
Abstract
Background
Pancreatic cancer continues to be one of the most aggressive malignant tumors. Work in recent years in cancer molecular biology has revealed that metabolic reprogramming is an additional hallmark of cancer that is involved in the pathogenesis of cancers, and is intricately linked to gene mutations.
Main text
However, though oncogenes such as KRAS and c-Myc play important roles in the process, and have been extensively studied, no substantial improvements in the prognosis of pancreatic cancer have seen. Therefore, some scientists have tried to explain the mechanisms of abnormal cancer metabolism from the perspective of tumor suppressor genes. In this paper, we reviewed researches about how metabolic reprogramming was regulated by tumor suppressor genes in pancreatic cancer and their clinical implications.
Conclusion
Abnormal metabolism and genetic mutations are mutually causal and complementary in tumor initiation and development. A clear understanding of how metabolic reprogramming is regulated by the mutated genes would provide important insights into the pathogenesis and ultimately treatment of pancreatic cancer.
Similar content being viewed by others

Background
Pancreatic cancer is one of the most aggressive forms of cancer and the fourth leading cause of cancer-related mortality both in men and women [1]. Although lots of money and efforts has been invested to study it, the results are disappointing. The overall 5-year survival rates of pancreatic cancer according to the latest data was only about 8%, even in the US where the best hospitals and cancer research institutions in the world are situated [1]
An increasing body of research suggests that an additional hallmark of cancer is involved in the pathogenesis of cancer, that is, the capability to modify, or reprogram, cellular metabolism in order to support tumor proliferation [2]. For example, under aerobic conditions, normal cells process glucose, first to pyruvate via glycolysis in the cytoplasm and thereafter to oxidative phosphorylation in the mitochondria; while cancer cells consume glucose avidly but they only use a small amount for tricarboxylic acid (TCA) even in the presence of ample oxygen [3, 4]. This anomalous characteristic of cancer cell energy metabolism was first observed by Otto Warburg and is termed “aerobic glycolysis” and also known as “the Warburg effect” [5]. Another remarkable metabolic feature of tumor cells is glutaminolysis; the process by which glutamine is metabolized to α-ketoglutarate by glutamate. Glutamine is the most abundant amino acid in human serum, and through glutaminolysis it can provide a ready supply of carbon for TCA anaplerosis and other cellular pathways [6, 7]. For tumor cells, enhanced reliance on this cascade leads to glutamine dependency for cell growth and survival. The metabolic alterations and adaptations of cancer cells create a phenotype that is essential for tumor cell growth and survival, altering flux along key metabolic pathways such as glycolysis and glutaminolysis. Some authors even divided pancreatic cancer into four subtypes according to phenotypes with distinct types of energy metabolism [8], and there is increasing evidence for the therapeutic potential of targeting cancer metabolic reprogramming [9].
Work in recent years in cancer molecular biology has revealed that metabolic regulation is intricately linked to gene mutations, promoted by oncogenes and inhibited by tumor suppressors, and drives cancer progression, indicating that it is intrinsically associated with oncogenic transformation [10]. This is particularly evident in the initiation and development of pancreatic cancer. Evidence suggests that pancreatic cancer is actually a genetic disease. The successive accumulation of mutations in key genes leads to pancreatic cancer that once established is a complex, heterogeneous and genetically unstable disease [11]. Given that human pancreatic cancer is characterized by profound gene mutations [12], a clear understanding of how the mutated genes participate in the initiation and development would provide important insights into disease pathogenesis and ultimately treatment. Among the complex mutational landscape, they can be roughly divided into two categories: oncogenes and tumor suppressor genes. Oncogenes such as KRAS, c-Myc and so on play important roles in tumor initiation and development, and have been extensively studied. Nevertheless, the frustrating outcomes of a series of subsequent explorations and clinical trials told us that pancreatic cancer is much more complicated than we had thought [13, 14]. Especially for KRAS, the most frequent mutation in pancreatic cancer. All clinical trials failed so far either due to its negative safety profiles or the slight effect in clinical trials which is significantly attenuated compared with the effects in preclinical trials [15,16,17]. While on the other hand, more and more research indicated that a series of tumor suppressor genes may provide new ideas in effective pancreatic cancer treatments [18,19,20,21]. Therefore, in this paper, we reviewed recent research about metabolic reprogramming by tumor suppressor genes in pancreatic cancer, and hope to identify novel molecular targets for the development of chemotherapeutic approaches in PDAC (pancreatic ductal adenocarcinoma).
Main text
Regulation of metabolic reprogramming by tumor suppressor genes
TP53
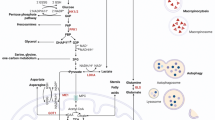
TP53, first described in 1979, was the first and most famous tumor-suppressor gene to be identified. It plays a critical role in maintaining genomic stability and preventing tumorigenesis, hence the reference to TP53 as “guardian of the genome” [22]. TP53 is mutated in more than 50% of PDAC cases, generally by missense alterations of the DNA-binding domain. Major advances in our understanding of p53 in cancer biology have been made by investigating its activities in regulating the cell cycle, senescence, apoptosis, and genomic stability [22]. In recent years, the roles of p53 in cancer metabolism have been increasingly recognized (Fig. 1).

The mechanisms of glucose metabolism alteration regulated by p53 in pancreatic cancer. The positive regulations are shown in arrows, and the negative regulations are shown in T-bar lines. TIGAR, TP53-induced glycolysis and apoptosis regulator; RRAD, Ras-related associated with diabetes; SCO2, synthesis of cytochrome oxidase 2; COX, cytochrome c oxidase; Fru-2,6-P2: fructose-2,6-bisphosphate; GLUT, glucose transporter; MCT: monocarboxylate transporter; MPC: mitochondrial pyruvate carrier
The roles of TP53 in glycolysis of pancreatic cancer cells
The loss of TP53 can alter metabolism in pancreatic cancer cells by inhibition of mitochondrial respiration and concurrent stimulation of glycolysis. This effect is mediated by regulating the cytochrome c oxidase (COX) complex through the downstream mediator Synthesis of Cytochrome c Oxidase (SCO)2, which is the major site of oxygen utilization in eukaryotic cells [23]. TP53 loss can also result in increased glycolysis by downregulating TIGAR (TP53-induced glycolysis and apoptosis regulator), whose expression lowers fructose-2,6-bisphosphate levels in cells which is a potent positive allosteric effector of 6-phosphofructo-1-kinase (PFK) that can stimulate glycolysis [24]. Butera et al. reported that mutant p53 can support glycolysis by preventing the nuclear translocation of the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) stabilizing its cytoplasmic localization in pancreatic cancer cells [25]. Lactate dehydrogenase-A (LDH-A) is an enzyme which promote pyruvate to metabolize to lactate during the process of aerobic glycolysis. Rajeshkumar et al. reported that FX11, a small-molecule inhibitor of LDH-A, can significantly suppress the growth of patient-derived mouse xenograft (PDX) models of pancreatic cancer harboring TP53 mutation [26]. Mutation of TP53 can also alter metabolism of pancreatic cancer by regulating the transmembrane (GLUT) proteins, which mediate glucose uptake in eukaryotic cells and are involved in the first step of the glucose utilization cascade. Fabiana et al. [27] found that in normal conditions, p53 has repressive effect on transcriptional activity of the GLUT1 and GLUT4 gene promoters, when it is mutated, the repressive effect was lost, and thereby resulting in increased glucose transportation and cell energy supply. It is also reported that p53 can inhibit the translocation of GLUT1 and represses glycolysis under hypoxic conditions by inducing RRAD (Ras-related associated with diabetes), the Ras-related small GTPase [28]. Kawauchi et al. found in p53-deficient primary mouse embryonic fibroblasts (MEFs), the activity of NF-κB (nuclear factor kappa-B) was enhanced. When NF-κB expression was absent, the oncogenic Ras-induced cell transformation and acceleration of aerobic glycolysis were suppressed in p53-deficient cells, but can be restored by GLUT3 expression, indicating that TP53 mutation can facilitate the glycolysis by upregulating the expression of GLUT3 through NF-κB pathway [29]. p53 can also regulate metabolic reprogramming through post-transcriptional mechanisms. For example, Kim et al. reported that p53 can suppress glycolysis through the regulation of microRNA-34a (miR-34a), a microRNA which targets multiple glycolytic enzymes, including hexokinase 1, hexokinase 2, glucose-6-phosphate isomerase and PDK1 in PDAC cells [30].
Recent research has identified some other roles of p53 in glycolysis. Tumor cells are in low glucose state due to the greatly increased consumption of glucose. Low-glucose conditions normally activate AMP-activated protein kinase (AMPK), whose activation induces phosphorylation of p53 at serine 15, and this phosphorylation is required to initiate AMPK-dependent cell-cycle arrest, which further causes cells to stop dividing or proliferating. However, when TP53 is mutated, the AMPK-dependent cell-cycle arrest in low-glucose state is abolished, thus leading to unlimited abnormal proliferation of cells that contribute to tumor initiation [31]. Lactic acid is a significant byproduct of cancer cell metabolism. Tumor cells need to deal with high levels of lactate due to elevated glycolytic flux, to remove excess carbon and maintain cellular NADPH stores. A large family of monocarboxylate transporters (MCTs) are described as H+/lactate symporters capable of bidirectional transport of lactic acid across the plasma membrane [32]. Among them, MCT1 is the most ubiquitous. Romain et al. found that p53-deficiency in tumors allows them to adapt to metabolic needs by facilitating lactate export or import depending on the glucose availability by MCT1 elevation [33].
The roles of TP53 in glutamine metabolism of pancreatic cancer cells
TP53 can also exert its tumor suppressor function by regulating glutamine metabolism. Glutaminase 2 (GLS2) is an enzyme which plays a key role in conversion of glutamine to glutamate, and thus regulating glutathione (GSH) synthesis and energy production. It was reported that p53 can upregulate GLS2, which facilitate glutamine metabolism and lower intracellular ROS levels, and the authors found that GLS2 suppress tumor cell growth and was lower expressed in liver tumors than normal tissues, indicating GLS2 has the potential tumor-suppressor role [34]. Tran et al. reported that when TP53 was mutated, the level of endogenous mutant p53 protein can affect cell sensitivity to glutamine withdrawal in human lymphoma cells by regulating the transactivation of p53-target gene CDKN1, thus triggering cell cycle arrest and promoting cell survival [35].
TP53 can also promote cancer cell proliferation by the regulation of metabolic reprogramming
However, as a tumor suppressor gene, p53 can also promote cancer cell proliferation and survival under glutamine starvation. Tajan et al. reported that p53 can help colon cancer cell line HCT116 cells survive in the absence of extra cellular glutamine by inducing the expression of SLC1A3 (solute carrier family 1 member 3), an aspartate/glutamate transporter [36]. Lowman et al. found that p53 can promote MEF cells adapt to glutamine deprivation by inducing the expression of an arginine transporter SLC7A3, and the significant influx of arginine serves as an effector for mTORC1 (mammalian target of rapamycin complex 1) activation which promote cell growth [37]. And it was reported recently that wild-type p53 can reduce pyruvate uptake of mitochondrion and increase glycolysis by promoting PUMA (p53 up-regulated apoptosis regulators), which inhibits mitochondrial pyruvate uptake by disrupting the function of mitochondrial pyruvate carrier (MPC) through PUMA-MPC interaction in hepatocellular carcinoma [38].
Therefore, though p53 is a classical tumor suppressor gene which inhibits the initiation of cancers in most cases, it could also have a counterintuitive effect to promote tumorigenesis, which illustrates the complexity of genes and metabolisms in the initiation and development of tumors.
CDKN2A
CDKN2A, also called p16 or INK4A, can lose its functions by mutation, deletion, or promoter hypermethylation, which occurs in 80–95% of sporadic PDAC, and is generally seen in moderately advanced lesions and highly associated with familial pancreatic cancer [11, 39]. Normally, CDKN2A inhibits complexes of cyclin D and the cyclin-dependent kinases CDK4/6, which mediate phosphorylation of RB tumor suppressor protein, thereby blocking entry into the S (DNA synthesis) phase of the cell cycle. Baschnagel et al. reported that GLUT1 expression was significantly higher in CDKN2A-negative head and neck squamous cell carcinomas, and high GLUT1 expressing tumors were associated with worse local control and disease-free survival. This indicates that CDKN2A may exert its tumor suppressor function by inhibiting expression of GLUT1 [40] Ju et al. reported that in case of Kras activation, p16 loss can accelerate oxidation of NADH and support increased glycolysis by generating NAD + , a substrate for GAPDH-mediated glycolytic reaction through Rb-E2F-NOX4 pathway, thereby promoting PDAC cell growth [41].
However, like the role of p53, CDKN2A also has the opposite roles in metabolism. For example, Aharon et al. reported that CDKN2A can enhance glucose uptake by upregulating genes associated with glucose metabolism, including Aldob, which encodes the glycolytic enzyme fructose-bisphosphate aldolase B, and Gck, which encodes glucokinase, the enzyme that controls glucose uptake and glycolysis rates, and the glucose uptake rate was 1.7-fold higher in cells isolated from p16-expressing islets than that in control islets [42]. There are still no relevant reports about the roles of CDKN2A in glutamine metabolism of pancreatic cancer.
SMAD4
Loss of the SMAD4/DPC4 tumor suppressor is another frequent event associated with PDAC progression. SMAD4 is targeted for deletion or intragenic point mutations in about 50% of PDAC cases, and serves as a central component in the transforming growth factor (TGF-β) signaling cascade [11]. Correspondingly, the mechanism by which SMAD4 loss contributes to tumorigenesis is likely to involve its central role in TGF-β mediated growth inhibition. There is recent evidence that SMAD4 deficiency may inhibit TGF-β induced cell cycle arrest and cell migration, while not affecting epithelial–mesenchymal transition (EMT), thereby shifting the balance of TGF-β signaling from tumor suppression to tumor promotion [43]. Li et al. reported that inhibition of SMAD4 in pancreatic beta-cells conferred mild but significant improvements in glucose levels and glucose tolerance in high fat diet-induced obese mice [44], which often occurs in pancreatic cancer. SMAD4 can also function as a negative regulatory element in the glucose transport system, targeted by specific miRNAs, such as miR-29a and miR-23a [45]. Liang et al. found loss of SMAD4 can enhance glycolysis and aggressive tumor behavior by upregulating phosphoglycerate kinase 1 (PGK1) in PDAC [19]. The relationship between SMAD4 and glutamine metabolism in tumors remains to be further studied.
RB
Since the retinoblastoma target gene RB was sequenced by Lee and colleagues in the 1980s, it has been regarded as a well-characterized tumor suppressor that exerts its function by inhibiting cell-cycle progression from G0/G1 to S phase [46]. The genetic alterations of RB have been found in many kinds of tumors, including osteosarcoma, renal cell carcinoma, soft-tissue sarcoma, and breast, lung (small cell) and prostate cancer, indicating an extensive role for RB dysfunction in the initiation and development of human tumors [47, 48]. In PDAC, it has been shown that RB is mutated or deleted in about 5% of patients [49]. Recent reports suggest that RB plays important roles in multiple biochemical pathways required for tumorigenesis, including cell cycle control, apoptosis, angiogenesis, metastasis, as well as cellular metabolism. It is reported that the expression and activity of glycolytic enzymes such as hexokinase isozyme II (HK2) [50] and lactic dehydrogenase (LDH) [51], are increased in retinoblastoma as well as other RB-deleted cancers. RB can recruit selective corepressor complexes, such as histone deacetylases, to silence gene transcription when bounded with the E2F activator transcription factors. And it is also well established that c-Myc can stimulate glycolytic flux to lactate via its control of glycolytic mRNAs including GLUT1, HK2, PKM2, and LDH-A [52]. Although the precise mechanisms for that have not been completely defined, considering that derepressed E2F-1 activity leads to increased expression of c-Myc through direct transcriptional activation of the Myc promoter, and the special association between RB and E2F, we can infer that RB could inhibit glycolysis of cancer cells through E2F-Myc-HK2/LDH pathway. In addition, it is reported that loss of RB function can cause a significant increase in activated Ras, which induces expression of 6-phosphofructo-2-kinase 3 (PFKFB3). PFKFB3 synthesizes fructose 2,6-bisphosphate (F2,6P2), which allosterically activates 6-phosphofructo-1-kinase (PFK-1), thus resulting in increased glycolytic metabolism in cancer cells [53]. Furthermore, while RB is the repressor of E2F-dependent transcription, which can directly induce the gene encoding pyruvate dehydrogenase kinase (PDK) 4, a key nutrient sensor and modulator of glucose homeostasis, thereby inhibiting glucose oxidation. Michael et al. demonstrated that loss of RB function can trigger enrichment of E2F1 occupancy onto the PDK4 promoter, thereby resulting in enhanced glucose uptake [54].
RB can also affect tumor growth by regulating glutamine metabolism. It is reported that glutamine uptake was significantly increased in immortalized mouse embryonic fibroblasts lacking RB family, and was mediated in part through increased expression of the glutamine transporter, ASCT2 (alanine-serine-cysteine transporter 2) and GLS1 [55]. Beyond the enhanced glutamine uptake, loss of the RB family leads to increased incorporation of glutamine into the TCA metabolite aspartate for mitochondrial function, and significantly reduces GSH levels, which is essential for maintaining redox homeostasis and facilitation of certain enzymatic reactions [55]. All these mechanisms render cancer cells glutamine “addicted” when RB function was suppressed. In addition, several genes have been found to be directly suppressed by RB-E2F-1, including subunits of ATP synthase, cytochrome c oxidase, ubiquinol-cytochrome c reductase, and the succinate dehydrogenase complex which are involved in electron transport chain activity and oxidative phosphorylation [56].
PTEN
The tumor suppressor gene PTEN (phosphatase and tensin homologue) is frequently mutated or deleted in many types of tumors including PDAC [57,58,59] and germline mutations of PTEN is associated with multiple hamartoma disorders. There are no exact data on the mutation rate of PTEN in pancreatic cancer, Wartenberg et al. reported that PTEN expression was lost in 60% PDAC cases, 27.8% in pancreatic intraepithelial neoplasia (PanINs) and 13.7% in non-neoplastic pancreatic tissues [60]. As a lipid phosphatase, PTEN dephosphorylates phosphatidylinositol 3,4,5-trisphosphate, the second messenger produced by phosphatidylinositol 3-kinase (PI3K), and therefore negatively regulates the PI3K/AKT signaling pathway [61]. AKT is a critical regulator of the glycolytic pathway and has been shown to enhance glycolysis by several ways including: (1) inducing translocation of glucose transporters, which is the first rate-limiting step for glucose metabolism to the plasma membrane; (2) activating glycolytic enzymes, such as HK2 and PFK; (3) phosphorylating and inactivating directly tumor suppressor tuberous sclerosis protein 2, a negative regulator of mammalian target of rapamycin complex (mTORC) 1, which functions as a key metabolic integration point and promotes glycolysis in cells. Martin et al. observed that PTEN loss can increase the expression of pAKT and enhance glucose metabolism in PTEN null prostate cancer cell lines [62]. Shinde et al. reported that PTEN can suppress the vesicular trafficking of GLUT1, an important glucose transporter in cancer cells, by physically interacting with the retromer complex named SNX27 (sorting nexin 27) which recycles transmembrane receptors [63]. Garcia-Cao et al. reported that PTEN can suppress the uptake of glucose, and redirect a greater fraction of glycolytic products into mitochondrial oxidative phosphorylation by regulating pyruvate kinase (PK) through mTORC1 [64]. Zhao et al. reported that oroxylin A, a natural active flavonoid, can induce the downregulation of mouse double minute 2 (MDM2) transcription by promoting the lipid phosphatase activity of PTEN, and further suppress the MDM2-mediated degradation of p53, thereby inhibiting glycolysis in MCF-7 and HCT116 cells [65]. In addition, PTEN can also negatively affect glycolysis by regulating PFKFB3, an essential enzyme and control point in glycolysis though the E3 ubiquitin ligase APC/Cdh1 complex. And in the same way, PTEN elevation can suppress gultaminolysis through enhancing degradation of GLS1, the first rate-limiting enzyme in glutaminolysis [64].
FBW7
F-box and WD repeat domain-containing (FBW) 7 is the substrate recognition component for the Skp1-Cul1-F-box (SCF) ubiquitin ligase complex and also a tumor suppressor; the regulatory network of which is perturbed in many human malignancies, including breast carcinoma, and colon, gastric and pancreatic cancer [66,67,68,69]. Overall, approximately 6% of human tumors harbor FBW7 mutations, and we previously reported that fewer than 2% of pancreatic cancer samples harbored FBW7 mutations, according to sequencing analysis [68]. FBW7 can bind to key regulators of cell division and growth after they have been phosphorylated within conserved phospho-degron motifs, including cyclin E, MYC, JUN and Notch. Most FBW7 substrates are proto-oncogenes that are broadly implicated in the pathogenesis of human cancers, and thus the loss of FBW7 function can lead to chromosomal instability and tumor initiation [70]. Researches have also found that FBW7 has intimate relationships with tumor metabolism. Ji et al. reported that almost all the enzymes related to glucose transportation (GLUT1, GLUT4, HK2, LDHA, and LDHB) decreased dramatically in FBW7-overexpressing PDAC cells compared with the control cells, and FBW7 negatively regulated the metabolism of glucose through regulation of the c-Myc/TXNIP (Thioredoxin-Binding Protein) axis in pancreatic cancer [71]. However, a recent report from Davis et al. found that though FBW7 mutation is closely associated with genes involved in mitochondrial function, FBW7 mutations shift cellular metabolism toward oxidative phosphorylation which was usually inhibited in cancer cells, and promote cell growth through the unique mitochondrial functions in anabolic metabolism [72]. It is also reported that FBW7 can negatively regulate HIF-1α through proteasomal degradation to modulate cell growth and migration [73, 74], given the critical role of HIF-1α in cell metabolism, further research are warranted to determine the significances of FBW7/HIF-1α pathway in metabolic regulation. In addition, the relationship between FBW7 and glutamine metabolism is also warranted to study.
LKB1
LKB1(liver kinase B1), also known as STK11, is a tumor suppressor gene whose mutation usually cause a familial cancer syndrome called Peutz-Jeghers syndrome which is associated with a > 40-fold increase in PDAC incidence [75]. Although there is some evidence that the rates of inactivation are high in intraductal papillary mucinous neoplasm (IPMN), which is identified as one of three PDAC precursor lesions [76], somatic mutation of LKB1 in sporadic PDAC appears to be rare, and was detected in only 4–6% of sporadic cases examined [77].
LKB1 encodes a serine/threonine kinase that is involved in regulation of diverse processes such as cell polarity and metabolism. A large amount of evidences indicate that LKB1 plays an important role in the metabolism of various tumor and non-tumor cells, such as pancreatic cancer, cervical carcinoma, breast cancer and liver cancer cells [78,79,80]. LKB1 regulates cell metabolism mainly through directly phosphorylating and activating AMPK, which is a central metabolic switch found in all eukaryotes that governs glucose and lipid metabolism in response to alterations in nutrients and intracellular energy levels [81]. It is reported that loss of LKB1 can increase glucose consumption and glycolysis in cervical cancer cells. This may be related to the enhanced expression of HK-2 in the glycolytic pathway through elevated c-Myc [82]. Dupuy et al. found that loss of LKB1 induces increased glycolytic metabolism in breast cancer both in vivo and in vitro, and demonstrated that this was regulated through the Akt/mTOR pathway [83]. Faubert et al. reported that loss of LKB1 promotes the metabolism of both glucose and glutamine through HIF-1α-dependent way, thus stimulates aerobic glycolysis and lowers reliance on OXPHOS (oxidative phosphorylation) of non-small cell lung cancer (NSCLC) cells [84]. And this was confirmed again in Parker’s study that functional LKB1 expressing NSCLC cells exhibited higher flux through oxidative mitochondrial pathways compared to those deficient in LKB1 [85]. In addition, Wang et al. found specific phosphorylation of LKB1 at Threonine 189 enhanced glucose uptake by promoting GLUT4 translocation to the plasma membrane [86]. Galan-Cobo et al. reported that LKB1 loss can enhance glutamine dependence and vulnerability to glutaminase inhibition by regulating the levels of intracellular reactive oxygen species and ATP, NADPH/NADP ratio, and glutathione [87]. Recently, it was reported that loss of LKB1 renders cells dependent on glutamine for growth in polycystic kidney disease, and metabolomics analysis suggested that LKB1 mutant kidneys require glutamine for non-essential amino acid and glutathione metabolism [88]. This indicates that there is also a link between LKB1 and glutamine metabolism in tumor cells.
BAP1
BAP1, also known as BRCA-associated protein 1, is a deubiquitinating enzyme that regulates various activities by forming multi-protein complexes. And it has been demonstrated that its mutation is associated with the initiation and development of multiple tumors including pancreatic cancer [89, 90]. Recently, Lee et al. demonstrated BAP1 exert a tumor suppressor function in pancreatic cancer by deubiquitinating LATS2 (large tumor suppressor, homolog 2), the negative regulator of Hippo pathway which could activate oncoproteins YAP and TAZ [89]. In terms of metabolism reprograming, Bononi et al. found the aerobic glycolysis and lactate secretion are increased, while mitochondrial respiration and ATP production are reduced both in primary fibroblasts and human mesothelial cells when BAP1 was mutated or downregulated [91]. Yang’s team demonstrated that BAP1 can promote gluconeogenesis by reducing the degradation of PGC-1α to improve glucose homeostasis in mouse liver cells [92]. In addition, it is also reported that BAP1 loss increased pancreatitis biomarkers but reduced mitochondria related proteins, and glucose and hexose metabolic pathways were also repressed in liver-specific BAP1 knockout mice [93].
Other tumor suppressor genes participate in metabolic reprogramming of pancreatic cancer alone or in cooperation
Apart from the several genes mentioned above (Table 1), many other tumor suppressor genes participating the initiation and development of pancreatic cancer are associated with the metabolic reprograming. For example, it was reported that SIRT4 can lead to mitochondrial glutamine metabolism repression, and the loss of it result in tumorigenic phenotypes including glutamine dependent proliferation and stress-induced genomic instability [94]. And pancreatic cancer, like most other cancers, arises from stepwise accumulation of genetic perturbations. Therefore, there are often multiple genes mutated at the same time co-contributing to the initiation of PDAC. In a mouse model for pancreatic cancer initiation in which one copy of BRCA2 is inactivated from birth, loss of heterozygosity (LOH) before acquisition of further mutations is not sufficient to drive tumorigenesis, instead promoting chromosomal instability. Intriguingly, even in the presence of KRAS activation, LOH at BRCA2 inhibits tumor formation as long as wild-type p53 remains. When p53 is mutated, however, loss of the second copy of BRCA2 accelerates pancreatic tumorigenesis in a KRAS-independent manner [95, 96]. The interactions between different genes promote the initiation and development of tumors, but at the same time provide a new strategy for us to the treatment of cancer. For example, Caiola et al. reported that co-occurring mutation of KRAS and LKB1 in NSCLC cells showed more efficient glycolysis and oxidative phosphorylation compared to cells with either single mutation genotype, however the enhanced metabolic activity renders cells with both genetic lesions more sensitive to nutrient limitation, suggesting the possibility to kill cancer cells through energy stress which induced by nutrition restriction regimens [97].
Therapies targeting metabolic reprogramming regulated by tumor suppressor genes
Metabolic reprogram regulated by tumor suppressor genes constitute an essential factor which facilitate the initiation and development of tumor, and this in turn provide us some targets to treat the depressing disease. More and more agents targeting metabolic alteration by tumor suppressor genes achieved significant tumor suppression effect (Table 2). For example, Sablina et al. reported that the mutation of TP53 tumor suppressor gene is associated with the excessive intracellular ROS which could cause DNA damage and genetic instability and thus leads to the initiation of cancer. While dietary supplementation of antioxidant N-acetylcysteine (NAC) could prevent the frequent lymphomas characteristic and slow down the growth of lung cancer xenografts in TP53 mutation mice [98]. In addition, it is reported that the low-molecular-weight compound APR-246 which reactivate mutant p53 can suppress tumor growth by inhibiting the oxidoreductase enzyme thioredoxin reductase 1 (TRXR1) and converting the enzyme to a pro-oxidant NADPH oxidase, thereby inducing oxidative stress and endoplasmic reticulum stress by its redox effects in osteosarcoma cells with TP53 mutation [99, 100]. Liu et al. reported that APR-246 can deplete glutathione (GSH) and thereby inducing lipid peroxidative cell death in oesophageal cancer [101]. Ali et al. reported that APR-246 can increase expression of genes that are related to oxidative stress including haeme oxygenase 1 (HMOX1) and so on in acute myeloid leukaemia [102]. The metabolic reprogramming driven by LKB1 and the KEAP1/NRF2 pathways enhanced sensitivity of lung adenocarcinoma to the glutaminase inhibitor both in vitro and in vivo, suggesting the clinical application of glutaminase inhibitor in subsets of KRAS-mutant tumors [87]. And in LKB1-deficient tumors, mTORC1 and hypoxia inducible factor (HIF) signaling are hyper-activated which, in turn, stimulates aerobic glycolysis and lowers reliance on OXPHOS [84]. While Whang et al. reported that loss of LKB1 which leads to dysfunctional mitochondria and metabolic dysregulation can also render LKB1-deficient tumors hyper-sensitive to pharmacological agents which induce energy stress [103]. Also it is important to evaluate the metabolism of the specific tumor that is selected for this therapy strategy, as the roles of some genes in cancer metabolism are complex, and sometimes even opposite. For example, we should be prudential when try to activate WTp53 with small molecules, or to restore normal function of mutant p53 in human cancers carrying p53 mutations, as the opposite roles in tumor development mentioned above [36,37,38].
Discussion
A large number of genes have been found to be closely related to the initiation and development of tumors, and have brought about some revolutionary changes in cancer treatment. For example, in breast cancer patients with HER-2 positive and germline mutations in BRCA1/2, the molecularly targeted drugs Trastuzumab and Talazoparib, have greatly improved the patients’ prognosis [104, 105]. And there are evidences that poly-ADP-ribose polymerase (PARP) inhibitors Olaparib can improve the progression-free survival of patients with a germline BRCA mutation and metastatic pancreatic cancer [106]. A lot of researches including whole genome sequencing (WGS) and whole exome sequencing (WES) of large sample groups have been done to try to find new target genes in pancreatic cancer treatment [107,108,109] And accumulating molecular data in recent years divided pancreatic cancer into different subgroups with distinct biology and provided potential subtype-specific therapeutic targets [110]. Apart from that, new tumor-associated genes are still being discovered every year. Some of these genes are oncogenes, some are tumor suppressor genes, and some have different effects of tumor suppression and promotion at different stages of tumorigenesis. This indicates that there is an inextricable and complex relationship between genetic changes and the development of tumors. In this review, we summarized the important roles of tumor suppressor gene mutations in the initiation and progression of pancreatic cancer from the perspective of metabolic reprogramming. For example, in the alterations of glucose metabolism, almost all the tumor suppressor genes mentioned above participated in the process, mainly by inhibiting the enzyme activities of oxidative phosphorylation such as SCO2 and PK, [23, 64] while upregulating enzyme activity of aerobic glycolysis such as PFK and HK2 [24, 50]. And in the other core alteration of glutamine metabolism, tumor suppressor genes mainly regulated the enzymes involved in glutamine conversion such as GLS2 and GLS1, and glutamine transporter such as ASCT2 [34, 55].
However, we should also note that while genes greatly alter tumor metabolism and facilitate tumor growth, abnormal tumor metabolism can also greatly affect the gene mutations through influencing the microenvironment. For example, the elevated glycolysis of tumor cells could cause increased generation of reactive oxygen species (ROS), which will induce the instability and accumulation of mutations and deletions leading to cancer [111, 112]. In addition, the accelerated anaerobic glycolysis in cells leaded the environment to become acidic which can induce the gene alterations. For example, the acid environment induces the expression of HIF-1α and VEGF to promote the neovascularization in ovarian cancer cells [113]. Enzo et al. found that YAP/TAZ, key transcription factors regulating tumor cell proliferation and aggressiveness, can be fully activated when cells actively incorporate glucose and route it through glycolysis. While when glycolysis is reduced, YAP/TAZ transcriptional activity is significantly decreased [114]. Ye et al. found that on one hand, the high expression of VCAM-1 (vascular cell adhesion molecule-1) in TAMs (tumor-associated macrophages) can induce glycolysis in pancreatic cancer cells, on the other hand, the enhanced aerobic glycolysis yield large amounts of lactate which activate macrophages to a TAM-like phenotype and lead to low immunity [115]. Due to the dense stroma and hypo-vascularization, which lead to nutrient and oxygen-poor microenvironment in pancreatic cancer, the above conditions may be particularly true during the tumor initiation and development.
Therefore, abnormal metabolism and genetic mutations are mutually causal and complementary in tumor initiation. In the process, the abnormal metabolic regulations of glucose and glutamine are at the core, but there are many other metabolic alterations that are essential for tumor growth, such as lipids and amino acids like serine, tryptophan and arginine that should not be ignored by us. By using genetically engineered mouse models and primary pancreatic epithelial cells, and performing transcriptional, proteomics, and metabolic analyses, Kottakis et al. found that LKB1 loss can cooperate with KRAS activation to support tumorigenic growth by induction of the serine-glycine-one-carbon pathway coupled to S-adenosylmethionine generation, and thus sensitizes cells and tumors to inhibition of serine biosynthesis [116]. And as mentioned before, p53 can promote the adaption to glutamine deprivation by increasing arginine uptake through upregulating SLC7A3 in MEFs [37]. Parker et al. found that PDAC cells lacking of SLC38A2 were unable to concentrate intracellular alanine and occurred a profound metabolic crisis which lead to markedly impaired tumor growth [117]. In addition, LKB1 can activate AMP-activated protein kinase (AMPK) and inhibit Acetyl-CoA carboxylase (ACC), which is a product in the first step of fatty acid (FA) synthesis, thus inhibiting FA synthesis and tumor growth in lung cancer mouse models [118, 119]. And as p53 can upregulate AMPK expression, it can suppress tumor growth by inhibiting de novo fatty acids synthesis through the inactivation of ACC [120]. Beyond that, p53 can also suppress adipogenesis by repressing Coactivator-associated arginine methyltransferase 1 (CARM1) in 3T3L1 preadipocytes [121]. Loss of PTEN can lead to aberrant accumulation of cholesteryl ester which is frequently occurred in pancreatic cancer. By inhibiting acyl-CoA cholesterol acyltransferase (ACAT), the proliferation of pancreatic cancer was attenuated both in vitro and in vivo [122]. In particular, when studies on glucose and glutamine metabolism fail to bring about breakthroughs in cancer treatment, alternative approaches that focus on interfering with the relationship between these non-core but important molecular metabolisms and gene-driven tumor development may be able to provide novel therapeutic avenues for pancreatic cancer [123].
In the long history of struggle with pancreatic cancer, especially in recent decades, scientists and clinicians have made great efforts to discover a variety of molecules and signaling pathways, but the treatment of pancreatic cancer has not achieved any substantial breakthrough, it is still the most lethal disease to human beings so far. This is like a fable in ancient China: our understanding of pancreatic cancer may have been the same as a blind man feeling an elephant, only touching one part of it, and concluding what the elephant is like. We look at pancreatic cancer from many different aspects, but may have never recognized the essence of tumor as a whole. Therefore, although pancreatic cancer is being explored and recognized from more and more aspects, how to integrate the scattered information in a complete form is the real challenge.
Conclusion
In conclusion, tumor suppressor genes play essential roles in the initiation and progression of pancreatic cancer by regulating the metabolic reprogramming of various substrates. And we believe that the study of gene mutations and reprogrammed metabolisms in pancreatic cancer will move forward rapidly and provide novel strategies in its treatment. Further original researches are warranted to elucidate the therapeutic values of these mechanisms and reasonable clinical trials should be designed to evaluate their effects on this lethal disease.
Availability of data and materials
The data supporting the conclusions of this review is included within the article.
Abbreviations
- ACC:
-
Acetyl-CoA carboxylase
- CARM1:
-
Coactivator-associated arginine methyltransferase 1
- COX:
-
Cytochrome c oxidase
- HK2:
-
Hexokinase isozyme II
- HIF:
-
Hypoxia inducible factor
- LATS2:
-
Large tumor suppressor, homolog 2
- LDH:
-
Lactate dehydrogenase
- MCTs:
-
Monocarboxylate transporters
- MPC:
-
Mitochondrial pyruvate carrier
- OXPHOS:
-
Oxidative phosphorylation
- PARP:
-
Poly-ADP-ribose polymerase
- PDAC:
-
Pancreatic duct adenocarcinoma
- PDK:
-
Pyruvate dehydrogenase kinase
- PGC-1α:
-
Peroxisome proliferator-activated receptor-C coactivator-1α
- PGK1:
-
Phosphoglycerate kinase 1
- PFK:
-
6-phosphofructo-1-kinase
- PFK-1:
-
6-phosphofructo-1-kinase
- PFKFB:
-
6-phosphofructo-2-kinase 3
- PUMA:
-
p53 up-regulated apoptosis regulators
- ROS:
-
Reactive oxygen species
- RRAD:
-
Ras-related associated with diabetes
- SCO2:
-
Synthesis of Cytochrome c Oxidase 2
- SNX27:
-
Sorting nexin 27
- TAMs:
-
Tumor-associated macrophages
- TCA:
-
Tricarboxylic acid
- TIGAR:
-
tP53-induced glycolysis and apoptosis regulator
- VCAM-1:
-
Vascular cell adhesion molecule-1
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269–70.
Som P, Atkins HL, Bandoypadhyay D, Fowler JS, MacGregor RR, Matsui K, et al. A fluorinated glucose analog, 2-fluoro-2-deoxy-D-glucose (F-18): nontoxic tracer for rapid tumor detection. J Nucl Med. 1980;21(7):670–5.
Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11(5):325–37.
Eagle H. The minimum vitamin requirements of the L and HeLa cells in tissue culture, the production of specific vitamin deficiencies, and their cure. J Exp Med. 1955;102(5):595–600.
Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619–34.
Liang C, Qin Y, Zhang B, Ji S, Shi S, Xu W, et al. Energy sources identify metabolic phenotypes in pancreatic cancer. Acta Biochim Biophys Sin (Shanghai). 2016;48(11):969–79.
Mehla K, Singh PK. Metabolic subtyping for novel personalized therapies against pancreatic cancer. Clin Cancer Res. 2020;26(1):6–8.
Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23(5):537–48.
Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20(10):1218–49.
Liang C, Qin Y, Zhang B, Ji S, Shi S, Xu W, et al. Metabolic plasticity in heterogeneous pancreatic ductal adenocarcinoma. Biochim Biophys Acta. 2016;1866(2):177–88.
Aslan M, Shahbazi R, Ulubayram K, Ozpolat B. Targeted therapies for pancreatic cancer and hurdles ahead. Anticancer Res. 2018;38(12):6591–606.
Zhang Y, Yang C, Cheng H, Fan Z, Huang Q, Lu Y, et al. Novel agents for pancreatic ductal adenocarcinoma: emerging therapeutics and future directions. J Hematol Oncol. 2018;11(1):14.
van Geel R, van Brummelen EMJ, Eskens F, Huijberts S, de Vos F, Lolkema M, et al. Phase 1 study of the pan-HER inhibitor dacomitinib plus the MEK1/2 inhibitor PD-0325901 in patients with KRAS-mutation-positive colorectal, non-small-cell lung and pancreatic cancer. Br J Cancer. 2020;122(8):1166–74.
Bodoky G, Timcheva C, Spigel DR, La Stella PJ, Ciuleanu TE, Pover G, et al. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest New Drugs. 2012;30(3):1216–23.
Infante JR, Somer BG, Park JO, Li CP, Scheulen ME, Kasubhai SM, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer. 2014;50(12):2072–81.
Hill R, Rabb M, Madureira PA, Clements D, Gujar SA, Waisman DM, et al. Gemcitabine-mediated tumour regression and p53-dependent gene expression: implications for colon and pancreatic cancer therapy. Cell Death Dis. 2013;4:e791.
Liang C, Shi S, Qin Y, Meng Q, Hua J, Hu Q, et al. Localisation of PGK1 determines metabolic phenotype to balance metastasis and proliferation in patients with SMAD4-negative pancreatic cancer. Gut 2019.
Shen L, Sun X, Fu Z, Yang G, Li J, Yao L. The fundamental role of the p53 pathway in tumor metabolism and its implication in tumor therapy. Clin Cancer Res. 2012;18(6):1561–7.
Lacroix M, Riscal R, Arena G, Linares LK, Le Cam L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Molecular metabolism. 2020;33:2–22.
Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137(3):413–31.
Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science. 2006;312(5780):1650–3.
Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–20.
Butera G, Pacchiana R, Mullappilly N, Margiotta M, Bruno S, Conti P, et al. Mutant p53 prevents GAPDH nuclear translocation in pancreatic cancer cells favoring glycolysis and 2-deoxyglucose sensitivity. Biochim Biophys Acta. 2018;1865(12):1914–23.
Rajeshkumar NV, Dutta P, Yabuuchi S, de Wilde RF, Martinez GV, Le A, et al. Therapeutic targeting of the warburg effect in pancreatic cancer relies on an absence of p53 Function. Can Res. 2015;75(16):3355–64.
Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64(7):2627–33.
Zhang C, Liu J, Wu R, Liang Y, Lin M, Liu J, et al. Tumor suppressor p53 negatively regulates glycolysis stimulated by hypoxia through its target RRAD. Oncotarget. 2014;5(14):5535–46.
Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008;10(5):611–8.
Kim HR, Roe JS, Lee JE, Cho EJ, Youn HD. p53 regulates glucose metabolism by miR-34a. Biochem Biophys Res Commun. 2013;437(2):225–31.
Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18(3):283–93.
Draoui N, Feron O. Lactate shuttles at a glance: from physiological paradigms to anti-cancer treatments. Dis Model Mech. 2011;4(6):727–32.
Boidot R, Vegran F, Meulle A, Le Breton A, Dessy C, Sonveaux P, et al. Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res. 2012;72(4):939–48.
Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA. 2010;107(16):7461–6.
Tran TQ, Lowman XH, Reid MA, Mendez-Dorantes C, Pan M, Yang Y, et al. Tumor-associated mutant p53 promotes cancer cell survival upon glutamine deprivation through p21 induction. Oncogene. 2017;36(14):1991–2001.
Tajan M, Hock AK, Blagih J, Robertson NA, Labuschagne CF, Kruiswijk F, et al. A Role for p53 in the adaptation to glutamine starvation through the expression of SLC1A3. Cell Metab. 2018;28(5):721–36.
Lowman XH, Hanse EA, Yang Y, Ishak Gabra MB, Tran TQ, Li H, et al. p53 promotes cancer cell adaptation to glutamine deprivation by upregulating Slc7a3 to increase arginine uptake. Cell reports. 2019;26(11):3051–60.
Kim J, Yu L, Chen W, Xu Y, Wu M, Todorova D, et al. Wild-Type p53 promotes cancer metabolic switch by inducing puma-dependent suppression of oxidative phosphorylation. Cancer cell. 2019;35(2):191–20.
Hustinx SR, Leoni LM, Yeo CJ, Brown PN, Goggins M, Kern SE, et al. Concordant loss of MTAP and p16/CDKN2A expression in pancreatic intraepithelial neoplasia: evidence of homozygous deletion in a noninvasive precursor lesion. Mod Pathol. 2005;18(7):959–63.
Baschnagel AM, Wobb JL, Dilworth JT, Williams L, Eskandari M, Wu D, et al. The association of (18)F-FDG PET and glucose metabolism biomarkers GLUT1 and HK2 in p16 positive and negative head and neck squamous cell carcinomas. Radiother Oncol. 2015;117(1):118–24.
Ju HQ, Ying H, Tian T, Ling J, Fu J, Lu Y, et al. Mutant Kras- and p16-regulated NOX4 activation overcomes metabolic checkpoints in development of pancreatic ductal adenocarcinoma. Nat Commun. 2017;8:14437.
Helman A, Klochendler A, Azazmeh N, Gabai Y, Horwitz E, Anzi S, et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med. 2016;22(4):412–20.
Levy L, Hill CS. Smad4 dependency defines two classes of transforming growth factor beta (TGF-{beta}) target genes and distinguishes TGF-{beta}-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol Cell Biol. 2005;25(18):8108–25.
Li HY, Oh YS, Lee YJ, Lee EK, Jung HS, Jun HS. Amelioration of high fat diet-induced glucose intolerance by blockade of Smad4 in pancreatic beta-cells. Exp Clin Endocrinol Diabetes. 2015;123(4):221–6.
Raychaudhuri S. MicroRNAs overexpressed in growth-restricted rat skeletal muscles regulate the glucose transport in cell culture targeting central TGF-beta factor SMAD4. PLoS ONE. 2012;7(4):e34596.
DiCiommo D, Gallie BL, Bremner R. Retinoblastoma: the disease, gene and protein provide critical leads to understand cancer. Semin Cancer Biol. 2000;10(4):255–69.
Comisso E, Scarola M, Rosso M, Piazza S, Marzinotto S, Ciani Y, et al. OCT4 controls mitotic stability and inactivates the RB tumor suppressor pathway to enhance ovarian cancer aggressiveness. Oncogene. 2017;36(30):4253–66.
Vormer TL, Wojciechowicz K, Dekker M, de Vries S, van der Wal A, Delzenne-Goette E, et al. RB family tumor suppressor activity may not relate to active silencing of E2F target genes. Cancer Res. 2014;74(18):5266–76.
Gerdes B, Ramaswamy A, Ziegler A, Lang SA, Kersting M, Baumann R, et al. p16INK4a is a prognostic marker in resected ductal pancreatic cancer: an analysis of p16INK4a, p53, MDM2, an Rb. Ann Surg. 2002;235(1):51–9.
Beemer FA, Vlug AM, Rijksen G, Hamburg A, Staal GE. Characterization of some glycolytic enzymes from human retina and retinoblastoma. Can Res. 1982;42(10):4228–322.
Dias PL, Shanmuganathan SS, Rajaratnam M. Lactic dehydrogenase activity of aqueous humour in retinoblastoma. Br J Ophthalmol. 1971;55(2):130–2.
Batchu RB, Gruzdyn OV, Bryant CS, Qazi AM, Kumar S, Chamala S, et al. Ritonavir-Mediated Induction of Apoptosis in Pancreatic Cancer Occurs via the RB/E2F-1 and AKT Pathways. Pharmaceuticals (Basel). 2014;7(1):46–57.
Zhu W, Ye L, Zhang J, Yu P, Wang H, Ye Z, et al. PFK15, a Small Molecule Inhibitor of PFKFB3, Induces Cell Cycle Arrest, Apoptosis and Inhibits Invasion in Gastric Cancer. PLoS ONE. 2016;11(9):e0163768.
Hsieh MC, Das D, Sambandam N, Zhang MQ, Nahle Z. Regulation of the PDK4 isozyme by the Rb-E2F1 complex. J Biol Chem. 2008;283(41):27410–7.
Reynolds MR, Lane AN, Robertson B, Kemp S, Liu Y, Hill BG, et al. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene. 2014;33(5):556–66.
Blanchet E, Annicotte JS, Lagarrigue S, Aguilar V, Clape C, Chavey C, et al. E2F transcription factor-1 regulates oxidative metabolism. Nat Cell Biol. 2011;13(9):1146–52.
Sarker D, Reid AH, Yap TA, de Bono JS. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin Cancer Res. 2009;15(15):4799–805.
Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1(6–7):315–22.
Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010;2(53):53ra75.
Wartenberg M, Centeno I, Haemmig S, Vassella E, Zlobec I, Galvan JA, et al. PTEN alterations of the stromal cells characterise an aggressive subpopulation of pancreatic cancer with enhanced metastatic potential. Eur J Cancer. 2016;65:80–90.
Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273(22):13375–8.
Martin PL, Yin JJ, Seng V, Casey O, Corey E, Morrissey C, et al. Androgen deprivation leads to increased carbohydrate metabolism and hexokinase 2-mediated survival in Pten/Tp53-deficient prostate cancer. Oncogene. 2017;36(4):525–33.
Shinde SR, Maddika S. PTEN regulates glucose transporter recycling by impairing SNX27 Retromer assembly. Cell Rep. 2017;21(6):1655–66.
Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VC, et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell. 2012;149(1):49–62.
Zhao K, Zhou Y, Qiao C, Ni T, Li Z, Wang X, et al. Oroxylin A promotes PTEN-mediated negative regulation of MDM2 transcription via SIRT3-mediated deacetylation to stabilize p53 and inhibit glycolysis in wt-p53 cancer cells. J Hematol Oncol. 2015;8:41.
Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001;413(6853):316–22.
Huang LY, Zhao J, Chen H, Wan L, Inuzuka H, Guo J, et al. SCF(FBW7)-mediated degradation of Brg1 suppresses gastric cancer metastasis. Nat Commun. 2018;9(1):3569.
Ji S, Qin Y, Shi S, Liu X, Hu H, Zhou H, et al. ERK kinase phosphorylates and destabilizes the tumor suppressor FBW7 in pancreatic cancer. Cell Res. 2015;25(5):561–73.
Hu Q, Qin Y, Zhang B, Liang C, Ji S, Shi S, et al. FBW7 increases the chemosensitivity of pancreatic cancer cells to gemcitabine through upregulation of ENT1. Oncol Rep. 2017;38(4):2069–77.
Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8(2):83–93.
Ji S, Qin Y, Liang C, Huang R, Shi S, Liu J, et al. FBW7 (F-box and WD repeat domain-containing 7) negatively regulates glucose metabolism by targeting the c-Myc/TXNIP (Thioredoxin-Binding Protein) Axis in Pancreatic Cancer. Clin Cancer Res. 2016;22(15):3950–60.
Davis RJ, Gonen M, Margineantu DH, Handeli S, Swanger J, Hoellerbauer P, et al. Pan-cancer transcriptional signatures predictive of oncogenic mutations reveal that Fbw7 regulates cancer cell oxidative metabolism. Proc Natl Acad Sci USA. 2018;115(21):5462–7.
Cassavaugh JM, Hale SA, Wellman TL, Howe AK, Wong C, Lounsbury KM. Negative regulation of HIF-1alpha by an FBW7-mediated degradation pathway during hypoxia. J Cell Biochem. 2011;112(12):3882–900.
Flugel D, Gorlach A, Kietzmann T. GSK-3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1alpha. Blood. 2012;119(5):1292–301.
Grover S, Syngal S. Hereditary pancreatic cancer. Gastroenterology 2010, 139(4): 1076–1080, 1080 e1071–1072.
Sahin F, Maitra A, Argani P, Sato N, Maehara N, Montgomery E, et al. Loss of Stk11/Lkb1 expression in pancreatic and biliary neoplasms. Mod Pathol. 2003;16(7):686–91.
Su GH, Hruban RH, Bansal RK, Bova GS, Tang DJ, Shekher MC, et al. Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am J Pathol. 1999;154(6):1835–40.
Imai K, Inukai K, Ikegami Y, Awata T, Katayama S. LKB1, an upstream AMPK kinase, regulates glucose and lipid metabolism in cultured liver and muscle cells. Biochem Biophys Res Commun. 2006;351(3):595–601.
Wu Q, Li J, Sun S, Chen X, Zhang H, Li B, et al. YAP/TAZ-mediated activation of serine metabolism and methylation regulation is critical for LKB1-deficient breast cancer progression. Biosci Rep 2017, 37(5).
Nafz J, De-Castro Arce J, Fleig V, Patzelt A, Mazurek S, Rosl F. Interference with energy metabolism by 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside induces HPV suppression in cervical carcinoma cells and apoptosis in the absence of LKB1. Biochem J. 2007;403(3):501–10.
Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1(1):15–25.
Zeng Q, Chen J, Li Y, Werle KD, Zhao RX, Quan CS, et al. LKB1 inhibits HPV-associated cancer progression by targeting cellular metabolism. Oncogene. 2017;36(9):1245–55.
Dupuy F, Griss T, Blagih J, Bridon G, Avizonis D, Ling C, et al. LKB1 is a central regulator of tumor initiation and pro-growth metabolism in ErbB2-mediated breast cancer. Cancer Metab. 2013;1(1):18.
Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc Natl Acad Sci USA. 2014;111(7):2554–9.
Parker SJ, Svensson RU, Divakaruni AS, Lefebvre AE, Murphy AN, Shaw RJ, et al. LKB1 promotes metabolic flexibility in response to energy stress. Metab Eng. 2017;43(Pt B):208–17.
Wang F, Yang X, Lu Y, Li Z, Xu Y, Hu J, et al. The natural product antroalbol H promotes phosphorylation of liver kinase B1 (LKB1) at threonine 189 and thereby enhances cellular glucose uptake. J Biol Chem. 2019;294(27):10415–27.
Galan-Cobo A, Sitthideatphaiboon P, Qu X, Poteete A, Pisegna MA, Tong P, et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res 2019.
Flowers EM, Sudderth J, Zacharias L, Mernaugh G, Zent R, DeBerardinis RJ, et al. Lkb1 deficiency confers glutamine dependency in polycystic kidney disease. Nat Commun. 2018;9(1):814.
Lee HJ, Pham T, Chang MT, Barnes D, Cai AG, Noubade R, et al. The tumor suppressor BAP1 regulates the Hippo pathway in pancreatic ductal adenocarcinoma. Cancer Res 2020.
Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43(10):1022–5.
Bononi A, Yang H, Giorgi C, Patergnani S, Pellegrini L, Su M, et al. Germline BAP1 mutations induce a Warburg effect. Cell Death Differ. 2017;24(10):1694–704.
Ruan HB, Han X, Li MD, Singh JP, Qian K, Azarhoush S, et al. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1alpha stability. Cell Metab. 2012;16(2):226–37.
Baughman JM, Rose CM, Kolumam G, Webster JD, Wilkerson EM, Merrill AE, et al. NeuCode proteomics reveals Bap1 regulation of metabolism. Cell reports. 2016;16(2):583–95.
Jeong SM, Xiao C, Finley LW, Lahusen T, Souza AL, Pierce K, et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell. 2013;23(4):450–63.
Rowley M, Ohashi A, Mondal G, Mills L, Yang L, Zhang L, et al. Inactivation of Brca2 promotes Trp53-associated but inhibits KrasG12D-dependent pancreatic cancer development in mice. Gastroenterology. 2011;140(4):1303–13.
Morton JP, Steele CW, Sansom OJ. Timing is everything: Brca2 and p53 mutations in pancreatic cancer. Gastroenterology. 2011;140(4):1143–6.
Caiola E, Falcetta F, Giordano S, Marabese M, Garassino MC, Broggini M, et al. Co-occurring KRAS mutation/LKB1 loss in non-small cell lung cancer cells results in enhanced metabolic activity susceptible to caloric restriction: an in vitro integrated multilevel approach. J Exp Clin Cancer Res. 2018;37(1):302.
Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11(12):1306–13.
Peng X, Zhang MQ, Conserva F, Hosny G, Selivanova G, Bykov VJ, et al. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell death & disease. 2013;4:e881.
Lambert JM, Moshfegh A, Hainaut P, Wiman KG, Bykov VJ. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene. 2010;29(9):1329–38.
Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, et al. Inhibiting the system xC(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nature communications. 2017;8:14844.
Ali D, Mohammad DK, Mujahed H, Jonson-Videsater K, Nore B, Paul C, et al. Anti-leukaemic effects induced by APR-246 are dependent on induction of oxidative stress and the NFE2L2/HMOX1 axis that can be targeted by PI3K and mTOR inhibitors in acute myeloid leukaemia cells. Br J Haematol. 2016;174(1):117–26.
Whang YM, Park SI, Trenary IA, Egnatchik RA, Fessel JP, Kaufman JM, et al. LKB1 deficiency enhances sensitivity to energetic stress induced by erlotinib treatment in non-small-cell lung cancer (NSCLC) cells. Oncogene. 2016;35(7):856–66.
Litton JK, Rugo HS, Ettl J, Hurvitz SA, Goncalves A, Lee KH, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379(8):753–63.
Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr, Davidson NE, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353(16):1673–84.
Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance Olaparib for Germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381(4):317–27.
Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–6.
Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501.
Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–52.
Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2019;16(4):207–20.
Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008;270(1):1–9.
Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res. 2001;477(1–2):7–21.
Favaro E, Nardo G, Persano L, Masiero M, Moserle L, Zamarchi R, et al. Hypoxia inducible factor-1alpha inactivation unveils a link between tumor cell metabolism and hypoxia-induced cell death. Am J Pathol. 2008;173(4):1186–201.
Enzo E, Santinon G, Pocaterra A, Aragona M, Bresolin S, Forcato M, et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 2015;34(10):1349–70.
Ye H, Zhou Q, Zheng S, Li G, Lin Q, Wei L, et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018;9(5):453.
Kottakis F, Nicolay BN, Roumane A, Karnik R, Gu H, Nagle JM, et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature. 2016;539(7629):390–5.
Parker SJ, Amendola CR, Hollinshead KER, Yu Q, Yamamoto K, Encarnacion-Rosado J, et al. Selective alanine transporter utilization creates a targetable metabolic niche in pancreatic cancer. Cancer Discov 2020.
Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med. 2016;22(10):1108–19.
Sunami Y, Rebelo A, Kleeff J. Lipid metabolism and lipid droplets in pancreatic cancer and stellate cells. Cancers. 2017;10:1.
Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, et al. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Can Res. 2007;67(7):3043–53.
Behera AK, Bhattacharya A, Vasudevan M, Kundu TK. p53 mediated regulation of coactivator associated arginine methyltransferase 1 (CARM1) expression is critical for suppression of adipogenesis. FEBS J. 2018;285(9):1730–44.
Li J, Gu D, Lee SS, Song B, Bandyopadhyay S, Chen S, et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene. 2016;35(50):6378–88.
Li L, Mao Y, Zhao L, Li L, Wu J, Zhao M, et al. p53 regulation of ammonia metabolism through urea cycle controls polyamine biosynthesis. Nature. 2019;567(7747):253–6.
Acknowledgments
We thank Long Zhang for his insightful advices and help of the illustrations.
Funding
This work was supported by National Science Fund for Distinguished Young Scholars [Grant Numbers 81625016], National Natural Science Foundation [Grant Numbers 81502031, 81372651 and 81602085] and Shanghai Sailing Program [Grant Number 16YF1401800].
Author information
Authors and Affiliations
Contributions
ML and WL contributed equally to this work, collected and analyzed the literatures, and drafted the manuscript. YQ, XX and XY reviewed and contributed to the revision of the manuscript. SJ and QZ designed the study and provided critical suggestions. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, M., Liu, W., Qin, Y. et al. Regulation of metabolic reprogramming by tumor suppressor genes in pancreatic cancer. Exp Hematol Oncol 9, 23 (2020). https://doi.org/10.1186/s40164-020-00179-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-020-00179-x