. 2019;19(1):203.
Chen R, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ, et al. 5-year survival and durability results of brentuximab vedotin in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2016;128(12):1562–6.
Wang CM, Wu ZQ, Wang Y, Guo YL, Dai HR, Wang XH, et al. Autologous T cells expressing CD30 chimeric antigen receptors for relapsed or refractory Hodgkin lymphoma: an open-label phase I trial. Clin Cancer Res. 2017;23(5):1156–66.
Cao Y, Xiao Y, Wang N, Wang G, Huang L, Hong Z, et al. CD19/CD22 chimeric antigen receptor T cell cocktail therapy following autologous transplantation in patients with relapsed/refractory aggressive B cell lymphomas. Transpl Cell Ther. 2021;27(11):910.e1.
Wrzesinski C, Restifo NP. Less is more: lymphodepletion followed by hematopoietic stem cell transplant augments adoptive T-cell-based anti-tumor immunotherapy. Curr Opin Immunol. 2005;17(2):195–201.
Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–48.
Gauthier J, Bezerra ED, Hirayama AV, Fiorenza S, Sheih A, Chou CK, et al. Factors associated with outcomes after a second CD19-targeted CAR T-cell infusion for refractory B-cell malignancies. Blood. 2021;137(3):323–35.
Kuruvilla J, Ramchandren R, Santoro A, Paszkiewicz-Kozik E, Gasiorowski R, Johnson NA, et al. Pembrolizumab versus brentuximab vedotin in relapsed or refractory classical Hodgkin lymphoma (KEYNOTE-204): an interim analysis of a multicentre, randomised, open-label, phase 3 study. Lancet Oncol. 2021;22(4):512–24.
Acknowledgements
We thank the faculty and staff of the Clinical and Laboratory Unit of the Department of Hematology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, for their clinical and technical support. We also appreciate Wuhan Bio-Raid Biotechnology Co., Ltd. for their services in cell manufacturing and quality control.
Funding
This work was supported by funding from the Key Program of the National Natural Science Foundation of China (81830008 to Dr. J.Z.), the National Natural Science Foundation of China (82070211 and 81670152 to Dr. L.H.), the National High Technology Research and Development Program of China (863 program 2014AA020532, to Dr. L.H.), the Milstein Medical Asian American Partnership Foundation (2018 MMAAP Foundation Hematology Fellowship Award, to Dr. L.H.) and the Young Top-notch Talent Cultivation Program of Hubei Province (to Dr. L. H.).
Author information
Authors and Affiliations
Contributions
JZ and LH designed and supervised the clinical study; TZ, HZ, and SZ supervised CAR T-cell production; HZ and SZ conducted preclinical validation and quality control; JZ, LH, YC, JW, JW, PZ, MZ, ZM, DW, YX, and YZ enrolled patients and took care of the patients; LC, JW, JW, PZ, MZ ZM, and DW contributed to the laboratory tests and response assessment of the patients; PZ, XY, LH, and DW collected and analyzed the clinical data; PZ, XY, and LH performed statistical analyses; LH, XY, PZ, and YC drafted and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was conducted in compliance with the principles of the Declaration of Helsinki. Ethics approval from the Institutional Review Board of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, was obtained before the start of the study. Written informed consent was obtained from all participants. This trial was registered in the Chinese Clinical Trial Registry (ChiCTR, number ChiCTR2100053662).
Consent for publication
Written informed consent for publication was obtained.
Competing interests
Authors H.Z., S.Z., and T.Z. were employed by Wuhan Bio-Raid Biotechnology Co., Ltd.. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplemental Materials.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, P., Yang, X., Cao, Y. et al. Autologous stem cell transplantation in tandem with Anti-CD30 CAR T-cell infusion in relapsed/refractory CD30+ lymphoma. Exp Hematol Oncol 11, 72 (2022). https://doi.org/10.1186/s40164-022-00323-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-022-00323-9