
Abstract
Background
Carcass traits are crucial indicators of meat production efficiency. However, the molecular regulatory mechanisms associated with these traits remain unclear.
Results
In this study, we conducted comprehensive transcriptomic and genomic analyses on 399 Tiannong partridge chickens to identify key genes and variants associated with carcass traits and to elucidate the underlying regulatory mechanisms. Based on association analyses with the elastic net (EN) model, we identified 12 candidate genes (AMY1A, AP3B2, CEBPG, EEF2, EIF4EBP1, FGFR1, FOXD3, GOLM1, LOC107052698, PABPC1, SERPINB6 and TBC1D16) for 4 carcass-related traits, namely live weight, dressed weight, eviscerated weight, and breast muscle weight. SERPINB6 was identified as the only overlapping gene by 3 analyses, EN model analysis, weighted gene co-expression network analysis and differential expression analysis. Cell-level experiments confirmed that SERPINB6 promotes the proliferation of chicken DF1 cells and primary myoblasts. Further expression genome-wide association study and association analysis indicated that rs317934171 is the critical site that enhances SERPINB6 expression. Furthermore, a dual-luciferase reporter assay proved that gga-miR-1615 targets the 3′UTR of SERPINB6.
Conclusions
Collectively, our findings reveal that SERPINB6 serves as a novel gene for chicken carcass traits by promoting fibroblast and myoblast proliferation. Additionally, the downstream variant rs317934171 regulates SERPINB6 expression. These results identify a new target gene and molecular marker for the molecular mechanisms of chicken carcass traits.
Similar content being viewed by others

Background
The worldwide attention given to food safety, particularly that of meat, eggs and milk, which are important sources of protein, has increased [1]. Meat is the most readily available source of various amino acids that the human body requires. In China, chicken is the second most widely consumed meat [2], and the demand for chicken meat is increasing annually. Local chicken breeds are preferred by native consumers over fast-growing chickens due to their delicate meat quality, including flavor and tenderness [3]. Additionally, carcass traits serve as important indicators of meat yield, which is a crucial economic trait of meat-type chickens. However, local chicken breeds have low meat production efficiency and their carcass trait regulatory mechanisms are largely unknown, making it difficult for them to meet the current demand for chicken meat.
Carcass traits are regulated by polygenic effects [4, 5]. Transcriptome sequencing (RNA-seq) revealed differential expression of CRELD1 and DNAJC30 in the breast muscle of white recessive rock chickens compared to Xinghua chickens [6]. RNA-seq analysis identified PRKG2 as an important candidate gene associated with the carcass traits of Chinese Ningdu yellow chickens [7]. At present, transcriptomics is the most commonly used method for identifying candidate genes based on gene expression levels. However, at the genomic level, genome-wide association studies (GWAS) are frequently used to identify major genes or genetic markers [8,9,10,11]. For instance, a quantitative trait locus (QTL) region was identified on chromosome 4 of Beijing-You chickens. The region includes 7 significant single-nucleotide polymorphisms (SNPs) and 4 candidate genes (LCORL, LAP3, LDB1 and TAPT1). These genes were found to be related to the dressed weight (DW) and eviscerated weight (EW) through a 60 K SNP chip and association analysis of 16 traits [12]. The results of a GWAS showed that the major-effect candidate gene ADGRG6 was associated with the whole-thigh percentage and thigh percentage, while the major-effect candidate gene DRD1 was associated with carcass traits such as carcass weight, EW, and thigh weight [13]. Numerous studies have demonstrated that combining GWAS and RNA-seq can aid in identifying candidate genes, mutations, or biological pathways [14] that affect complex traits. Tan et al. [15], found that the causal gene SOX6 influenced the breast muscle yield of fast-growing broilers, based on genomic and transcriptomic evidence.
This study analyzed nearly 400 Tiannong partridge hens using RNA-Seq and whole-genome resequencing (WGS). Candidate genes were identified by combining large-scale transcriptomic data with multiple carcass traits, including live weight (LW), DW, EW, and breast muscle weight (BMW), through 3 analyses. Key loci affecting carcass traits were identified through expression genome-wide association study (eGWAS) and association analysis.
Methods
Ethics statement
The chickens were fed and handled in accordance with relevant national and international guidelines. The study and all experimental protocols were approved by the Laboratory Animal Welfare and Animal Experimental Ethical Inspection Board of Foshan University (No. 18091801).
Animals and sample collection
All animals used in this study were provided by Guangdong Tinoo’s Foods Group Co., Ltd. All birds were raised on one of the company’s farms according to commercial feeding standards. After weighing, a total of 399 female Tiannong partridge chickens were slaughtered at 126 d using a mechanized slaughter line with moderate scalding water (61 °C). The left pectoral muscle was completely stripped and weighed, and samples of the same part of the right pectoral muscle were collected uniformly. The samples were quickly frozen in liquid nitrogen and stored at −80 °C for subsequent RNA and DNA isolation. We determined the DW, EW and BMW according to the performance terminology and measurements for poultry [16].
RNA extraction and sequencing
Total RNA from breast muscle was isolated with TRIzol reagent (Invitrogen, Carlsbad, CA, USA). After quality control, the qualified RNA samples were subjected to reverse transcription and cDNA library construction as previously reported [17]. Briefly, RNA-seq was performed on an Illumina NovaSeq 6000 (Illumina, San Diego, CA, USA), and paired-end 150 bp reads were generated. FastQC v0.11.9 [18] was used to verify the quality of the raw sequence data for each sample. The trimmed transcriptome data was aligned to the chicken reference genome GRCg6a (GCA_000002315.5) using HISAT2 v2.1.0 [19]. Normalization of gene expression levels was carried out using DESeq2 v1.24.0 [20].
Elastic net-1 standard error model for association analysis
Four statistical models, namely linear mixed model (LMM), least absolute shrinkage and selection operator (LASSO), ridge regression (RR), and elastic net (EN), were used to analyze the associations between gene expression levels and carcass traits of 381 chickens. 18 individuals with phenotypic deletions were eliminated prior to analysis [21]. According to our previous research, the simulation study results showed that the elastic net-1 standard error (EN-1SE) model, which does not involve significance testing, was the best method for analyzing the associations between expression levels and BMW. This was true in terms of both the type I error rate and the detection power [21]. Therefore, we used the EN-1SE model [22] to analyze the association between gene expression and LW, DW, and EW. Briefly, candidate genes for 4 traits were detected using EN with tuning parameters determined based on the one standard error rule (based on the results of the simulation study). All data, including phenotypic records and gene expression levels, were scaled to a mean of zero and one unit of variance prior to association analyses for both the simulation and the empirical studies. The EN models are multi-locus models in which the expression levels of all genes are analyzed together, and EN is classified as regularized linear regression model, which is the basic linear regression model:
where µ is the population mean, 1 is a vector of 1s, the independent variable Xi is a vector of expression values of the ith gene, and bi is the effect of the ith gene on the trait under study.
The EN model uses a mixture of l1 and l2 penalties and can be formulated as follows:
where the value of the second parameter, α, is also determined via cross-validation. The Wald test was used to test whether the estimates of bi from the 4 models were significantly different from zero. Under the null hypothesis that bi = 0, the Wald test statistic is:
and follows approximately a one-degree of freedom Chi-square distribution. The GLMNET/R package was used to calculate EN-1SE [23]. In addition, EN automatically performs variable selection. We simply selected non-zero effect genes as candidate genes in the analyses using EN-1SE model.
Weighted gene co-expression network analysis (WGCNA)
A weighted gene co-expression network was constructed in the R environment using the standard pipeline, based on gene expression levels. A total of 350 individuals were used for WGCNA [24], and outlier samples were removed based on the criterion mean ± 3 standard deviations. Briefly, the soft threshold (β = 6) was determined according to the scale-free distribution (R2 > 0.85). To determine if 2 genes have similar expression patterns, a soft threshold is typically set for screening. Genes with expression levels above the soft threshold are considered similar. The soft threshold is not a fixed value, but rather an idempotent that reflects the importance of a numerical value based on its magnitude. WGCNA uses correlation coefficient weighting, which involves raising the gene correlation coefficient to the Nth power to ensure that the connections between genes in the network follow a scale-free distribution. The gene network was constructed using step-by-step and dynamic cutting methods to detect modules (minModuleSize = 50, mergeCutHeight = 0.25). The module with the highest correlation coefficient was identified as the target module. The module membership (MM) and gene significance (GS) were calculated in the target module [25]. Within the module, the hub genes were defined according to the criteria GS > 0.2 and MM > 0.6.
Differential expression analysis and pathway enrichment
To further screen for differentially expressed genes (DEGs) related to the 4 carcass traits, we separately sorted 350 individuals based on high to low values for the LW, DW, EW and BMW, and a total of 60 birds were divided into the high group (n = 30) and the low group (n = 30) (Additional file 1: Fig. S1). The genes with a fold change > 1.5 and P < 0.05 were identified as DEGs. Similarly, DEGs from the chicken embryonic fibroblast line (DF1 cells), were screened using the criteria (fold change > 1.5 and P < 0.05). Simultaneously, all of these DEGs were subjected to perform Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) enrichment analyses using KOBAS 3.0 (http://kobas.cbi.pku.edu.cn). A P value of 0.05 was used as the threshold for significant pathway enrichment.
Cell culture
DF1 cells (from the cell bank of the Chinese Academy of Sciences) and human embryonic kidney 293 cells (HEK293 cells, from ATCC) were cultured in high-glucose DMEM medium (Gibco, CA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, New Zealand) and 1% penicillin‒streptomycin (Gibco, CA, USA) under 5% CO2 at 37 °C. In addition, the chicken primary myoblasts (CPMs) were isolated from the breast muscle tissue of healthy less than 10-day-old commercial chickens in a sterile environment. Briefly, the breast muscle tissue was washed no less than 3 times with PBS, and the breast muscle tissue was cut to the size of rice grains and digested with collagenase. At the same time, erythrocyte lysates were used to remove erythrocytes. The isolated CPMs were cultured in DMEM/F12 medium (Gibco, CA, USA) supplemented with 10% FBS and 1% penicillin–streptomycin. CPMs were used in subsequent experiments within 5 generations.
RNA oligonucleotides, plasmid construction and cell transfection
SERPINB6 small interfering RNAs (siRNAs) were provided by RiboBio Co., Ltd. (Guangzhou, China). The miR-1615 mimic, mimic negative control (mimic NC), miR-1615 inhibitor and inhibitor negative control (inhibitor NC) were synthesized by GenePharma Co., Ltd. (Shanghai, China), based on oligonucleotide sequences (Additional file 2: Table S1).
For the construction SERPINB6 overexpression plasmid, the full-length sequence of SERPINB6 was amplified from chicken breast muscle cDNA by PCR and cloned and inserted into the empty expression plasmid pcDNA3.1 using the NheI and XhoI restriction sites. For the pGL4.18 dual-luciferase reporter vector, the segment sequence of the 3′UTR of SERPINB6 containing the putative gga-miR-1615 binding sequence was amplified by PCR and then subcloned and inserted between the XhoI and HindIII restriction sites in the pGL4.18 dual-luciferase reporter vector. The SERPINB6 3′UTR mutant plasmid was generated by changing the binding site of gga-miR-1615 from AGCTGCCA to AGCTACCA. PCR amplification was performed for mutagenesis and DpnI digestion to remove the parental DNA.
Transfection was performed when the cells were at high confluence (> 80%), and in 96-well plates when the cell confluence was approximately 70%. Transient transfection of plasmids and oligonucleotides was performed using Lipofectamine™ 3000 reagent (Invitrogen, CA, USA). Liposomes, plasmids, and oligonucleotides were diluted with modified minimal essential medium (Opti-MEM, Gibco, CA, USA). The diluted liposomes were mixed with diluted plasmids or oligonucleotides, and then incubated at room temperature for 15 min to prepare the transfection solution. For RNA oligonucleotides, a concentration of 150 nmol/L was used. The transfection solution was added to the cell medium and incubated for 6–8 h to complete the transfection, after which the cell medium was changed. TransIT-TKO transfection reagent (Mirus Bio, Germany) was used only for the transfection of siRNA into CPMs. The qPCR was performed at 24 h after cell transfection.
Gene expression analysis of SERPINB6
Total cellular RNA was extracted using TRIzol according to the manufacturer’s instructions, and the concentration and integrity of the RNA was measured using an Agilent 2100 Nano and by gel electrophoresis. The RNA obtained from SERPINB6 overexpressing DF1 cells was subjected to RNA-seq according to the methods described above. cDNA was generated from 2 μg of RNA using FastKing gDNA Dispelling RT SuperMix (TIANGEN Biotech Co., Ltd., Beijing, China). The obtained reverse transcription products were subsequently mixed with PowerUp™ SYBR™ Green Master Mix (Applied Biosystems, Waltham, MA, USA), primers, and double-distilled water (ddH2O) to produce a 10 μL RT‒PCR system. Primers were designed using the National Center for Biotechnology Information (NCBI) database based on the coding sequence of SERPINB6 (F: 5′–CCTCAGAACTGCTAACCGACTT–3′, R: 5′–CTTCTCTTCCACCCAGCCATT–3′) and flanking genes (Additional file 3: Table S2) in the 5′ to 3′ direction. Three technical replicates were performed for each sample. The RT–PCR conditions were as follows: 94 °C for 3 min, followed by 40 cycles of 94 °C for 3 s and an annealing temperature for 34 s. GAPDH and RPL32 were selected as the internal controls (Additional file 3: Table S2). According to the RT–PCR results, the expression level was calculated using the 2−ΔΔCT method [26]. Three independent biological replicates were used for each experimental group.
CCK8 and 5-ethynyl-2-deoxyuridine (EdU) proliferation assays
Cell proliferation was measured using Cell Counting Kit-8 (CCK8) and EdU assays. DF1 cells and CPMs were seeded in 96-well plates for 24 h, 10 μL of CCK8 reagent was added to each well at 12, 24, 36, and 48 h post-transfection, and the plates were then incubated for 2 h. The absorbance value of each sample was measured using a microplate reader (Thermo, Waltham, MA, USA) at 450 nm. EdU assays were performed using the Cell-LightTM EdU Apollo 488 In Vitro kit (RiboBio Co., Ltd., Guangzhou, China) according to the manufacturer’s instructions. Briefly, at 24 h after transfection, the cells were exposed to 50 mmol/L EdU for 2 h. The cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. Subsequently, the cells were incubated in Apollo reaction solution for 1 h and stained with Hoechst 33342 for 0.5 h. The cells were further analyzed by calculating the ratio of EdU-positive cells to the total number of cells. Three independent biological replicates were used for each experimental group. Three fields were randomly selected from each well, and the images were captured using a DMI3000B microscope (Leica, Wetzlar, Germany).
Whole-genome sequencing
Genomic DNA from each individual was extracted from breast muscle samples using the phenol–chloroform method. The DNA quality was assessed by agarose gel electrophoresis. A DNA library (paired-end, 2 × 150 bp) was constructed for each DNA sample, and all libraries were sequenced on the Illumina Nova 6000 sequencing platform. Overall, approximately 2.4 Tb raw data was generated. Quality control criteria for the reference panel included an individual call rate ≥ 90%, SNP call rate ≥ 90%, and minor allele frequency (MAF) ≥ 0.05, as implemented in PLINK v1.9 [27]. We obtained 11,766,230 filtered SNPs on chromosomes 1–33, Z and W in 357 chickens.
Variant calling, quality control, and annotation
Raw reads were first trimmed using Trimmomatic v0.36 with default parameters [28], and only paired quality reads were retained for the subsequent analysis. The filtered reads for all individuals were aligned to the chicken reference genome (GRCg6a) using Burrows‒Wheeler Aligner (BWA) v0.7.17 [29]. The resulting Sequence Alignment Map (SAM) files were converted to Binary Alignment Map (BAM) format and then sorted by coordinates and indexed using SAMtools v1.12 [30]. Polymerase chain reaction (PCR) duplicates were removed using PICARD v2.26. SNPs were first called using the HaplotypeCaller function in Genome Analysis Toolkit (GATK) v4.2.2 [31], and the genomic variant call format (gVCF) file was acquired for each chicken was acquired. The functions CombineGVCFs and GenotypeGVCFs were used to combine all the gVCF files and to jointly call SNPs together. In addition, SNPs were removed using GATK with specific standards: Quality Score < 30.0, QualByDepth < 2.0, FisherStrand > 60.0, RMSMappingQuality < 40.0, StrandOddsRatio > 3.0, MappingQualityRankSumTest < −12.5, and ReadPosRankSum < −8.
Expression GWAS and association analysis
Association analysis of SERPINB6 expression was performed using the mixed linear model in Genome-wide Efficient Mixed Model Association (GEMMA) v0.98.4 [32]. An eGWAS of SERPINB6 expression was performed according to the following formula:
where y is the vector of phenotypic values, α is the corresponding coefficient including the intercept, X is the genotype vector, β is the SNP effect vector, u is the random effect, and ε is the residual. Briefly, a Bonferroni correction was applied to adjust for multiple testing. The genome-wide significance threshold was 0.05/11,766,230, and the genome-wide potential significance threshold was 1/11,766,230. Visualization of the eGWAS results was performed using the qqman package of the R software.
We extracted all SNPs of SERPINB6, including those in the promoter region spanning 2 kb upstream of the gene. Based on the general linear model (GLM), correlation analyses of 4 traits and all SNPs of SERPINB6 were performed using PLINK v1.9 [27]. No fixed effects (e.g., sex, batch) were included in this model. Significant SNPs affecting each trait were screened using P < 0.05 as a threshold.
Target mRNA prediction
To further explore the underlying molecular mechanisms by which SERPINB6 regulates carcass traits, its upstream and downstream targets were predicted using bioinformatics software, including PROMO software (http://alggen.lsi.upc.es/) and miRDB software (http://www.mirdb.org/mirdb/index.html).
Luciferase reporter assays
HEK293 cells were seeded in 24-well plates. After co-transfection with the pGL4.18-WT-SERPINB6-3′UTR (wild type, WT) or pGL4.18-MT-SERPINB6-3′UTR (mutant type, MT) plasmid and the gga-miR-1615 mimic or mimic NC, firefly and Renilla luciferase activities were measured at 36 h post-transfection using a Dual-GLO Luciferase Assay System Kit (Promega, Madison, Wisconsin, WI, USA) according to the manufacturer’s instructions. Luminescence was measured using a Fluorescence/Multi-Detection Microplate Reader (BioTek, Vinusky, VT, USA), and firefly luciferase activity was normalized to Renilla luminescence in each well. Each sample included 3 technical replicates, and 3 independent biological replicates were performed.
Statistical analysis
Statistical analyses were performed using SPSS 26.0 software or a custom script in the R environment. The Kruskal‒Wallis test was used to determine the differences in SNPs between the 3 genotypes. The GLM was used to compare the differences in carcass trait phenotypes and SERPINB6 expression, with P < 0.05 was set as the significance threshold. Spearman correlation analysis was used to estimate the relationships between 4 carcass traits (LW, DW, EW, and BMW). All the data are presented as the mean ± 3 standard deviations from 3 biological replicates.
Results
The 4 carcass traits are highly correlated
Descriptive statistics of the carcass traits of 381 Tiannong partridge chickens with phenotypes are shown in Table 1. At 126 d of age, the LW, DW, EW and BMW were 1,513.41 g (CV = 11.98%), 1,343.99 g (CV = 12.19%), 1,014.29 g (CV = 12.41%) and 167.36 g (CV = 13.92%), respectively. In addition, the 4 carcass traits showed a highly significant correlation, such as the strong correlation between the EW and BMW (r = 0.74, P < 0.01), and the close relationships between the EW and LW and between the EW and DW are also reflected in the heatmap (Additional file 4: Fig. S2).
The 12 candidate genes were identified using EN-1SE model
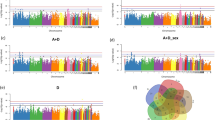
In this study, a total of 15,092 coding genes (excluding non-coding genes) were selected from 399 chickens for subsequent analysis. To identify candidate genes for the traits analyzed, we estimated effect sizes for each trait. The estimated effect sizes of genes between traits and expression levels are shown based on the EN-1SE model for phenotypic associations in Fig. 1. We identified 400 genes in total that had non-zero effects on 4 carcass traits using the EN-1SE model, among which 12 genes had non-zero effects on the LW, DW, EW and BMW (Additional file 5: Table S3), i.e., AMY1A, AP3B2, CEBPG, EEF2, EIF4EBP1, FGFR1, FOXD3, GOLM1, LOC107052698, PABPC1, SERPINB6 and TBC1D16. Therefore, these 12 genes were identified as candidate genes based on the EN-1SE model (Additional file 6: Fig. S3 and Table 2). Additionally, we found that SERPINB6 and TBC1D16 had large effects and were among the top 5 genes affecting the EW and BMW (Fig. 1C and D).

Identification of candidate genes for (A) live weight (LW), B dressed weight (DW), C eviscerated weight (EW) and D breast muscle weight (BMW) based on association analyses using EN-1SE. The estimated effect sizes and Manhattan plot of the genes are shown. The parameters of EN-1SE were determined based on the one standard error rule. Each dot represents a gene
Hub genes were screened by WGCNA
To determine the biological relationships and possible functions of the hub genes, we constructed networks using WGCNA. First, we set the height to 1,200,000 based on the basis of the gene expression level and eliminated 6 outlier samples (Additional file 7: Fig. S4A). The optimal soft threshold was determined (β = 6) by taking R2 > 0.85 as the standard, and 24 modules were obtained (Additional file 7: Fig. S4B). Then, the modules with a similarity greater than 0.7 were clustered according to the module eigengenes (Additional file 7: Fig. S4C). Finally, 20 modules were obtained (Additional file 7: Fig. S4D). The correlation coefficients and P values between the modules and the 4 carcass traits were obtained. We found that the target module was the green module, which was considered to be the most clearly relevant module. The green module had the highest correlation with the LW, EW and BMW (r = −0.22, P = 5.0 × 10–5; r = −0.25, P = 3.0 × 10–6; r = −0.25, P = 3.0 × 10–6) (Fig. 2A). A total of 101 hub genes of the target modules were determined using the criteria GS > 0.2 and MM > 0.6 (Additional file 8: Table S4). Twenty-six hub genes were screened for the LW, EW and BMW, including CREBBP, SERPINB6 and ZNF335 (Fig. 2B and Table 2).

A candidate functional gene, SERPINB6, was identified by 3 analyses, including EN-1SE model analysis, WGCNA and differential expression analysis. A Correlation analysis between the module and carcass traits (n = 350). Red represents a positive correlation, blue represents a negative correlation, and a darker color indicates a stronger the correlation. B Venn diagram of 26 overlapping hub genes associated with carcass traits based on WGCNA. C Histogram of differential expression of overlapping genes identified from the differential expression analysis, EN-1SE model or WGCNA. Low indicates the low group, and High indicates the high group. *: P < 0.05, **: P < 0.01, ***: P < 0.001. D Venn diagram of 1 overlapping gene (SERPINB6) associated with the EW based on the genes with non-zero effects, hub genes and DEGs. EN-1SE: elastic net with parameters determined based on the one standard error rule, WGCNA: weighted gene co-expression network analysis, DEGs: differentially expressed genes, LW: live weight, DW: dressed weight, EW: eviscerated weight, BMW: breast muscle weight
SERPINB6 is a candidate gene for carcass traits
Furthermore, we screened hundreds of DEGs in the high and low groups for each phenotype (Additional file 9: Fig. S5, Additional file 10: Table S5). Only 5 of the 12 candidate genes (AP3B2, EIF4EBP1, FGFR1, LOC107052698 and SERPINB6) based on the EN-1SE model were DEGs that were differentially expressed in at least one phenotype (Fig. 2C). The expression of SERPINB6 was higher in the high EW group than in the low EW group. Among the 3 analyses mentioned above, SERPINB6 was the only overlapping gene for the EW (Fig. 2D). Therefore, SERPINB6 was confirmed as the key candidate gene for carcass traits.
SERPINB6 promotes cell proliferation
To determine whether exogenous expression of SERPINB6 could promote cell proliferation, we performed CCK8 and EdU assays. First, a SERPINB6 was successfully overexpressed using a SERPINB6 overexpression vector (Additional file 11: Fig. S6A), and SERPINB6-si-3 (si-SERPINB6) had the highest interference efficiency among the 3 siRNAs in DF1 cells (Additional file 11: Fig. S6B). SERPINB6 overexpression significantly improved the proliferation of DF1 cells (Fig. 3A), while a lower absorbance value was observed after si-SERPINB6 transfection compared to the control in the CCK8 assay (Fig. 3B). SERPINB6 overexpression resulted in increased EdU positivity compared to the control, but SERPINB6 knockdown significantly decreased EdU incorporation (Fig. 3C and D). Therefore, SERPINB6 promotes the proliferation of DF1 cells. Moreover, we used CPMs as a cellular model for further validation, and the results were consistent with those obtained for DF1 cells (Fig. 3E–H and Additional file 11: Fig. S6C–D).

Verification the function of SERPINB6 at the cellular level. A and B Cell growth after the transfection of DF1 cells with the empty vector pcDNA3.1 and pcDNA3.1-SERPINB6 or si-NC and si-SERPINB6. *: P < 0.05, **: P < 0.01. C and D The proliferation of transfected DF1 cells assessed by EdU incorporation. **: P < 0.01, ***: P < 0.001. E and F Cell growth following the transfection of CPMs with the empty vector pcDNA3.1 and pcDNA3.1-SERPINB6 or si-NC and si-SERPINB6. *: P < 0.05, **: P < 0.01, ***: P < 0.001. G and H Proliferation of transfected CPMs was assessed by EdU incorporation. **: P < 0.01. I The top 15 KEGG pathways enriched in DEGs in SERPINB6-overexpressing cells. J Heatmap of DEGs. N indicates pcDNA3.1, and O indicates pcDNA3.1-SERPINB6. DEGs: differentially expressed genes
After overexpressing SERPINB6 at the cellular level, we investigated the role of this gene in this pathway. We found 1,576 DEGs (1,023 upregulated and 553 downregulated) (Additional file 11: Fig. S6E). Further enrichment analyses revealed that 38 KEGG pathways and 456 GO terms were enriched (Additional file 12: Table S6). Moreover, all of these enriched pathways, which included KEGG pathways (tight junction, ECM-receptor interaction, MAPK signaling and focal adhesion) and GO terms (extracellular space, cytoplasm and cytosol) (Fig. 3I and Additional file 11: Fig. S6F), were involved in the proliferative function of SERPINB6. Among them, 20 major DEGs were enriched in the above-mentioned pathways and promoted or inhibited cell proliferation at the transcriptional level (Fig. 3J). The 20 DEGs revealed potential interactions between ACTA2, COL6A1 and SERPINB6 at the protein level (Additional file 13: Fig. S7).
Three loci were screened by eGWAS and association analysis
To identify the key variants affecting the expression of SERPINB6, we performed an eGWAS to detect the molecular markers. A ~11.59 kb region located on chicken (Gallus gallus) chromosome 2 (31.67–43.26 kb) showed a significant peak for SERPINB6 expression (Fig. 4A). Within this region, rs317934171 and rs313806168 were identified as being above the genome-wide significance threshold (P = 5.12 × 10–9). No population stratification was found based on the calculated genomic inflation factor (1.019) in the Q‒Q plot (Fig. 4B). A total of 8 SNPs over the suggestive threshold were observed. The lead SNP (rs317934171) had the highest allele frequency, and was located in the functional region which was the 3′UTR of SERPINB6 (Table 3). To further evaluate the relationships between genotype and phenotype (4 carcass traits), we used the GLM for association analysis. Fifteen SNPs were significantly associated with the 4 carcass traits in 357 samples (Additional file 14: Table S7). Among them, rs317934171, rs14200392 and rs731580755 were overlapping SNPs (Fig. 4C).

eGWAS of SERPINB6 expression and effects of different genotypes of rs317934171. A and B Manhattan plots and quantile‒quantile (Q‒Q) plots of eQTLs for SERPINB6. Each dot represents a SNP. The horizontal red and blue lines indicate the genome-wide significant (P = 5.12 × 10–9) and suggestive (P = 1.02 × 10–7) thresholds, respectively. C Venn diagram of 3 overlapping SNPs (rs317934171, rs14200392 and rs731580755) based on the GLM and eGWAS. GLM: general linear model, eGWAS: expression genome-wide association study. D and E Differences in the EW and BMW between different genotypes of rs317934171. ns: P > 0.05, **: P < 0.01, BMW: breast muscle weight, EW: eviscerated weight. F Differences in the SERPINB6 expression level between different genotypes of rs317934171. ns: P > 0.05, **: P < 0.01, ***: P < 0.001, ****: P < 0.0001
rs317934171 is a variant of SERPINB6
In Tiannong partridge chickens, individuals with the CC genotype had significantly higher eviscerated weight and breast muscle weight than did those with the TT genotype (Fig. 4D and E). Moreover, SERPINB6 expression showed the same trend that SERPINB6 expression was greater in the CC genotype than those in the TT genotype (Fig. 4F). Accordingly, rs317934171 was considered a key SNP associated with SERPINB6 expression based on the results of eGWAS and association analysis. In addition, we found that the CC genotype increased the promoter activity of SERPINB6 using dual-luciferase assays (Additional file 15: Fig. S8). Considering that the SNP is located in the 3′UTR, it was surprising that the transcription factor Adf-1 was only associated with the CC genotype of rs317934171, as determined by PROMO software (Additional file 16: Fig. S9). Therefore, we believe that the binding of the Adf-1 transcription factor to the key variant rs317934171 may be the key to improving SERPINB6 expression and meat production.
gga-miR-1615 binds to SERPINB6
Based on the identified key variants located in the 3′UTR, we asked whether miRNAs binding blocks translation of the SERPINB6 gene. Using the miRDB software, we predicted that gga-miR-1615 would act on the 3′UTR of SERPINB6 according to the principle of complementary base pairing. Coincidentally, the seed sequence “AGCTGCCA” contains rs317934171 (Fig. 5A). We confirmed the authenticity of rs317934171 by Sanger sequencing (Fig. 5B). To further verify the relationship between gga-miR-1615 and SERPINB6, we constructed a dual-luciferase reporter vector. After co-transfecting the dual luciferase reporter plasmids (pGL4.18 and WT/MT) and gga-miR-NC/gga-miR-1615, we found that gga-miR-1615 significantly decreased the dual luciferase activity compared with that obtained with gga-miR-NC (Fig. 5C). However, the expression of SERPINB6 did not change significantly after transfection with 4 siRNAs, namely, mimic-NC, miR-1615-mimic, inhibitor-NC and miR-1615-inhibitor, in DF1 cells and CPMs (Additional file 17: Fig. S10). Therefore, our results indicate that gga-miR-1615 affects the translation of SERPINB6 and that other miRNAs may also play a role in this process.

gga-miR-1615 directly targets SERPINB6. A Complementary pairing of gga-miR-1615 with the wild type or mutant 3′UTR of the target gene, SERPINB6. B Validation of the wild type and mutant type by Sanger sequencing. WT indicates wild type and MT indicates mutant type. C HEK293 cells were co-transfected with the wild type or mutant SERPINB6 3′UTR dual-luciferase vector and the miR-1615 mimic or mimic NC. The data represent the mean ± 3 standard deviations from 3 biological replicates. *: P < 0.05
Discussion
The Tiannong partridge chicken is a commercially available 3-way cross chicken produced from 3 pure lines of Qingyuan partridge chicken and Guangxi partridge chicken; this chicken is an important indigenous Chinese slow-growing yellow-feathered chicken and is well known for its superior meat quality [21]. Few key candidate genes for carcass traits have been identified, and the molecular mechanisms regulating carcass traits are unclear. Here, based on large-scale RNA-seq and WGS, we identified key genes and molecular markers associated with carcass traits, particularly EW (Fig. 6).

Map of a novel functional gene, SERPINB6, for carcass traits and a key variant, rs317934171, located in the 3′UTR of the gene. A Venn diagram of 1 overlapping gene (SERPINB6) associated with the EW based on RNA-seq data. EN-1SE with parameters determined based on the one standard error rule, WGCNA: weighted gene co-expression network analysis, DEGs: differentially expressed genes. B SERPINB6 promotes DF1 cell and CPMs proliferation. C Interaction between gga-miR-1615 and SERPINB6. D A key molecular marker (rs317934171) in the 3′UTR has significant effects on increasing SERPINB6 expression, EW and BMW, as revealed by GLM and eGWAS. And Adf-1 only binds to the CC genotype of rs317934171. TF: transcription factor, GLM: general linear model, WGS: whole-genome sequencing
Advantages of the EN-1SE model for screening candidate genes
At present, one or two analyses are generally used to identify candidate genes by RNA-seq [33]. However, the main advantage of this study is that candidate genes were screened by 3 methods, namely EN-1SE model, WGCNA and differential expression analysis. First, our previous research showed the following: LMM, LASSO-1SE and EN-1SE had the lowest type I error rates, which were exactly zero; EN-Min minimized cross-validated mean squared prediction error [34] with an additional significance test; and LASSO-Min showed type I error rates of less than 0.02. EN-Min without significance testing and RR-1SE had high type I error rates, although these were lower than those of RR-Min. Taken together, the results of the simulation study demonstrated that EN-1SE without significance testing was the optimal method for analyzing associations of expression levels with traits in terms of both the type I error rate and the detection power [21].
Based on the advantages of EN-1SE, 12 genes were found to be significantly associated with 4 carcass traits. Some genes were reported to be associated with carcass traits or muscle development. For example, AMY1A polymorphisms were found to be associated with growth, carcass traits and feed intake efficiency in chickens, including daily gain, average daily feed intake, leg muscle weight and abdominal fat (P < 0.05) [35]. CEBPG is a transcription factor that binds to the promoter and enhancer regions of target genes [36], and ATF5 in combination with CEBPG only reduces the size of glycolytic fibers and tends to increase the proportion of oxidative fibers [37]. EIF4EBP1, which is resistant to mTORC1-mediated inhibition, preserves muscle function by increasing mitochondrial activities [38]. FGFR1 can bind to LBP1C-2 to activate satellite cells and promote satellite cell self-renewal through upregulation of SPAY1 [39], and may play a role regulating postnatal skeletal muscle maintenance [40].
Characteristics of WGCNA screening for hub genes
The second method, WGCNA, is a systematic analysis method used to describe gene association patterns [41, 42]. This method can divide thousands of genes into multiple modules according to the gene expression patterns and identify hub genes in the modules and is instrumental for improving the accuracy and reducing the number of calculations. Among the 26 hub genes (e.g., CREBBP, ZNF335, and SERPINB6) we identified, the regulatory mechanism for genes associated with carcass traits is largely unknown. According to previous reports, the CREBBP gene is ubiquitously expressed and is involved in the transcriptional co-activation of many different transcription factors [43]. ZNF335 is essential for self-renewal of neural progenitors, neurogenesis, and neuronal differentiation [44].
Function of SERPINB6
We found that SERPINB6 was the only overlapping gene between the 3 analyses. SERPINB6 encodes a member of the serine proteinase inhibitor (serpin) superfamily and the ovalbumin serpin subfamily, which are involved in cell migration, cell differentiation and apoptosis [45]. SERPINB6 is abundantly expressed by mast cells in all organs and by the human mast cell line HMC-1 [46]. Further studies have shown that SERPINB6 can regulate the activity of endogenous trypsin in the cytoplasm and is significantly negatively correlated with deer meat shear force and intramuscular fat, with correlation coefficients of −0.734 and −0.816, respectively [47]. Recent research has shown that SERPINB6 is a target protein of dutomycin. For the first time, dutomycin was shown to directly bind to SERPINB6, activate an intracellular serine protease and induce autophagy [48].
SERPINB6 promotes cell proliferation
To date, SERPINB6 has not been reported to be significantly associated with carcass traits, and the molecular mechanism by which SERPINB6 affects carcass traits is also unknown. We then selected DF1 cells and CPMs to verify the new function of SERPINB6 in chickens. Here, we constructed an experimental model for the proliferation of DF1 cells and CPMs after SERPINB6 knockdown or overexpression. Through a series of in vitro experiments, CCK8 and EdU assays indicated that SERPINB6 overexpression promoted the proliferation of DF1 cells and CPMs, and SERPINB6 knockdown inhibited the proliferation of DF1 cells and CPMs.
SERPINB6 overexpression results in the formation of a complex regulatory network
Moreover, thousands of DEGs (e.g., MYC [49], ACTA2, and MYH10 [50]) were enriched mainly after SERPINB6 overexpression, mainly in tight junction, ECM-receptor interaction, focal adhesion, and MAPK signaling pathway and so on. Tight junction proteins contribute to the control of cell proliferation [51]. Transcriptome analysis revealed that the ECM-receptor interaction pathway is associated with meat quality in broiler chickens [52] and can directly or indirectly control cellular activities such as adhesion, migration, differentiation, proliferation, and apoptosis. The focal adhesion pathway plays an essential role in important biological processes such as cell motility, cell proliferation, cell differentiation and gene expression regulation [53, 54]. Therefore, SERPINB6 promotes cell proliferation through a complex regulatory network.
eGWAS identifies associations between genetic variants and SERPINB6 expression
We identified associations between genetic variation and SERPINB6 expression through eGWAS that is to screen for key SNPs and laying the foundation for further elaboration of the mechanism affecting gene expression levels [55]. Our results indicated that eight variants associated with SERPINB6 (67,630,787–67,640,291) were located in this region (67,631,671–67,643,258), and these SNPs were the cis-eQTLs.
We finally identified 3 molecular markers (rs317934171, rs14200392 and rs731580755) by eGWAS and association analysis, and rs317934171 happened to be located in the 3′UTR of SERPINB6. With the SNP serving as a molecular marker, we found that the SNP increased SERPINB6 expression. The EW and BMW of the CC genotype were significantly higher than those of the TT genotype, with increases of 4.60% and 5.12%, respectively. Notably, the proportion of SERPINB6 in the CC genotype was 26.28% higher than that in the TT genotype. At the same time, the wild type (CC genotype) significantly enhanced luciferase activity. Therefore, the CC genotype has a greater effect on rs317934171. The CC genotype is predicted to bind to the Adf-1 transcription factor, which can enhance the promoter activity of SERPINB6 and promote its expression. Adf-1 has also been reported to be a key modulator of the dynamic organization of the actin cytoskeleton [56]. Further validation of the interaction between Adf-1 and rs317934171 or SERPINB6 is needed.
gga-miR-1615 binds to SERPINB6
In general, the 3′UTR of genes often binds to miRNAs. For example, miR-21-5p regulates skeletal muscle satellite cell proliferation and differentiation by targeting KLF3 in the 3′UTR in chickens [57]. Cai et al. [58], confirmed that miR-16-5p, which significantly decreased luciferase activity, targeted SESN1 to regulate the p53 signaling pathway, affecting myoblast proliferation and apoptosis. We found that gga-miR-1615 significantly decreased double luciferase activity, which is consistent with previous research reports [57, 58].
Moreover, transfection of the gga-miR-1615 mimic and gga-miR-1615 inhibitor did not significantly change the SERPINB6 expression levels in DF1 cells, but the SERPINB6 expression levels were significantly reduced after transfection of the gga-miR-1615 inhibitor in CPMs. Multiple miRNAs may silence gene expression to further induce phenotypic changes, or both gga-miR-1615 and Adf-1 may change SERPINB6 expression. More studies on this article will be performed in the future.
Conclusion
Overall, we identified SERPINB6 as a novel functional gene that is significantly associated with carcass traits by association analysis, including the EN-1SE model, WGCNA and differential expression analysis, based on RNA-seq data. Furthermore, our results suggested that rs317934171 of the CC genotype, as identified through eGWAS and association analysis, significantly increases SERPINB6 expression levels, and that SERPINB6 promoted cell proliferation in DF1 cells and CPMs. Furthermore, a direct target region relationship was found between gga-miR-1615 and SERPINB6. We explored the biological function and preliminary regulatory mechanism of SERPINB6 and identified its molecular markers.
Availability of data and materials
The datasets analyzed during the current study are available from the corresponding author upon reasonable request, and we’ve uploaded them to Genome Sequence Archive (https://ngdc.cncb.ac.cn/gsa) under the BioProject no. CRA014989 and CRA015111.
Abbreviations
- BMW:
-
Breast muscle weight
- CCK8:
-
Cell Counting Kit-8
- CV:
-
Coefficient of variation
- DEGs:
-
Differentially expressed genes
- DF1:
-
Chicken embryonic fibroblasts line
- DW:
-
Dressed weight
- EdU:
-
5-Ethynyl-2-deoxyuridine
- EN-1SE:
-
Elastic net-1 standard error
- eGWAS:
-
Expression genome-wide association study
- FBS:
-
Fetal bovine serum
- GLM:
-
General linear model
- GO:
-
Gene Ontology
- GS:
-
Gene significance
- GWAS:
-
Genome-wide association study
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LW:
-
Live weight
- MAF:
-
Minor allele frequency
- MM:
-
Module membership
- MT:
-
Mutant type
- RNA-seq:
-
Transcriptome sequencing
- RT‒PCR:
-
Real time-polymerase chain reaction
- SD:
-
Standard deviation
- siRNAs:
-
Small interfering RNAs
- SNP:
-
Single-nucleotide polymorphism
- WGCNA:
-
Weighted gene co-expression network analysis
- WGS:
-
Whole-genome sequencing
- WT:
-
Wild type
References
McClements DJ, Grossmann L. The science of plant-based foods: Constructing next-generation meat, fish, milk, and egg analogs. Compr Rev Food Sci Food Saf. 2021;20(4):4049–100. https://doi.org/10.1111/1541-4337.12771.
FAOSTAT. 2022. https://www.fao.org/faostat/en/#data/QCL.
Jin Y, Cui H, Yuan X, Liu L, Liu X, Wang Y, et al. Identification of the main aroma compounds in Chinese local chicken high-quality meat. Food Chem. 2021;359:129930. https://doi.org/10.1016/j.foodchem.2021.129930.
Claire D’Andre H, Paul W, Shen X, Jia X, Zhang R, Sun L, et al. Identification and characterization of genes that control fat deposition in chickens. J Anim Sci Biotechnol. 2013;4:43. https://doi.org/10.1186/2049-1891-4-43.
Van Laere AS, Nguyen M, Braunschweig M, Nezer C, Collette C, Moreau L, et al. A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature. 2003;425(6960):832–6. https://doi.org/10.1038/nature02064.
Zhou Z, Cai D, Wei G, Cai B, Kong S, Ma M, et al. Polymorphisms of CRELD1 and DNAJC30 and their relationship with chicken carcass traits. Poult Sci. 2023;102(1):102324. https://doi.org/10.1016/j.psj.2022.102324.
Xiong X, Zhou M, Zhu X, Tan Y, Wang Z, Gong J, et al. RNA sequencing of the pituitary gland and association analyses reveal PRKG2 as a candidate gene for growth and carcass traits in Chinese Ningdu yellow chickens. Front Vet Sci. 2022;9:892024. https://doi.org/10.3389/fvets.2022.892024.
Ramdas S, Judd J, Graham SE, Kanoni S, Wang Y, Surakka I, et al. A multi-layer functional genomic analysis to understand noncoding genetic variation in lipids. Am J Hum Genet. 2022;109(8):1366–87. https://doi.org/10.1016/j.ajhg.2022.06.012.
Tan X, He Z, Fahey AG, Zhao G, Liu R, Wen J. Research progress and applications of genome-wide association study in farm animals. Anim Res One Health. 2023;1(1):56–77. https://doi.org/10.1002/aro2.14.
Li W, Zheng M, Zhao G, Wang J, Liu J, Wang S, et al. Identification of QTL regions and candidate genes for growth and feed efficiency in broilers. Genet Sel Evol. 2021;53(1):13. https://doi.org/10.1186/s12711-021-00608-3.
Li W, Liu R, Zheng M, Feng F, Liu D, Guo Y, et al. New insights into the associations among feed efficiency, metabolizable efficiency traits and related QTL regions in broiler chickens. J Anim Sci Biotechnol. 2020;11:65. https://doi.org/10.1186/s40104-020-00469-8.
Liu R, Sun Y, Zhao G, Wang F, Wu D, Zheng M, et al. Genome-wide association study identifies Loci and candidate genes for body composition and meat quality traits in Beijing-You chickens. PLoS One. 2013;8(4):e61172. https://doi.org/10.1371/journal.pone.0061172.
Yang X, Sun J, Zhao G, Li W, Tan X, Zheng M, et al. Identification of major loci and candidate genes for meat production-related traits in broilers. Front Genet. 2021;12:645107. https://doi.org/10.3389/fgene.2021.645107.
Velez-Irizarry D, Casiro S, Daza KR, Bates RO, Raney NE, Steibel JP, et al. Genetic control of longissimus dorsi muscle gene expression variation and joint analysis with phenotypic quantitative trait loci in pigs. BMC Genom. 2019;20:3. https://doi.org/10.1186/s12864-018-5386-2.
Tan X, Liu R, Zhao D, He Z, Li W, Zheng M, et al. Large-scale genomic and transcriptomic analyses elucidate the genetic basis of high meat yield in chickens. J Adv Res. 2024;55:1–16. https://doi.org/10.1016/j.jare.2023.02.016.
Performance terminology and measurements for poultry. NY/T 823-2020. CN-NY. https://www.nssi.org.cn/nssi/front/112331443.html.
Chen F, Wu P, Shen M, He M, Chen L, Qiu C, et al. Transcriptome analysis of differentially expressed genes related to the growth and development of the Jinghai yellow chicken. Genes (Basel). 2019;10(7):539. https://doi.org/10.3390/genes10070539.
Andrews S. FastQC: A quality control tool for high throughput sequence data. 2014. https://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60. https://doi.org/10.1038/nmeth.3317.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. https://doi.org/10.1186/s13059-014-0550-8.
Kang H, Zhao D, Xiang H, Li J, Zhao G, Li H. Large-scale transcriptome sequencing in broiler chickens to identify candidate genes for breast muscle weight and intramuscular fat content. Genet Sel Evol. 2021;53(1):66. https://doi.org/10.1186/s12711-021-00656-9.
Zou H, Hastie T. Regularization and variable selection via the elastic net. J R Stat Soc B Stat Methodol. 2005;67(2):301–20. https://doi.org/10.1111/j.1467-9868.2005.00503.x.
Waldmann P, Mészáros G, Gredler B, Fuerst C, Sölkner J. Evaluation of the lasso and the elastic net in genome-wide association studies. Front Genet. 2013;4:270. https://doi.org/10.3389/fgene.2013.00270.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. https://doi.org/10.1186/1471-2105-9-559.
Zheng PF, Chen LZ, Guan YZ, Liu P. Weighted gene co-expression network analysis identifies specific modules and hub genes related to coronary artery disease. Sci Rep. 2021;11:6711. https://doi.org/10.1038/s41598-021-86207-0.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–8. https://doi.org/10.1006/meth.2001.1262.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. https://doi.org/10.1086/519795.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. https://doi.org/10.1093/bioinformatics/btu170.
Heng L. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint. 2013. https://doi.org/10.48550/arXiv.1303.3997.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10(2):giab008. https://doi.org/10.1093/gigascience/giab008.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. https://doi.org/10.1101/gr.107524.110.
Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44(7):821–4. https://doi.org/10.1038/ng.2310.
Silva-Vignato B, Coutinho LL, Poleti MD, Cesar ASM, Moncau CT, Regitano LCA, et al. Gene co-expression networks associated with carcass traits reveal new pathways for muscle and fat deposition in Nelore cattle. BMC Genomics. 2019;20:32. https://doi.org/10.1186/s12864-018-5345-y.
Engebretsen S, Bohlin J. Statistical predictions with glmnet. Clin Epigenetics. 2019;11(1):123. https://doi.org/10.1186/s13148-019-0730-1.
Zhang Z, Zhong H, Lin S, Liang L, Ye S, Xu Z, et al. Polymorphisms of AMY1A gene and their association with growth, carcass traits and feed intake efficiency in chickens. Genomics. 2021;113(2):583–94. https://doi.org/10.1016/j.ygeno.2020.10.041.
Davydov IV, Bohmann D, Krammer PH, Li-Weber M. Cloning of the cDNA encoding human C/EBP gamma, a protein binding to the PRE-I enhancer element of the human interleukin-4 promoter. Gene. 1995;161(2):271–5. https://doi.org/10.1016/0378-1119(95)00271-7.
Brearley-Sholto MC, Loczenski-Brown DM, Jones S, Daniel Z, Ebling FJP, Parr T, et al. Effect of AAV-mediated overexpression of ATF5 and downstream targets of an integrated stress response in murine skeletal muscle. Sci Rep. 2021;11:19796. https://doi.org/10.1038/s41598-021-99432-4.
Ang SJ, Crombie EM, Dong H, Tan KT, Hernando A, Yu D, et al. Muscle 4EBP1 activation modifies the structure and function of the neuromuscular junction in mice. Nat Commun. 2022;13:7792. https://doi.org/10.1038/s41467-022-35547-0.
Meng J, Lv Z, Chen X, Sun C, Jin C, Ding K, et al. LBP1C-2 from Lycium barbarum maintains skeletal muscle satellite cell pool by interaction with FGFR1. iScience. 2023;26(5):106573. https://doi.org/10.1016/j.isci.2023.106573.
Eash J, Olsen A, Breur G, Gerrard D, Hannon K. FGFR1 inhibits skeletal muscle atrophy associated with hindlimb suspension. BMC Musculoskelet Disord. 2007;8:32. https://doi.org/10.1186/1471-2474-8-32.
Kadarmideen HN, Watson-Haigh NS, Andronicos NM. Systems biology of ovine intestinal parasite resistance: disease gene modules and biomarkers. Mol Biosyst. 2011;7(1):235–46. https://doi.org/10.1039/c0mb00190b.
Kogelman LJ, Cirera S, Zhernakova DV, Fredholm M, Franke L, Kadarmideen HN. Identification of co-expression gene networks, regulatory genes and pathways for obesity based on adipose tissue RNA Sequencing in a porcine model. BMC Med Genomics. 2014;7:57. https://doi.org/10.1186/1755-8794-7-57.
Nonaka M, Kim R, Sharry S, Matsushima A, Takemoto-Kimura S, Bito H. Towards a better understanding of cognitive behaviors regulated by gene expression downstream of activity-dependent transcription factors. Neurobiol Learn Mem. 2014;115:21–9. https://doi.org/10.1016/j.nlm.2014.08.010.
Yang YJ, Baltus AE, Mathew RS, Murphy EA, Evrony GD, Gonzalez DM, et al. Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell. 2012;151(5):1097–112. https://doi.org/10.1016/j.cell.2012.10.043.
Benarafa C, Remold-O’Donnell E. The ovalbumin serpins revisited: perspective from the chicken genome of clade B serpin evolution in vertebrates. Proc Natl Acad Sci U S A. 2005;102(32):11367–72. https://doi.org/10.1073/pnas.0502934102.
Strik MC, Wolbink A, Wouters D, Bladergroen BA, Verlaan AR, van Houdt IS, et al. Intracellular serpin SERPINB6 (PI6) is abundantly expressed by human mast cells and forms complexes with beta-tryptase monomers. Blood. 2004;103(7):2710–7. https://doi.org/10.1182/blood-2003-08-2981.
López-Pedrouso M, Franco D, Serrano MP, Maggiolino A, Landete-Castillejos T, De Palo P, et al. A proteomic-based approach for the search of biomarkers in Iberian wild deer (Cervus elaphus) as indicators of meat quality. J Proteomics. 2019;205:103422. https://doi.org/10.1016/j.jprot.2019.103422.
Jang M, Hara S, Kim GH, Kim SM, Son S, Kwon M, et al. Dutomycin induces autophagy and apoptosis by targeting the serine protease inhibitor SERPINB6. ACS Chem Biol. 2021;16(2):360–70. https://doi.org/10.1021/acschembio.0c00889.
Bretones G, Delgado MD, León J. Myc and cell cycle control. Biochim Biophys Acta. 2015;1849(5):506–16. https://doi.org/10.1016/j.bbagrm.2014.03.013.
Ye M, Ye F, He L, Luo B, Yang F, Cui C, et al. Transcriptomic analysis of chicken Myozenin 3 regulation reveals its potential role in cell proliferation. PLoS One. 2017;12(12):e0189476. https://doi.org/10.1371/journal.pone.0189476.
Díaz-Coránguez M, Liu X, Antonetti DA. Tight junctions in cell proliferation. Int J Mol Sci. 2019;20(23):5972. https://doi.org/10.3390/ijms20235972.
San J, Du Y, Wu G, Xu R, Yang J, Hu J. Transcriptome analysis identifies signaling pathways related to meat quality in broiler chickens - the extracellular matrix (ECM) receptor interaction signaling pathway. Poult Sci. 2021;100(6):101135. https://doi.org/10.1016/j.psj.2021.101135.
Shen J, Cao B, Wang Y, Ma C, Zeng Z, Liu L, et al. Hippo component YAP promotes focal adhesion and tumour aggressiveness via transcriptionally activating THBS1/FAK signalling in breast cancer. J Exp Clin Cancer Res. 2018;37(1):175. https://doi.org/10.1186/s13046-018-0850-z.
Nardone G, Oliver-De La Cruz J, Vrbsky J, Martini C, Pribyl J, Skládal P, et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat Commun. 2017;8:15321. https://doi.org/10.1038/ncomms15321.
Nica AC, Dermitzakis ET. Expression quantitative trait loci: present and future. Philos Trans R Soc Lond B Biol Sci. 2013;368(1620):20120362. https://doi.org/10.1098/rstb.2012.0362.
Yu Y, Qiao L, Chen J, Rong Y, Zhao Y, Cui X, et al. Arabidopsis REM16 acts as a B3 domain transcription factor to promote flowering time via directly binding to the promoters of SOC1 and FT. Plant J. 2020;103(4):1386–98. https://doi.org/10.1111/tpj.14807.
Zhang D, Ran J, Li J, Yu C, Cui Z, Amevor FK, et al. miR-21–5p regulates the proliferation and differentiation of skeletal Muscle satellite cells by targeting KLF3 in chicken. Genes (Basel). 2021;12(6):5972. https://doi.org/10.3390/genes12060814.
Cai B, Ma M, Chen B, Li Z, Abdalla BA, Nie Q, et al. MiR-16-5p targets SESN1 to regulate the p53 signaling pathway, affecting myoblast proliferation and apoptosis, and is involved in myoblast differentiation. Cell Death Dis. 2018;9(3):367. https://doi.org/10.1038/s41419-018-0403-6.
Acknowledgements
The authors thank all the members of the Guangdong Provincial Key Laboratory of Animal Molecular Design and Precise Breeding for sample collection.
Funding
This research was supported by grants from the Key Projects of National Natural Science Foundation of China (No. 32230101), the Project of Qingyuan Science and Technology (2020A01, 2021SJXM011), the Agriculture Research System (CARS-41) and the Core Breed Source Research Project JBGS (2021) 107.
Author information
Authors and Affiliations
Contributions
DZ analyzed the data, performed the functional assays and drafted the manuscript. RRL contributed to the design of the study, interpretation of the data, and manuscript preparation. XDT analyzed the genomic data. HMK performed the association analysis with the EN model. ZM extracted the genomic DNA. JW, HQZ and HX participated in sample collection. ZFZ provided the chicken samples. HL contributed to chicken sampling and offered financial support. GPZ contributed to the design of the study, the acquisition of financial support, the interpretation of the data, and the preparation of the manuscript. All authors contributed to the article and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal experiments were conducted in accordance with the protocols approved by the Laboratory Animal Welfare and Animal Experimental Ethical Inspection Board of Foshan University (No. 18091801). The birds were raised under the same environmental conditions, and food and water were provided ad libitum. To ensure animal welfare, the birds were humanely slaughtered, and all efforts were made to minimize suffering.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest. Thus, the authors declare that they have no competing interests.
Supplementary Information
Additional file 1:
Fig. S1. Low group and high group for 4 carcass traits.
Additional file 2:
Table S1. RNA oligonucleotides used in this study.
Additional file 3:
Table S2. Primer pairs for RT‒PCR.
Additional file 4:
Fig. S2. Spearman correlation analysis of the 4 carcass traits (n = 381).
Additional file 5:
Table S3. Genes with non-zero effects on each carcass trait for the EN-1SE model.
Additional file 6:
Fig. S3. Venn diagram of 12 overlapping genes with non-zero effects on carcass traits based on the EN-1SE model.
Additional file 7:
Fig. S4. Weighted gene co-expression network analysis.
Additional file 8:
Table S4. Hub genes for target modules.
Additional file 9:
Fig. S5. Volcano map of 4 carcass traits.
Additional file 10:
Table S5. DEGs associated with carcass traits.
Additional file 11:
Fig. S6. Transfection efficiency of SERPINB6 in DF1 cells and CPMs.
Additional file 12: Table S6.
Thirty-eight KEGG pathways and 456 GO terms were enriched.
Additional file 13: Fig. S7.
Protein–protein interaction network of DEGs in DF1 cells.
Additional file 14: Table S7.
Comparison of the phenotypes of different genotypes with significant association sites of SERPINB6.
Additional file 15: Fig. S8.
rs317934171 enhances promoter activity.
Additional file 16: Fig. S9.
Prediction of transcription factors before and after mutation of rs317934171.
Additional file 17: Fig. S10.
mRNA expression levels of SERPINB6 after transfection of mimic-NC, miR-1615-mimic, inhibitor-NC and miR-1615-inhibitor in DF1 cells and CPMs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhao, D., Liu, R., Tan, X. et al. Large-scale transcriptomic and genomic analyses reveal a novel functional gene SERPINB6 for chicken carcass traits. J Animal Sci Biotechnol 15, 70 (2024). https://doi.org/10.1186/s40104-024-01026-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40104-024-01026-3