
Abstract
Background
Bovine milk is an important source of nutrition for human consumption, and its quality is closely associated with the microbiota and metabolites in it. But there is limited knowledge about the milk microbiome and metabolome in cows with subacute ruminal acidosis.
Methods
Eight ruminally cannulated Holstein cows in mid lactation were selected for a 3-week experiment. The cows were randomly allocated into 2 groups, fed either a conventional diet (CON; 40% concentrate; dry matter basis) or a high-concentrate diet (HC; 60% concentrate; dry matter basis).
Results
The results showed that there was a decreased milk fat percentage in the HC group compared to the CON group. The amplicon sequencing results indicated that the alpha diversity indices were not affected by the HC feeding. At the phylum level, the milk bacteria were dominated by Proteobacteria, Actinobacteria, Bacteroidetes, and Firmicutes both in the CON and HC groups. At the genus level, the HC cows displayed an improved proportion of Labrys (P = 0.015) compared with the CON cows. Results of both the principal components analysis and partial least squares of discriminant analysis of milk metabolome revealed that samples of the CON and HC groups clustered separately. A total of 31 differential metabolites were identified between the two groups. Of these, the levels of 11 metabolites decreased (α-linolenic acid, prostaglandin E2, L-lactic acid, L-malic acid, 3-hydroxysebacic acid, succinyladenosine, guanosine, pyridoxal, L-glutamic acid, hippuric acid, and trigonelline), whereas the levels of the other 20 metabolites increased in the HC group with respect to the CON group (P < 0.05).
Conclusion
These results suggested that subacute ruminal acidosis less impacted the diversity and composition of milk microbiota, but altered the milk metabolic profiles, which led to the decline of the milk quality.
Similar content being viewed by others


Background
As an important food source for human consumption, bovine milk contains high levels of nutrients such as proteins, fatty acids, phospholipids, vitamins, and minerals [1], and can be further processed into dairy products such as cream, butter, yogurt, ice cream, and cheese [2]. With the improvement of people’s living standards, the demand for milk and dairy products is gradually increasing. However, forage often does not contain enough energy to support the high milk production, and therefore high-concentrate (HC) diets are widely used in modern dairy production, which conversely leads to a high incidence of subacute ruminal acidosis (SARA) [3,4,5].
Previous studies revealed that cows might develop mastitis during a grain-based SARA challenge, accompanied by the alteration of microbial composition in the milk [6, 7]. The microbiota composition of raw milk is an important consideration for mammary gland health [8]. Moreover, the microbiota count is one of the most important quality indicators related to the milk price. In Germany and other European countries, the microbial load of class 1 raw milk should be not more than 100,000 CFU/mL according to the European Regulation No 853/2004 [9]. In the United States, the microbial load of grade ‘A’ milk should be less than 100,000 CFU/mL according to the Pasteurized Milk Ordinance [10].
Besides milk fat, milk protein, and milk lactose, the milk also contains a variety of different small-molecule metabolites and micronutrients [11]. These metabolites are primarily derived from the metabolism of the mammary epithelial cells [12], and can be used as the mirror of the mammary gland function and milk quality [13]. As mentioned above, SARA could impact the health of mammary gland, which might further affect its metabolism. However, limited knowledge is available on the changes in the milk metabolites during SARA.
Therefore, the objectives of our study were to explore the bacterial changes and metabolic profiles in the milk of cows with SARA by using the 16S rRNA gene sequencing and high-performance liquid chromatography-mass spectrometry (HPLC-MS)-based metabolomics analyses, and finally, to clarify the effects of SARA on mammary gland health and milk quality in lactating dairy cows.
Methods
Animals, diets, and experimental design
Eight healthy multiparous lactating Holstein cows (2 to 3 parity) with an initial body weight of 582 ± 50 kg were chosen for the experiment, and were housed in individually tethered stalls with good ventilation. On average, the cows were in 120 ± 6 d in milk, with a mean milk yield of 18.2 ± 2.66 kg/d at the beginning of the experiment. All the cows were healthy and had free access to clean water throughout the 3-week experiment period. The cows were randomly allocated into two groups and fed either a conventional (CON; 40% concentrate; DM basis; n = 4) diet or a HC (60% concentrate; DM basis; n = 4) diet (Table 1). The diets had the same CP content and were formulated to meet or exceed the energy and milk production requirements of the cows according to NRC (2001) [14]. The diet was supplied twice daily at 08:00 and 19:00, with approximately 10% feed refusal. The cows were milked twice daily before feeding using a pipeline milking system.
Sample collection
The measurement of ruminal pH and dry matter intake (DMI) were described previously [16].
Milk production was recorded on the last 2 d of each week and milk samples were collected on the last day of each week for each cow. Before sample collection, teats were first dipped in iodine and followed by physical scrubbing with alcohol. Besides, the first 2 streams of milk were discarded to minimize possible contamination. Samples were immediately divided into two portions after collection. The first portion was added with potassium dichromate tablets (milk preservative) and mixed proportionally according to the milk yield of the morning and evening; then, the composite samples were stored at 4 °C for subsequent milk composition (milk fat, milk protein, and lactose) measurements using infrared analysis with a spectrophotometer (Foss-4000, Foss Electric A/S, Hillerød, Denmark). The second portion was immediately put into liquid nitrogen until the subsequent 16S rRNA sequencing and metabolomics analysis.
Microbial DNA isolation, 16S rRNA sequencing, and analyses
For each cow, 1–2 mL composite milk samples of morning and evening on the last day of each week were used for DNA isolation. After being fully vortexed, the milk samples were centrifuged at 13,000 × g for 10 min at 4 °C to remove the fat layer firstly [17]. Genomic DNA of the milk samples was extracted using a Water DNA Kit (Omega Bio-Tek, Norcross, GA, USA) according to the manufacturer’s protocols. The concentration and quality of the extracted DNA was assessed by a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Madison, USA) and the 1.0% agarose gel electrophoresis, respectively. All the extracted DNA samples were stored at −80 °C until subsequent processing.
The 16S rRNA genes were amplified using the 341F-806R primers (341F: 5′- CCTAYGGGRBGCASCAG-3′, 806R: 5′- GGACTACNNGGGTATCTAAT-3′) targeted the V3–V4 hypervariable region. The PCR mixtures consisted of 0.5 U of Taq polymerase (TransGen Biotech, Beijing, China) in 25 μL of 10 × PCR buffer, 200 μL each dNTP, 0.2 μmol/L each primer, and 2 μL of DNA (50 ng/μL). The amplification program was executed as follows: 95 °C for 2 min, 25 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, and a final extension at 72 °C for 5 min. We then visualized the PCR products using a 2% agarose gel, and bands between 400 and 450 bp were excised and next purified using the GeneJET Gel Extraction Kit (Thermo Scientific, Waltham, MA, USA). The paired-end sequencing (2 × 250 bp) was accomplished using an Illumina MiSeq PE 250 platform (Illumina Inc., San Diego, CA, USA) following standard Illumina sequencing protocols.
The raw sequences were first demultiplexed with an in-house Perl script and the low-quality sequences were filtered out based on the following criteria: the 250 bp reads were truncated at any site receiving an average quality score < 20 over a 10-bp sliding window, discarding the truncated reads that were shorter than 50 bp. The high-quality reads were then merged with a minimum overlap of 10-bp using FLASH v1.2.7 [18]. The sequences were screened to remove chimeras using the Vsearch software (v2.18.0) [19], and further followed by dereplication and amplicon sequence variant (ASV) feature table construction with DADA2 [20] plugin implemented in QIIME 2 v2021.08 [21]. Taxonomic assignment of the ASVs were performed using the naive Bayes classifier [22] trained against the SILVA v138 database [23] that trimmed to match the V3-V4 region sequenced. To control for inter-sample depth variability, all samples were rarefied to the size of the smallest sample (21,465 reads). Rarefaction curves were plotted to guarantee adequate sequencing depth (Fig. 1A). The analyses of α diversity, β diversity, and taxonomic classification were based on the rarefied ASV counts table. Alpha and beta diversity metrics were determined using the plugin q2-diversity in QIIME 2. Beta diversity was measured using Bray-Curtis dissimilarity and visualized with a principal coordinate analysis (PCoA) plot. Statistical significance of the PCoA was conducted by the “adonis” function in the R package “vegan” (v2.5–7) with 999 permutations.

A Rarefaction curves based on an amplicon sequence variant (ASV) are shown. Each curve represents one individual sample. B Principal component (PC) analysis of Bray-Curtis dissimilarities between the CON and HC diet groups. PERMANOVA results with 999 permutations are shown. CON = conventional diet; HG = high-concentrate diet
Milk metabolome analysis
The 24 milk samples used in the HPLC-MS-based metabolomics analyses were prepared as follows. Firstly, the samples were thawed at room temperature and 100 μL of each composite sample from the morning and evening were transferred into a centrifuge tube. Then, all samples were extracted with 300 μL of methanol, and 10 μL of internal standard (2.8 mg/mL, DL-o-chlorophenylalanine) was added. Next, the samples were vortexed for 30 s, and incubated for 1 h at −20 °C. Finally, the samples were centrifuged at 13,800 × g for 15 min at 4 °C, and 200 μL of the supernatant was transferred to the vial for subsequent HPLC-MS analysis.
The HPLC-MS was performed using an Ultimate 3000LC-Q-Exactive instrument (Thermo, California, USA) incorporating a Hyper gold C18 column (Thermo; 100 mm × 2.1 mm, 1.9 μm), and the column temperature was maintained at 40 °C. The injection volume was 10 μL and the autosampler was maintained at 4 °C. The gradient of mobile phase consisted of A [water + 5% (v/v) acetonitrile + 0.1% (v/v) formic acid] and B [acetonitrile + 0.1% (v/v) formic acid]. The flow rate was 0.3 mL/min. The elution procedure was designated as follows: 5% B for 1 min, 40% B at 2 min, 80% B at 7 min, 95% B at 11 min, and 5% B for 15.5–19.5 min. The mass spectrometric settings for positive and negative ion modes were as follows: heater temperature, 300 °C; sheath gas flow rate, 45 arb; aux gas flow rate, 15 arb; sweep gas flow rate, 1 arb; spray voltage, 3.0 KV/−3.2 KV; capillary temperature, 350 °C; and S-lens RF level, 30%/60%, respectively.
The raw data were conducted with feature extraction and preprocessing using Compound Discoverer 2.0 software (Thermo Scientific). Only ion peak data that were present in ≥ 50% of samples were retained. The main parameters were set as follows: intensity threshold, 300,000; m/z range, 70–1050; m/z width, 5 ppm; frame time width, 0.2 min; and retention time start and end values were 0.01–19.5 min, respectively. Then, the data were normalized according to the interior label and post-edited in Excel 2010 software. The KEGG database (http://www.genome.jp/kegg) and the Human Metabolome Database (http://www.hmdb.ca) were utilized to identify the metabolites through alignment of the molecular mass data. If the value between theoretical mass and observed mass was less than 10 ppm, the metabolites were reported. And the matched metabolites were further validated by isotopic distribution measurement.
The final metabolites data were imported into SIMCA-P software (Version 13, Umetrics AB, Sweden) for multivariate statistical analysis. The principal component analysis (PCA) and partial least squares-discriminate analysis (PLS-DA) were carried out to explore the differences of the metabolome profile between the two groups. Statistical significance of the PCA was performed using the “adonis” function in the R package “vegan” with 999 permutations. The PLS-DA models were validated based on variation interpretation (R2Y) and predictability (Q2) of the model in cross-validation and permutation tests with 200 iterations. The differential metabolites between the two groups were screened with a cut-off condition of the Benjamini-Hochberg adjusted P-value (Q) < 0.05 and the variable importance in the projection (VIP) of PLS-DA model > 1. Fold change (FC, HC vs. CON) and pathway analyses were processed with the web-based tool MetaboAnalyst 5.0 (http://www.metaboanalyst.ca). For the pathway analysis results, differences were regarded significant at P < 0.05. The correlation network between the milk composition and the differential milk metabolites was visualized using the Fruchterman-Reingold Algorithm in Gephi 0.9 software (https://gephi.org/) [24].
Statistical analyses
The milk yield, milk composition, and ruminal pH were analyzed by the linear mixed-effects models (MIXED) procedure of IBM SPSS statistics V25.0 (IBM Corp., Armonk, NY, USA). The treatment (CON or HC), day, and their interaction were treated as fixed factors. The cow was considered a random effect. The milk microbiota and metabolome data were analyzed using the non-parametric Scheirer-Ray-Hare extension of the Kruskal-Wallis test [25], which is a non-parametric analog of ANOVA based on ranked variates with two independent factors (diet and day) plus their interactions. Effects were deemed significant when P < 0.05.
Results
DMI and ruminal pH
The results for the DMI and rumen pH of the cows were reported previously [16]. Briefly, there was no significant difference in DMI between the CON and HC groups (23.79 vs. 22.40, P = 0.524), and the HC feeding resulted in an duration of a rumen pH of < 5.8 of 9.2 h/d in average.
Milk yield and composition
There were no significant differences in milk yield, 4% fat corrective milk, milk protein ratio, lactose ratio, nor in the yield of milk fat, milk protein, and lactose between the CON and HC groups (P > 0.05; Table 2). However, the milk fat percentage was significantly lowered in the HC group compared with the CON group (P = 0.034).
Structure and composition of the milk bacteria
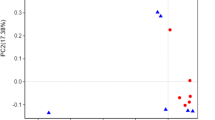
Across all the 24 milk samples, a total of 1,030,033 high-quality reads pairs were obtained, with an average of 42,918 per sample. The PCoA result based on the Bray-Curtis distance showed that principal coordinates 1 and 2 accounted for 26.33% and 23.10% of the total variance, respectively (Fig. 1B). The samples collected in the HC group did not separate from those in the CON group (PERMANOVA test, P = 0.743). The alpha diversity indices of the bacterial communities between the two groups are shown in Table 3, all of which showed no differences between the two groups (P > 0.05).
Forty-six bacterial phyla were identified among all the samples. Among them, Proteobacteria, Bacteroidetes, Actinobacteria, and Firmicutes were the predominant phyla, representing more than 95% of the bacterial community (97.78% in the CON group and 95.33% in the HC group). There were no significant differences in the relative abundance of all these 4 phyla between the two groups (P > 0.05; Table 4).
At the genus level, 16 predominant taxa whose relative abundance ≥ 1% in at least one group of the CON and HC groups was examined, and the HC group showed a higher abundance of Labrys compared to the CON group (P = 0.015; Table 5).
At the ASV level, we only analyzed the abundant ASVs whose proportions ≥ 0.1% in at least one of the 2 groups (Table S1). The results showed that there were 49 bacterial ASVs co-existed between the 2 groups, accompanied by 1 ASV existing independently in the CON group and 7 ASVs existing independently in the HC group. Compared with the CON group, the HC feeding resulted in a higher relative abundance of ASV44 (genus: Beijerinckiaceae_28-YEA-48) and ASV13 (genus: Labrys), whereas a lower relative abundance of ASV33 (genus: Rhodococcus) (P < 0.05; Table S1).
Identification and general characterization of the milk metabolites
In total, 188 compounds were identified from the milk samples of the CON and HC groups, consisting primarily of fatty acids, lipids, amino acids, sugars, organic acids, nucleotides, and vitamins.
The PCA results revealed a distinct difference in milk metabolic profiles between the CON and HC groups (P = 0.015 by PERMANOVA; Fig. 2A). Also, the PLS-DA results showed a clear separation between the two groups (Fig. 2B). The validity of the PLS-DA model was evaluated using R2Y and Q2 [26]. In our study, the R2Y and Q2 of the PLS-DA score plot was 0.991 and 0.836 respectively, signifying a proper goodness and a high predictive power of the model. Moreover, the Q2 intercept value of the permutation test was less than 0, indicating that the model was not overfitted (Fig. 2C).

Multivariate analysis of milk metabolome between the conventional (CON) and high-concentrate (HC) diet groups. A Principal component (PC) analysis scores plot. PERMANOVA results with 999 permutations are shown. B Partial least squares-discriminate analysis scores. PLS1 is the first principal component; PLS2 is the second principal component. C Permutation tests plots of 200 iterations. R2 and Q2 are fitness and predictive power of the model, respectively
Differences in milk metabolites between the CON and HC groups
With a threshold of VIP > 1 and Q < 0.05, a total of 31 differential metabolites were identified between the CON and HC groups. Compared with the CON group, the levels of 20 metabolites were up-regulated in the HC group, including cholic acid, L-palmitoylcarnitine, tetradecanoylcarnitine, stearoylcarnitine, decanoylcarnitine, LysoPA(8:0/0:0), glycocholic acid, L-octanoylcarnitine, LysoPE(0:0/18:0), 3-phospho-D-glycerate, β-D-fructose 6-phosphate, deoxyribose 1-phosphate, D-ribose 5-phosphate, N-acetyl-α-D-galactosamine 1-phosphate, phosphoenolpyruvic acid, flavine mononucleotide, 7-methylguanosine, niacinamide, acetylcholine, and sphingosine. On the contrary, the levels of 11 metabolites were down-regulated in the HC group, including α-linolenic acid (ALA), prostaglandin E2, L-lactic acid, L-malic acid, 3-hydroxysebacic acid, succinyladenosine, guanosine, pyridoxal, L-glutamic acid, hippuric acid, and trigonelline (Fig. 3). Pathway analysis indicated that these 31 metabolites were mainly enriched in 5 key differential metabolic pathways of pyruvate metabolism, pentose phosphate pathway, glycolysis/gluconeogenesis tricarboxylic acid (TCA) cycle, and riboflavin metabolism (P < 0.05; Fig. 4).

Differential metabolites identified in the milk between the conventional (CON) and high-concentrate (HC) diets feeding groups

Pathway analysis of KEGG for differential metabolites between the conventional (CON) and high-concentrate (HC) diets feeding groups
Correlation network of the phenotypic traits and the milk metabolites of the cows
The correlation network analysis was performed to visualize the relationships of the cows’ phenotypic traits (DMI, milk yield, and milk composition) and the differential milk metabolites (Table S2). Results showed that the correlation network was composed of 37 nodes and 127 edges, which included 77 positive correlations and 50 negative correlations (|r| > 0.75 and P < 0.05; Fig. 5). Among them, milk fat was positively correlated with hippuric acid, while hippuric acid was positively correlated with phosphoenolpyruvic acid (PEP) and acetylcholine. However, there were no significant correlations between milk yield, milk protein, lactose, and those differential milk metabolites.

Correlation networks of cow phenotypic traits (DMI, milk yield, and milk composition) and differential milk metabolites based on Spearman’s correlation coefficients (|r| > 0.75 and P < 0.05). Node size and color corresponds to the correlation degree and substance classification, respectively. Red lines denote positive correlations and green lines denote negative correlations
Discussion
In this study, we conducted the comparation of the milk bacterial and metabolic profiles between the cows feeding the CON and HC diets.
The composition and function of the bacterial communities that colonized in the teat apex, teat orifice, and teat canal lining could reflect the status of the udder health [27], and these bacteria play a major role in the development of intramammary infection [28]. Migrating from off-udder sites and the environment are the two major sources of milk bacteria, including from the cows’ teat surface, the milking canal, the milking machines (or other dairy equipment), the bedding material, the feces, the parlor air (stable and milking) and so on [29, 30]. In the current study, the milk bacteria were mainly dominated by Proteobacteria, Actinobacteria, Bacteroidetes, and Firmicutes regardless of diet, which was in line with previous studies on the milk microbiome [31]. At the genus level, there was an increased relative abundance of Labrys in the HC group. The Labrys belong to the order Rhizobiales, which were abundant in soil-associated environments [32]. Furthermore, the significantly changed abundance of ASV44 (genus: Beijerinckiaceae_28-YEA-48) and ASV33 (genus: Rhodococcus) between the CON and HC groups were also environment associated taxon [33, 34]. This result suggested that the enriched bacteria in the milk of HC feeding cows were mainly from the external environment, which was consisted with the earlier reported findings [35].
Moreover, one interesting observation from our study was the high abundance of Mycobacterium in both the CON and HC groups, and it did not present difference between the 2 groups (Table 5), which implied that its high percentage was not attributed to the HG feeding. Further analyses at the ASV level indicated that this taxon in the present study was composed of only ASV3 and it was aligned to uncultured bacterium at the species level. According to the literature, Mycobacterium are not among the most abundant bacterial taxa in studies on bovine milk microbiota [36, 37], and the majority of this genus are opportunistic pathogens [38]. Among them, Mycobacterium bovis (M. bovis), a pathogen that belongs to the Mycobacterium tuberculosis complex, is most commonly reported in researches on cows since it causes zoonotic tuberculosis (TB) disease [39, 40]. Recently, an increasing number of studies reported that M. bovis milk contamination is at a relatively wide prevalence as a result of contact between healthy cows and infected cows in modern intensive farming systems [41,42,43]. Milk-borne transmission of zoonotic TB by M. bovis always occurs through the consumption of unpasteurized or poor pasteurized contaminated dairy products [44, 45]. In the United States, about 1%–2% of TB cases are attributable to M. bovis infection [46]. Altogether, though we cannot confirm if the high-abundance Mycobacterium identified in our study are opportunistic pathogens, it still reminds us that certain pathogens contamination (such as M. bovis) can be a latent occurrence in raw milk and pasteurization is vitally important to people’s health.
The most common pathogenic bacteria attributed to the cows’ mastitis are members of genus Staphylococcus and Streptococcus [47, 48]. Additionally, a recent study revealed that Stenotrophomonas might be associated with the development of mastitis in the cows [7]. The possible mechanism for the cows’ mastitis under the HC feeding may due to the translocation of the rumen-derived lipopolysaccharide, which disrupts the blood-milk barrier and eventually leads to the translocation of pathogenic bacteria through blood [7, 49]. However, in the present study, we did not detect Staphylococcus, Streptococcus or Stenotrophomonas in either the CON or the HC groups, which might suggest that the cows did not develop mastitis during our experiment. The inconsistent results might be due to the relatively low concentrate proportion used [6, 7] and the relatively short experiment period adopted [49] in our study, accompanied by the individual variances of the host animals between different studies, which eventually did not cause the translocation of rumen lipopolysaccharide.
Among the 31 differential metabolites between the two groups, the concentrations of all the fatty acylcarnitines were increased in the HC group (Fig. 3). Fatty acylcarnitines are fatty acids esters formed when fatty acyl-CoA are shuttled into the mitochondrion for β-oxidation [50]; and their elevated concentrations here might imply a dysfunction of the lipid metabolism in the HC cows, which could traced to the changes in rumen fermentation (increased propionate concentration, altered lipid metabolism, and so on) caused by the shifts of rumen microbiota profiles (structure, composition, and function) under the HC feeding [16, 51]. The milk metabolite profiles are tightly associated with the coagulation properties of the milk [52, 53]. It revealed that the elevated milk carnitine concentrations would affect the coagulation properties of the milk and the carnitine levels were always lower in good-coagulating milk samples [53]. Moreover, earlier studies demonstrated that higher levels of long-chain acylcarnitines were related with liver diseases, obesity, and type 2 diabetes [54,55,56]. Therefore, the up-regulated fatty acylcarnitines in the HC diet feeding cows might suggest a lower milk quality in our research.
Compared to the CON group, the level of the milk cholic acid was greatly up-regulated under the HC feeding (FC = 4.27, HC vs. CON). Metabolites in the milk could originate from a variety of different sources, including being secreted by the mammary epithelial cells, being leaked from the damaged somatic cells, being transferred from the blood, or coming from the microbiota metabolism present in the milk [11, 57]. In our earlier study, we found a high level of cholic acid in plasma of the HC group (FC = 2.76, HC vs. CON) [58], so we speculate that the accumulation of cholic acid in the milk might derive from the increased concentration of cholic acid in the blood. Cholic acid is a naturally occurring, primary bile acid that synthesized from cholesterol in the liver [59]. Bile acids are known to have lipid-lowering effects [60], and it has been confirmed that treating gallstone patients with chenodeoxycholic acid could decrease the hepatic very-low-density lipoprotein production and the plasma triglyceride level in clinical trial [61]. The triglyceride concentration was also found to be elevated in plasma of the HC feeding cows in our earlier study (1.508:1.228 mmol/L, HC vs. CON, P = 0.014) [58], and it revealed that there was a positive correlation (Pearson r = 0.51, P = 0.011) between the levels of milk cholic acid and plasma triglyceride.
Besides, the levels of all of the sugars and the corresponding derivatives were up-regulated in the HC group, including 3-phospho-D-glycerate (FC = 2.58, HC vs. CON), β-D-fructose 6-phosphate (FC = 2.20, HC vs. CON), deoxyribose 1-phosphate (FC = 2.03, HC vs. CON), D-ribose 5-phosphate (FC = 1.83, HC vs. CON), and N-acetyl-α-D-galactosamine 1-phosphate (FC = 1.43, HC vs. CON; Fig. 3). Among them, 3-phospho-D-glycerate is a biochemically significant metabolic intermediate in glycolysis [62]. Beta-D-fructose 6-phosphate and D-ribose 5-phosphate, accompanied by the elevated level of an organic acid compound-PEP (FC = 2.13, HC vs. CON) for the HG cows are all involved in both pentose phosphate and glycolysis/gluconeogenesis pathways [63]. Their enrichment indicated that the pentose phosphate pathway and the glycolysis/gluconeogenesis pathway in the mammary gland were affected by the HC diet feeding. Deoxyribose-1-phosphate is a proangiogenic paracrine stimulus released by platelets and macrophages, which acts on endothelial cells to drive tissue repair [64]. Hence, the increased level of deoxyribose-1-phosphate in the HC group might hint a potential injury of the mammary epithelial cells.
In contrast, the HG diet feeding resulted in a decreased concentration of milk ALA (FC = 0.50, HC vs. CON). The ALA is an important precursor to the synthesis of docosahexaenoic acid, which is a major contributor to the visual acuity and brain development [65]. Furthermore, it has been reported that when the cows were fed an ALA-enriched diet, the expression of some local and systemic pro-inflammatory markers in vivo would be down-regulated [66]. Therefore, the decreased concentration of ALA in the milk of the HC feeding cows might reduce the levels of the beneficial ingredients in the milk, which finally impact the milk quality. Additionally, the decreased level of L-malic acid (FC = 0.76, HC vs. CON) in milk with the HC feeding might be explained by the changes in the energy metabolism. In detail, SARA could cause an impaired TCA cycle function, which in turn results in active secretion or leakage of L-malic acid from the mammary epithelial cells into the milk [67]. Glutamic acid can be converted into alpha-ketoglutarate, which was a fuel for the TCA cycle [68]. The declined level of L-glutamic acid in the HC group (FC = 0.38, HC vs. CON) might also be a mirror of the impaired TCA cycle.
As for the other differential metabolites between the 2 groups, the decreased levels of succinyladenosine (FC = 0.64, HC vs. CON) and guanosine (FC = 0.04, HC vs. CON), and the increased levels of flavine mononucleotide (FC = 2.57, HC vs. CON) and 7-methylguanosine (FC = 1.73, HC vs. CON) implied a dysfunction of nucleotide metabolism in the mammary gland of the HC cows. Hippuric acid is a benzoilglycine, a compound of benzoic acid and glycine, which could lead to the detoxication of benzoic acid and benzoates [69]. In the present study, we found that the concentration of hippuric acid was decreased in the HC cows (FC = 0.70, HC vs. CON) and it was closely correlated with milk fat, PEP and acetylcholine. Up to now, there is not any study that reports the association between hippuric acid and milk fat. However, hippuric acid has been found to be associated with diet, and it was proposed as a biomarker for organic feeding [70, 71]. The PEP is involved in the TCA cycle, therefore, the decreased concentration of hippuric acid in the HC group might mirror certain alterations in the energy metabolism of the cows. At the same time, it echoes the above-discussed result of the reduced concentration of L-malic acid in the HC cows which might be caused by the impaired TCA function. Acetylcholine is found in mesothelial, endothelial, glial, circulating blood cells (platelets, mononuclear cells), and alveolar macrophages [72]. Non-neuronal acetylcholine appears to be involved in the regulation of some important cell functions, such as mitosis, trophic functions, automaticity, locomotion, ciliary activity, cell-cell contact, cytoskeleton, and barrier and immune functions [73]. Hence, the increased level of acetylcholine in the HC cows (FC = 1.89, HC vs. CON) and its association with the decreased level of hippuric acid might imply a feedback regulation of the mammary gland against the damages caused by SARA.
Conclusions
The 16S rRNA gene sequencing and HPLC-MS methods were combined to analyze the differences in milk bacterial and metabolic profiles between the CON and HC groups. Taken together, our results indicated that the bacterial community of the milk was primarily dominated by Proteobacteria, Actinobacteria, Bacteroidetes, and Firmicutes at the phylum level, and the differential taxon between the 2 groups were mainly from the external environment. Besides, our results revealed a comprehensive profiling of the milk metabolome associated with SARA and suggested that HC diet feeding led to a decline of the milk quality. In addition, there were no direct or indirect utilization or productive relationships found between the milk bacteria and metabolites through our results. All these findings are beneficial to our subsequent research to explore the diagnostic biomarkers in the milk of cows with SARA and provide us with a new sight to further explore the occurrence and harms of SARA.
Availability of data and materials
Raw reads of milk 16S rRNA gene sequencing were deposited in NCBI SRA database under accession number PRJNA736538.
Abbreviations
- ALA:
-
α-linolenic acid
- ASV:
-
Amplicon sequence variant
- CON:
-
Conventional
- DMI:
-
Dry matter intake
- FC:
-
Fold change
- HC:
-
High-concentrate
- HPLC-MS:
-
High-performance liquid chromatography-mass spectrometry
- PCA:
-
Principal component analysis
- PCoA:
-
Principal coordinate analysis
- PEP:
-
Phosphoenolpyruvic acid
- PLS-DA:
-
Partial least squares-discriminate analysis
- SARA:
-
Subacute rumen acidosis
- TB:
-
Tuberculosis
- TCA:
-
Tricarboxylic acid
- VIP:
-
Importance in the projection
References
Lock AL, Bauman DE. Modifying milk fat composition of dairy cows to enhance fatty acids beneficial to human health. Lipids. 2004;39(12):1197–206.
Toral PG, Chilliard Y, Rouel J, Leskinen H, Shingfield KJ, Bernard L. Comparison of the nutritional regulation of milk fat secretion and composition in cows and goats. J Dairy Sci. 2015;98(10):7277–97.
Enemark JMD. The monitoring, prevention and treatment of subacute ruminal acidosis (SARA): a review. Vet J. 2008;176(1):32–43.
Abdela N. Sub-acute ruminal acidosis (SARA) and its consequence in dairy cattle: a review of past and recent research at global prospective. Achiev Life Sci. 2016;10(2):187–96.
Kleen JL, Cannizzo C. Incidence, prevalence and impact of SARA in dairy herds. Anim Feed Sci Technol. 2012;172(1–2):4–8.
Zhang R, Huo W, Zhu W, Mao S. Characterization of bacterial community of raw milk from dairy cows during subacute ruminal acidosis challenge by high-throughput sequencing. J Sci Food Agric. 2015;95(5):1072–9.
Hu X, Li S, Mu R, Guo J, Zhao C, Cao Y, et al. The rumen microbiota contributes to the development of mastitis in dairy cows. Microbiol Spectr. 2022;10(1):e0251221.
Kuehn JS, Gorden PJ, Munro D, Rong R, Dong Q, Plummer PJ, et al. Bacterial community profiling of milk samples as a means to understand culture-negative bovine clinical mastitis. PLoS One. 2013;8(4):e61959.
European Commission. Regulation (EC) No 853/2004 of the European Parliament and of the Council of 29 April 2004 laying down specific hygiene rules for food of animal origin. Off J Eur Union. 2004;47:22–82. https://www.legislation.gov.uk/eur/2004/853/pdfs/eurcs_20040853_en_001.pdf.
Martin NH, Boor KJ, Wiedmann M. Symposium review: effect of post-pasteurization contamination on fluid milk quality. J Dairy Sci. 2018;101(1):861–70.
Sundekilde U, Poulsen N, Larsen L, Bertram H. Nuclear magnetic resonance metabonomics reveals strong association between milk metabolites and somatic cell count in bovine milk. J Dairy Sci. 2013;96(1):290–9.
Silanikove N, Merin U, Shapiro F, Leitner G. Milk metabolites as indicators of mammary gland functions and milk quality. J Dairy Res. 2014;81(3):358–63.
Pyorala S. Indicators of inflammation in the diagnosis of mastitis. Vet Res. 2003;34(5):565–78.
NRC. Nutrient requirements of dairy cattle. 7th ed. Washington, DC: National Academy of Science; 2001.
MOA (Ministry of Agriculture of P.R. China). Feeding standard of dairy cattle (NY/T 34–2004). Beijing: MOA; 2004.
Mu YY, Qi WP, Zhang T, Zhang JY, Mao SY. Gene function adjustment for carbohydrate metabolism and enrichment of rumen microbiota with antibiotic resistance genes during subacute rumen acidosis induced by a high-grain diet in lactating dairy cows. J Dairy Sci. 2021;104(2):2087–105.
Cocolin L, Aggio D, Manzano M, Cantoni C, Comi G. An application of PCR-DGGE analysis to profile the yeast populations in raw milk. Int Dairy J. 2002;12(5):407–11.
Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–63.
Rognes T, Flouri T, Nichols B, Quince C, Mahe F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–3.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–7.
Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018;6(1):1–17.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2012;41(D1):D590–6.
Bastian M, Heymann S, Jacomy M. Editors. Gephi: an open source software for exploring and manipulating networks. Am J Sociol. 2009;3(1):361–2.
Sokal RR, Rohlf FJ. Biometry: the principles and practice of statistics in biological research. New York: WH Free- man & Co; 1995.
Mickiewicz B, Vogel HJ, Wong HR, Winston BW. Metabolomics as a novel approach for early diagnosis of pediatric septic shock and its mortality. Am J Respir Crit Care Med. 2013;187(9):967–76.
Haveri M, Hovinen M, Roslof A, Pyorala S. Molecular types and genetic profiles of Staphylococcus aureus strains isolated from bovine intramammary infections and extramammary sites. J Clin Microbiol. 2008;46(11):3728–35.
Derakhshani H, Fehr KB, Sepehri S, Francoz D, De Buck J, Barkema HW, et al. Invited review: microbiota of the bovine udder: contributing factors and potential implications for udder health and mastitis susceptibility. J Dairy Sci. 2018;101(12):10605–25.
Montel MC, Buchin S, Mallet A, Delbes-Paus C, Vuitton DA, Desmasures N, et al. Traditional cheeses: rich and diverse microbiota with associated benefits. Int J Food Microbiol. 2014;177:136–54.
Quigley L, O'Sullivan O, Beresford TP, Ross RP, Fitzgerald GF, Cotter PD. Molecular approaches to analysing the microbial composition of raw milk and raw milk cheese. Int J Food Microbiol. 2011;150(2–3):81–94.
Raats D, Offek M, Minz D, Halpern M. Molecular analysis of bacterial communities in raw cow milk and the impact of refrigeration on its structure and dynamics. Food Microbiol. 2011;28(3):465–71.
Albert RA, Waas NE, Langer S, Pavlons SC, Feldner JL, Rossello-Mora R, et al. Labrys wisconsinensis sp. nov., a budding bacterium isolated from Lake Michigan water, and emended description of the genus Labrys. Int J Syst Evol Microbiol. 2010;60(Pt 7):1570–6.
Gautam A, Sekaran U, Guzman J, Kovács P, Hernandez JLG, Kumar S. Responses of soil microbial community structure and enzymatic activities to long-term application of mineral fertilizer and beef manure. Environ Sustain Indic. 2020;8:100073.
Bell K, Philp J, Aw D, Christofi N. The genus rhodococcus. J Appl Microbiol. 1998;85(2):195–210.
Metzger SA, Hernandez LL, Skarlupka JH, Suen G, Walker TM, Ruegg PL. Influence of sampling technique and bedding type on the milk microbiota: results of a pilot study. J Dairy Sci. 2018;101(7):6346–56.
Oikonomou G, Addis MF, Chassard C, Nader-Macias MEF, Grant I, Delbès C, et al. Milk microbiota: what are we exactly talking about? Front Microbiol. 2020;11:60.
Addis M, Tanca A, Uzzau S, Oikonomou G, Bicalho R, Moroni P. The bovine milk microbiota: insights and perspectives from-omics studies. Mol BioSyst. 2016;12(8):2359–72.
Meehan CJ, Barco RA, Loh Y-HE, Cogneau S, Rigouts L. Reconstituting the genus Mycobacterium. Int J Syst Evol Microbiol. 2021;71(9):004922.
Forrellad MA, Klepp LI, Gioffré A, Sabio y Garcia J, Morbidoni HR, Santangelo MD, et al. Virulence factors of the Mycobacterium tuberculosis complex. Virulence. 2013;4(1):3–66.
Hlavsa MC, Moonan PK, Cowan LS, Navin TR, Kammerer JS, Morlock GP, et al. Human tuberculosis due to Mycobacterium bovis in the United States, 1995-2005. Clin Infect Dis. 2008;47(2):168–75.
Chidumayo NN. Estimation of Brucella and Mycobacterium bovis contamination in bovine milk in Africa. Int J One Health. 2021;7(1):37–41.
Collins ÁB, Floyd S, Gordon SV, More SJ. Prevalence of Mycobacterium bovis in milk on dairy cattle farms: an international systematic literature review and meta-analysis. Tuberculosis. 2022;132:102166.
Carneiro PA, Takatani H, Pasquatti TN, Silva CB, Norby B, Wilkins MJ, et al. Epidemiological study of Mycobacterium bovis infection in buffalo and cattle in Amazonas, Brazil. Front Vet Sci. 2019;6:434.
Girma K, Tilahun Z, Haimanot D. Review on milk safety with emphasis on its public health. World J Dairy Food Sci. 2014;9(2):166–83.
Angulo FJ, LeJeune JT, Rajala-Schultz PJ. Unpasteurized milk: a continued public health threat. Clin Infect Dis. 2009;48(1):93–100.
Gallivan M, Shah N, Flood J. Epidemiology of human Mycobacterium bovis disease, California, USA, 2003-2011. Emerg Infect Dis. 2015;21(3):435–43.
Ronco T, Klaas IC, Stegger M, Svennesen L, Astrup LB, Farre M, et al. Genomic investigation of Staphylococcus aureus isolates from bulk tank milk and dairy cows with clinical mastitis. Vet Microbiol. 2018;215:35–42.
Keefe G. Update on control of Staphylococcus aureus and Streptococcus agalactiae for management of mastitis. Vet Clin North Am Food Anim Pract. 2012;28(2):203–16.
Zhang K, Chang G, Xu T, Xu L, Guo J, Jin D, et al. Lipopolysaccharide derived from the digestive tract activates inflammatory gene expression and inhibits casein synthesis in the mammary glands of lactating dairy cows. Oncotarget. 2016;7(9):9652–65.
Rico J, Zang Y, Haughey N, Rius A, McFadden J. Circulating fatty acylcarnitines are elevated in overweight periparturient dairy cows in association with sphingolipid biomarkers of insulin resistance. J Dairy Sci. 2018;101(1):812–9.
Mao SY, Zhang RY, Wang DS, Zhu WY. Impact of subacute ruminal acidosis (SARA) adaptation on rumen microbiota in dairy cattle using pyrosequencing. Anaerobe. 2013;24:12–9.
Sundekilde UK, Gustavsson F, Poulsen NA, Glantz M, Paulsson M, Larsen LB, et al. Association between the bovine milk metabolome and rennet-induced coagulation properties of milk. J Dairy Sci. 2014;97(10):6076–84.
Sundekilde UK, Frederiksen PD, Clausen MR, Larsen LB, Bertram HC. Relationship between the metabolite profile and technological properties of bovine milk from two dairy breeds elucidated by NMR-based metabolomics. J Agric Food Chem. 2011;59(13):7360–7.
Zhou L, Wang Q, Yin P, Xing W, Wu Z, Chen S, et al. Serum metabolomics reveals the deregulation of fatty acids metabolism in hepatocellular carcinoma and chronic liver diseases. Anal Bioanal Chem. 2012;403(1):203–13.
Mihalik SJ, Goodpaster BH, Kelley DE, Chace DH, Vockley J, Toledo FG, et al. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity. 2010;18(9):1695–700.
Saiki S, Hatano T, Fujimaki M, Ishikawa K-I, Mori A, Oji Y, et al. Decreased long-chain acylcarnitines from insufficient β-oxidation as potential early diagnostic markers for Parkinson’s disease. Sci Rep. 2017;7(1):1–15.
Mansbridge R, Blake J. Nutritional factors affecting the fatty acid composition of bovine milk. Br J Nutr. 1997;78(1):S37–47.
Qi WP, Mu YY, Zhang T, Zhang JY, Mao SY. Plasma biochemical indexes and metabolomics profile changes of dairy cows with subacute ruminal acidosis. Acta Pratacult Sin. 2021;30(6):141.
Li T, Apte U. Bile acid metabolism and signaling in cholestasis, inflammation, and cancer. Adv Pharmacol. 2015;74:263–302.
Li T, Chiang JY. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev. 2014;66(4):948–83.
Schoenfield LJ, Lachin JM, Baum RA, Habig RL, Hanson RF, Hersh T, et al. Chenodiol (chenodeoxycholic acid) for dissolution of gallstones: the National Cooperative Gallstone Study: a controlled trial of efficacy and safety. Ann Intern Med. 1981;95(3):257–82.
Hitosugi T, Zhou L, Elf S, Fan J, Kang H-B, Seo JH, et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell. 2012;22(5):585–600.
Elson DL, Cox MM. Glycolysis, gluconeogenesis, and the pentose phosphate pathway. In: Lehninger principles of biochemistry. 4th ed. New York: W.H. Freeman; 2004.
Vara D, Watt JM, Fortunato TM, Mellor H, Burgess M, Wicks K, et al. Direct activation of NADPH oxidase 2 by 2-deoxyribose-1-phosphate triggers nuclear factor kappa B-dependent angiogenesis. Antioxid Redox Signal. 2018;28(2):110–30.
Richard C, Calder PC. Docosahexaenoic acid. Adv Nutr. 2016;7(6):1139–41.
Rezamand P, Hatch BP, Carnahan KG, McGuire MA. Effects of α-linolenic acid-enriched diets on gene expression of key inflammatory mediators in immune and milk cells obtained from Holstein dairy cows. J Dairy Res. 2016;83(1):20–7.
Xue Y, Guo C, Hu F, Liu J, Mao S. Hepatic metabolic profile reveals the adaptive mechanisms of ewes to severe undernutrition during late gestation. Metabolites. 2018;8(4):85.
Zhao X, Fu J, Du J, Xu W. The role of D-3-phosphoglycerate dehydrogenase in cancer. Int J Biol Sci. 2020;16(9):1495–506.
Toromanović J, Kovač-Bešović E, Šapčanin A, Tahirović I, Rimpapa Z, Kroyer G, et al. Urinary hippuric acid after ingestion of edible fruits. Bosn J Basic Med Sci. 2008;8(1):38.
Carpio A, Bonilla-Valverde D, Arce C, Rodríguez-Estévez V, Sánchez-Rodríguez M, Arce L, et al. Evaluation of hippuric acid content in goat milk as a marker of feeding regimen. J Dairy Sci. 2013;96(9):5426–34.
Boudonck KJ, Mitchell MW, Wulff J, Ryals JA. Characterization of the biochemical variability of bovine milk using metabolomics. Metabolomics. 2009;5(4):375–86.
Kirkpatrick CJ, Bittinger F, Nozadze K, Wessler I. Expression and function of the non-neuronal cholinergic system in endothelial cells. Life Sci. 2003;72(18–19):2111–6.
Wessler I, Kirkpatrick CJ, Racke K. Non-neuronal acetylcholine, a locally acting molecule, widely distributed in biological systems: expression and function in humans. Pharmacol Ther. 1998;77(1):59–79.
Acknowledgements
Not applicable.
Funding
This research was funded by the National Key R&D Program of China (2022YFD1301001) and Jiangsu Province Special Project for Carbon Peak & Carbon Neutral Science and Technology Innovation (BE2022309).
Author information
Authors and Affiliations
Contributions
SM and YM designed the experiments. YM, WQ, TZ, and JZ performed the experiments. YM and WQ analysed the data. YM wrote and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The experimental design and procedures of this study were approved by the Animal Care and Use Committee of Nanjing Agricultural University following the requirements of the Regulations for the Administration of Airs Concerning Experimental Animals (The State Science and Technology Commission of P. R. China, 1988. No. SYXK (Su) 2015–0656).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Supplementary Information
Additional file 1: Table S1.
Effects of feeding the conventional (CON) or high-concentrate (HC) diet on the relative abundance (%) of rumen bacteria at the amplicon sequence variant (ASV) level.
Additional file 2:
Table S2. Correlation networks of cow phenotypic traits (DMI, milk yield, and milk composition) and differential milk metabolites based on Spearman’s correlation coefficients.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mu, Y., Qi, W., Zhang, T. et al. Coordinated response of milk bacterial and metabolic profiles to subacute ruminal acidosis in lactating dairy cows. J Animal Sci Biotechnol 14, 60 (2023). https://doi.org/10.1186/s40104-023-00859-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40104-023-00859-8