
Abstract
Background
Diarrhea is a major cause of morbidity and mortality in young calves, resulting in considerable economic loss for dairy farms. To determine if some gut microbes might have resistance to dysbiotic process with calf diarrhea by dictating the microbial co-occurrence patterns from birth to post-weaning, we examined the dynamic development of the gut microbiota and diarrhea status using two animal trials, with the first trial having 14 Holstein dairy calves whose fecal samples were collected 18 times over 78 d from birth to 15 d post-weaning and the second trial having 43 Holstein dairy calves whose fecal samples were collected daily from 8 to 18 days of age corresponding to the first diarrhea peak of trial 1.
Results
Metataxonomic analysis of the fecal microbiota showed that the development of gut microbiota had three age periods with birth and weaning as the separatrices. Two diarrhea peaks were observed during the transition of the three age periods. Fusobacteriaceae was identified as a diarrhea-associated taxon both in the early stage and during weaning, and Clostridium_sensu_stricto_1 was another increased genus among diarrheic calves in the early stage. In the neonatal calves, Prevotella_2 (ASV4 and ASV26), Prevotella_9 (ASV43), and Alloprevotella (ASV14) were negatively associated with Clostridium_sensu_stricto_1 (ASV48), the keystone taxa of the diarrhea-phase module. During weaning, unclassified Muribaculaceae (ASV28 and ASV44), UBA1819 (ASV151), Barnesiella (ASV497), and Ruminococcaceae_UCG-005 (ASV254) were identified being associated with non-diarrheic status, and they aggregated in the non-diarrhea module of co-occurrence patterns wherein unclassified Muribaculaceae (ASV28) and Barnesiella (ASV497) had a direct negative relationship with the members of the diarrhea module.
Conclusions
Taken together, our results suggest that the dynamic successions of calf gut microbiota and the interactions among some bacteria could influence calf diarrhea, and some species of Prevotella might be the core microbiota in both neonatal and weaning calves, while species of Muribaculaceae might be the core microbiota in weaning calves for preventing calf diarrhea. Some ASVs affiliated with Prevotella_2 (ASV4 and ASV26), Prevotella_9 (ASV43), Alloprevotella (AVS14), unclassified Muribaculaceae (ASV28 and ASV44), UBA1819 (ASV151), Ruminococcaceae_UCG-005 (ASV254), and Barnesiella (ASV497) might be proper probiotics for preventing calf diarrhea whereas Clostridium_sensu_stricto_1 (ASV48) might be the biomarker for diarrhea risk in specific commercial farms.
Similar content being viewed by others


Background
Diarrhea is one of the most common causes of morbidity and mortality in young calves, especially dairy calves younger than one month [1, 2]. In the US, the National Animal Health Monitoring System estimated that more than half of the calf deaths were due to diarrhea and related diseases [3]. A similar mortality rate among dairy calves attributable to diarrhea was also reported in Korea [1]. Calf diarrhea not only incurs treatment cost but also lowers daily weight gain in calves [4], delays first conceptions [5], decreases milk production in the first lactation [6], consequently resulting in a great economic loss for dairy farms. Many pathogens have been implicated in calf diarrhea, including Escherichia coli K99+ (E. coli K99+), Salmonella spp., Clostridium perfringens, Cryptosporidium parvum, bovine rotavirus (BRV), and bovine coronavirus (BCoV) [1]. Non-infectious factors such as improper colostrum management [7], weaning stress [8], and poor feeding environments can also increase the risk of calf diarrhea [9]. Moreover, oftentimes uncertain causality of diarrhea occurrence and cross infection together make diarrhea difficult to prevent and treat.
Diarrhea is much more common among young animals, especially after birth to shortly after weaning than among adult animals because of higher risk of enteric pathogens colonization in young animals than in adult animals [10, 11]. The high risk of colonization by pathogens in young animals stems from their underdeveloped gut and gut microbiota. Indeed, Kim et al. found that the absence of some species of Clostridia in the gut microbiota of neonatal mice made them unable to resist the colonization by Salmonella enterica Typhimurium or Citrobacter rodentium [12]. In addition, dysbiosis of gut microbiota induced by certain factors such as antibiotics and diseases can also lower resistance and increase enteric pathogen infection. Wu et al. demonstrated that dysbiosis of gut microbiota in rat induced by antibiotics lowered the resistance to colonization by Salmonella [13]. Compared to uninfected calves, calves infected with rotavirus have a higher abundance of Escherichia, Clostridium_g21, Streptococcus, and Clostridium, but a lower abundance of Lactobacillus, Subdoligranulum, Blautia, and Coprococcus_g2 in the fecal microbiota [14].
Weaning is a stressful process for young animals and can lower resistance to pathogenic colonization. Haag et al. found that infant mice after weaning were deficient in preventing their gut from colonization by Campylobacter jejuni [15]. During weaning, dairy calves are stressed nutritionally, and their gut microbiota undergoes compositional changes [16]. Typically, during weaning, the gut microbiota increases its diversity, and some important taxa, such as Bacteroides, Blautia, Ruminococcus, and Succinivibrio, may change their abundance [17]. The nutritional stresses coupled with changes in gut microbiota during and immediately after weaning increase the risk of enteric diseases, particularly diarrhea [17, 18].
Interestingly, some calves are prone to diarrhea, whereas others are resistant to diarrhea. In the 56-day experiment of Ma et. al [19], 13 out 42 milk replacer fed calves never exhibited diarrhea, 18 calves exhibited diarrhea and recovered after treated with electrolyte, but 11 calves exhibited diarrhea and needed to be treated with therapeutic antimicrobials. Ma et al. defined the latter two diarrhea status as resistant to diarrhea-induced dysbiosis, and susceptible to diarrhea-induced dysbiosis, respectively. The age of first diarrhea of these calves varied from d 8 to 19. Using the random forest machine learning algorithm with the microbiota collected from health calves at 7, 14, and 21 days of age and diarrhea calves prior to the onset of diarrhea, Ma et al. suggest that diarrhea could be predicted by the microbiota shift in early life. Kim et al. [20] described the similar age (5–50 days of age) fecal microbiota transplantation could ameliorate calf diarrhea with increasing the family Porphyromonadaceae. Therefore, we hypothesized that the gut microbiota might strongly correlate to calf diarrhea for some gut microbes might have resistance to the dysbiotic process with calf diarrhea from birth to post-weaning. To test our hypothesis, the stages with high incidence of diarrhea were identified in cohort from birth to shortly after weaning, and the fecal microbiotas between diarrheic calves and non-diarrheic calves in stages of high incidence of diarrhea, and between diarrhea phases (pre-diarrhea, diarrhea and post-diarrhea) were compared, and the diarrheic status- and diarrhea phase-associated amplicon sequence variants (ASVs) and modules were identified.
Methods
Animal experiment and sample collection
Two animal trials were conducted on a commercial farm located in Shaoxing (more than 3800 cows in stock), Zhejiang Province, China from September to November 2017 (trial 1) and November to December 2017 (trial 2). In trial 1, 14 female Holstein calves (initial bodyweight = 38.2 ± 2.0 kg, mean ± SD) were enrolled at birth. Immediately after birth, the calves were separated from their dams and moved to individual pens (1.0 m × 1.2 m) bedded with dry wheat straw and each offered 4 L of thawed colostrum (pooled the colostrum from other mothers before the trial, and stored at – 80 ºC) via an esophageal tube within 2 h after birthing. All neonatal calves in this trial were fed the same colostrum. From 1 to 5 days of age, each calf was bottle-fed 2 L of whole milk each at 0700, 1330, and 1800 h. During these 5 d, 4 g each of tylosin tartrate and sulfadimidine soluble (both powder) was mixed with the morning milk and fed to each calf for prophylaxis of pneumonia. At d 6, all the calves were moved to one group pen (8.0 m × 10.0 m) bedded with a rubber mat and dry wheat straw. The straw was replaced every other day. The calves had free access to a preset volume of whole milk (Additional file 1: Fig. S1) with an automated milk feeder system (Förster-Technik, Engen, Germany). Briefly, the calves get 6 L/d of whole milk from d 6 and this amount increased evenly up to 8 L/d on d 22. From d 23 to 41, each calf received 8 L of whole milk daily, and from d 42 to 62, the milk allowance was decreased evenly from 8 to 2 L/d. Milk supply was denied using the automated milk feeder system once a calf consumed 2 L of milk within 2 h during each meal to avoid overconsumption of milk. All the calves were weaned off milk at d 63. Calf starter pellets and a hay mixture of oat and alfalfa were offered ad libitum in the group pen. All the calves had free access to clean drinking water throughout the trial. Fecal samples were collected directly via rectal stimulation from every calf of the trial at d 1, 3, 5, 7, 9, 12, 15, 18, 28, 38, 48, 58, 62, 63, 65, 68, 73, and 78 (18 times in total). The fecal samples were placed in 2 mL nuclease-free tubes, immediately frozen in liquid nitrogen, then each month the 15 L liquid nitrogen tank with samples was transported to the lab and samples were stored at – 80 °C. The DNA of all the stored fecal samples was extracted in one month after the trial. When fecal samples were collected, fecal scores were recorded based on fecal fluidity [21]: 1 = normal, 2 = soft, 3 = runny, or 4 = watery. Calves that had a fecal score of 3 or 4 were considered diarrheic. No additional antibiotics were offered to calves after the diarrhea episode.
In trial 2, 43 female Holstein calves (initial bodyweight = 36.6 ± 1.9 kg, mean ± SD) were enrolled at birth. The calves were fed, nursed, and sampled exactly the same as for those in trial 1, but fecal samples were collected daily from d 8 to 18, which corresponded to the age when the first diarrhea peak was observed in trial 1.
Metataxonomic analysis of the fecal microbiota
DNA was extracted from individual fecal samples according to the method of Zoetendal et al. with minimal modifications [22]. In brief, approximately 0.2 g of frozen fecal sample each was homogenized together with 1 mL of TE buffer using bead-beating (Biospec Products; Bartlesville, OK, United States) followed by phenol:chloroform-based DNA extraction. Agarose gel (1%) electrophoresis was performed to evaluate the DNA quality, and the DNA concentrations were determined using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, United States). The V3-V4 region of the 16S rRNA gene was amplified using primers 341F (5'-CCTAYGGGRBGCASCAG-3') and 806R (5'-GGACTACNNGGGTATCTAAT-3') to prepare amplicon libraries with each being labeled with a unique barcode sequence [23]. The amplicon libraries of all the samples were pooled in equal molar ratio and paired-end (2 × 250) sequenced on the Illumina HiSeq platform by Novogene Bioinformatics Technology Co., Ltd. (Tianjin, China). After demultiplexing, the paired-end sequencing reads were processed using the DADA2 package (version 1.8.0) in R and its pipeline [24]. Briefly, barcodes and primers were removed from the reads. Reads with more than 2 expected errors (maxN = 0, maxEE = c(2, 2), truncQ = 2) were filtered out. Dereplication and inference were performed using the DADA2 pipeline. After merging the paired reads and chimera filtering, an ASV table was constructed (to resolve bacteria at the species level [25]). The ASVs were taxonomically assigned based on the SILVA 16S rRNA gene database (version 132) [26] using a naive Bayesian classifier method [27] implemented in DADA2. The sequences of each sample were rarefied to same size with the minimum number of sequences in samples (35,137 sequences/sample in trial 1, and 19,986 sequences/sample in trial 2) using the ‘rarefy_even_depth' function in Phyloseq package (version 1.24.2) [28], and ASVs that appeared only once among all the samples were removed from the dataset. Alpha diversity metrics including observed species and Shannon diversity index were calculated using the Phyloseq package. Faith’s phylogenetic diversity (Faith’s PD) was calculated using the Picante package (version 1.8.2) in R, and evenness was calculated using the Microbiome package (version 1.12.0) in R. To minimize individual variance of calves, only the ASVs that were observed in at least 20% of calves at every single day (3 out of 14 calves in trial 1) or every single diarrheic status-associated phase (trial 2) were subjected to the downstream analysis.
Pathogen detection
Major pathogens that can lead to calf diarrhea were tested in the fecal samples of trial 1 when diarrhea (fecal score ≥ 3) was first noted in a calf. Salmonella spp. was detected by PCR with specific primers (F:5′-TCGTCATTCCATTACCTACC-3′ and R:5′-AAACGTTGAAAAACTGAGGA-3′ [29] using fecal DNA samples. A commercial ELISA kit (BIO K 315, Bio-X Diagnostics, Rochefort, Belgique) each was used to detect BRV, BCoV, and E. coli (through its F5 attachment factor) in the fecal samples.
Evaluation of the effects of age, diarrheic status, and diarrheic phases on fecal microbiota
A generalized linear mixed-effects model implemented in the nlme package (version 3.1.137) [30] in R was used to evaluate the effect of age and diarrhea on the alpha diversity metrics of the fecal microbiota, and Tukey’s all-pair comparison test using the ‘glht’ function in the multcomp package (version 1.4.8) [31] in R was used to do the multiple comparisons. In trial 1, the model included calf age and diarrheic status (diarrheic vs. non-diarrheic calves) among the calves as fixed effects and individual calves as random effect:
In trial 2, the model included diarrhea phases (pre-diarrhea, diarrhea, and post-diarrhea, see the results for the delineation of diarrhea phases) as fixed effect and individual calves as random effect:
where Yijk and Ymk represent the variable of interest; Ai is the fixed effect of calf age; Hj is the fixed effect of calf diarrheic status, defined as non-diarrheic or diarrheic status based on fecal score; Sm is the fixed effect of calf diarrhea phases, defined as pre-diarrhea phase, diarrhea phase, or post-diarrhea phase based on the temporal changes of fecal scores of the same calves; Ik is the random effect of individual calves; and εijk and εmk are the residual error. Non-parametric Kruskal–Wallis test was used to assess the effects of age and diarrhea on bacterial relative abundance, and Dunn’s all-pairs rank comparison test with P adjusted by false discovery rate was used to conduct multiple comparisons. A significant change was declared with P < 0.05.
Fecal microbiota comparison among ages and identification of age-associated genera of bacteria
In trial 1, the overall fecal microbiotas between two ages were pairwise compared using analysis of similarity (ANOSIM) implemented in the Vegan package (version 2.5.3) [32] in R. When P < 0.05, the fecal microbiotas between two ages were considered completely different (R-value > 0.75), different (0.5 < R-value < 0.75), or tended to be different (0.3 < R-value < 0.5). R-value < 0.3 was considered not different.
Random forest regression was used to identify the fecal bacterial genera that were associated with the age of the calves using the randomForest package (version 4.6.14) [33, 34] in R. The genus table of all the samples was the input data. The random forest algorithm was executed with the default parameters (ntree = 1000, default mtry of p/3, where p is the number of input genera (‘features’)). The importance of a genus was ranked in the order of its ‘feature importance’, with feature importance being the decrease in prediction accuracy (in percent) of the model when that genus was removed. To explore the age-associated microbiota development, cross-validation was performed to estimate the optimal number of top-ranking age-associated genera required for prediction using the rfcv function implemented in the randomForest package in R. The identified genera were shown in a heatmap with their relative abundance. These genera were considered microbial markers of the respective ages.
Fecal microbiota comparison between diarrheic statuses, between diarrhea phases, and identification of their associated ASVs and modules
Fecal microbiotas between the diarrheic and non-diarrheic calves in trial 1 and between diarrhea phases (pre-diarrhea, diarrhea, and post-diarrhea) in trial 2 were compared using principal coordinates analysis (PCoA) and permutational multivariate analysis of variance (PERMANOVA/adonis) based on Bray–Curtis dissimilarity. When the fecal microbiotas of diarrheic calves were significantly different from those of age-matched non-diarrheic calves, in trial 1, and when the fecal microbiotas of the calves with diarrhea differed significantly from those at their pre- or post-diarrhea phase in trial 2, Linear discriminant analysis Effect Size (LEfSe) [35] and significance test with DESeq2 [36] were used to identify the ASVs that might be associated with one of the diarrheic statuses or one of the diarrhea phases. Of the ASVs with an LDA score > 2 in LEfSe or an adjusted P-value < 0.05 in DESeq2, those with a log2 fold change > 1 (diarrheic/non-diarrheic calves, or diarrhea/pre- or post-diarrhea phase) were considered to be associated with diarrheic status or phase, whereas those with a log2 fold change < – 1 (defined the same as above) were considered associated with non-diarrheic status or phase.
Co-occurrence patterns of the fecal microbiotas of the diarrheic and non-diarrheic calves (see the results for the delineation of the stages) in trial 1, or the fecal microbiota of the calves at different diarrhea phases were examined using the SparCC algorithm [37] with ASV count table as the input data. The pattern was visualized using the igraph package (version 1.2.5) [38] in R, and correlations with a P < 0.05 and a co-efficient R ≥ 0.5 or ≤ – 0.5 being considered positive and negative correlations, respectively. Modules of the co-occurrence patterns were generated using the walktrap algorithm [39] implemented in igraph. Modules with less than 3 nodes were deleted from the co-occurrence patterns. The identified ASVs associated with a diarrheic status or diarrhea phase were highlighted in the patterns. The modules aggregated with ASVs that were associated with diarrhea in trial 1 or with the diarrhea phase in trial 2 were considered as diarrhea modules and diarrhea-phase modules, respectively, whereas the modules formed with ASVs that were not with diarrhea in trial 1 or associated with pre- or post-diarrhea phases in trial 2 were considered as non-diarrhea modules or pre- or post-diarrhea modules.
Results
Trial 1
Development of fecal bacterial microbiota of calves and temporal microbial successions
In total, 15,283,464 quality-filtered amplicon sequences were obtained from 251 fecal samples (the fecal sample of calf Y03 on d 5 was not analyzed due to contamination with wheat straw) with an average of 60,890 ± 7193 (mean ± SD) sequences per sample. The sequencing depth coverage reached > 99.96% on average (99.89% to 100.00%). In the fecal samples collected from 1 day of age, 345 ASVs (referred to as species hereafter) on average per sample were identified with a Shannon diversity index of 4.03 (Fig. 1A). The number of observed species, Faith’s PD, Shannon diversity index, and evenness decreased from d 1 to 7 but recovered at d 9. Then, observed species, Faith’s PD, Shannon index, and evenness gradually increased, though with fluctuation, to about 509, 28.77, 5.03 and 0.81, respectively, at d 78 (Fig. 1A). Over this period, age significantly (P < 0.05) affected all the diversity metrics, whereas diarrheic status did not affect (P > 0.05) any of these four metrics.

Dynamic changes of fecal bacterial microbiota of calves from birth to post-weaning (trial 1). Dynamic changes of alpha diversity metrics with the red boxes indicating the diarrhea peaks (A), bacterial phyla (B), and major genera (C) across ages. Only the phyla and genera each with a relative abundance > 1% in at least 60% of samples at any single age were shown. The relative abundance significantly differing from that of d 1 is indicated with a * (P < 0.05)
Collectively, the ASVs were classified into 200 genera within 12 phyla. Bacteroidota, Firmicutes, Proteobacteria, and Fusobacteria each had a relative abundance > 1% in more than 60% of the fecal samples at any day (Fig. 1B). Of the 200 bacterial genera identified across all the fecal samples, 15 genera each had a relative abundance > 1% in at least 60% of the fecal samples at a single day. These genera included Alloprevotella, Bacteroides, Escherichia/Shigella, Faecalibacterium, Fusobacterium, Acinetobacter, Prevotellaceae_UCG-003, Rikenellaceae_RC9_gut_group, Ruminococcaceae_UCG-005, Ruminococcaceae_UGC-010, Butyricicoccus, Lachnospiraceae_FCS020_group, Parabacteroides, Prevotella_9 and Sutterella. These predominant genera displayed temporal changes in relative abundance over the course of the trial (Fig. 1C). Alloprevotella, Faecalibacterium, Parabacteroides, and Sutterella increased their relative abundance (P < 0.05) and maintained a higher abundance around d 15 to 58 compared to d 1 and then decreasing towards the end of the trial. Compared to d 1, Bacteroides, Escherichia/Shigella, Fusobacterium, and Butyricicoccus increased and then decreased their relative abundance sharply (P < 0.05) at around d 10. On the contrary, Prevotellaceae_UCG-003, Rikenellaceae_RC9_gut_group, Ruminococcaceae_UCG-005, Ruminococcaceae_UGC-010, and Lachnospiraceae_FCS020_group increased their relative abundance (P < 0.05) during the later days of the trial and maintained or decreased their relative abundance until the end of the trial. Acinetobacter differed from all the other genera as it lost its initial high relative abundance dramatically by d 3 (P < 0.05) and never recovered. Prevotella_9 only increased (P < 0.05) on d 9.
Composition and distribution of age-associated bacterial genera from birth to post-weaning
Pair-wise comparison of the fecal microbiotas at the ASV level among the 18 time points using ANOSIM revealed three age periods, with the first, second, and third age periods being from d 1 to 12, 15 to 63, and 58 to 78, respectively. The fecal microbiotas were similar (R-value < 0.5) within each age period but different between most of the two age periods (Table 1).
Thirty-five age-associated bacterial genera were identified by random forest regression (Fig. 2A and B), and they were distributed in 4 clusters (Fig. 2C) each corresponding to one of the age periods (Table 1). Of these age-associated genera, Klebsiella, Escherichia/Shigella, Enterococcus, Bacteroides, Butyricicoccus and Megamonas in the first cluster were predominant at the early age (d 1 to 12); Alloprevotella, Faecalibacterium, Intestinimonas, Paraprevotella, UBA1819, Lachnospiraceae_UCG-010, Pygmaiobacter, and Subdoligranulum in the second cluster were predominant at d 15 to 58; Blautia, Breznakia, Agathobacter, Anaeroplasma, Romboutsia, Erysipelotrichaceae_UCG-004, Prevotella_9, and Succinivibrio in the third cluster and Candidatus_Stoquefichus, Parasutterella, Lachnospiraceae_NK4A136_group, Oscillibacter, Prevotellaceae_UCG-003, Family_XIII_AD3011_group, Lachnospiraceae_FCS020_group, Rikenellaceae_RC9_gut_group, Prevotellaceae_UCG-001, Ruminococcaceae_UCG-005, Ruminococcaceae_UCG-010, Negativibacillus, and Tyzzerella in the fourth cluster were predominant at d 48 to 68 and d 58 to 78, respectively.

Age-associated bacterial genera from birth to post-weaning (trial 1). The top 35 age-associated bacterial genera ranked by importance to the accuracy of the random forest regression model (A). Ten-fold cross-validation error as a function of the number of input genera was used to regress against the chronologic age of calves. The dotted line indicates the 35 genera used in the model (B). Heatmap of the top 35 age-associated genera and the clusters they formed based on their relative abundance across ages (C)
Diarrhea characteristics of the study cohort
Over the course of the trial 1, all the calves had fecal score ≥ 3 at least one sampling day (Fig. 3A). Based on the fecal scores of all the calves, the 78 d of trial was divided into five stages: stage 1: d 1 to 7, before the first diarrhea peak; stage 2: d 9 to 15, the first diarrhea peak; stage 3: d 18 to 38, the stage after the first peak but before the second diarrhea peak; stage 4: d 48 to 68, the second diarrhea peak; and stage 5: d 75 to 78, the stage after the second diarrhea peak. All the 14 calves were non-diarrheic in stages 1, 3, and 5. Pathogen detection showed that all the 20 diarrheic fecal samples (9 in the first and 11 in the second diarrhea peaks) were Salmonella spp. and BCoV negative, but eight were E. coli K99+ positive (2 in the first and 6 in the second diarrhea peaks). One of the diarrheic samples (in the second diarrhea peak) was both BRV and E. coli K99+ positive (Fig. 3A).

Development stages of the fecal microbiota based on fecal score and the comparison of the overall fecal microbiota from birth to post-weaning (trial 1). Development stages of the 14 calves (A). Principal coordinates analysis (PCoA) plots comparing the fecal microbiotas among the 5 stages (B) and between diarrheic and non-diarrheic calves at stage 2 (C) and stage 4 (D). Age refers to days after birth. The gradual weaning started at d 42 and ended at d 63
ASVs and modules associated with diarrheic status
The fecal microbiotas differed among the five stages (P < 0.001, Fig. 3B). The fecal microbiotas of diarrheic and non-diarrheic calves did not differ (P = 0.450) in stage 2 (Fig. 3C) but did differ (P = 0.004) in stage 4 (Fig. 3D). Based on fold change and LEfSe or DESeq2 analysis, 147 diarrheic status-associated ASVs were identified in stage 4 including 91 diarrhea-associated ASVs and 56 non-diarrhea-associated ASVs (Additional file 2: Fig. S2). The diarrhea-associated ASVs mainly consisted of Bacteroides (15 ASVs), Ruminococcaceae_UCG-005 (8 ASVs), Ruminococcaceae_UCG-010 (6 ASVs), Ruminococcaceae_UCG-013 (4 ASVs) and Lachnospiraceae_FCS020_group (4 ASVs). The non-diarrhea-associated ASVs mainly consisted of Ruminococcaceae_UCG-005 (8 ASVs), Flavonifractor (4 ASVs) and Bacteroides (3 ASVs).
The co-occurrence pattern for stage 4 had 195 nodes, 603 edges, and 17 modules (Fig. 4A, Additional file 3: Table S1). Nineteen diarrhea-associated ASVs and 5 non-diarrhea-associated ASVs showed up in the co-occurrence pattern. The diarrhea-associated ASVs including Prevotellaceae_UCG-003 (ASV11, ASV43), Ruminococcaceae_UCG-010 (ASV427, ASV574), Butyricicoccus (ASV130), Lachnospiraceae_FCS020_group (ASV234) and unclassified Ruminococcaceae (ASV557) were scattered without aggregation in any of the modules. Bacteroides (ASV170, ASV206), Rikenellaceae_RC9_gut_group (ASV33), Ruminococcaceae_UCG-010 (ASV196), Lachnospiraceae_FCS020_group (ASV174), unclassified Bacteroidales (ASV177), unclassified Barnesiellaceae (ASV22) and unclassified Ruminococcaceae (ASV340) were aggregated in diarrhea module 1 (D-M1), while Bacteroides (ASV39, ASV41, ASV145, and ASV259) were aggregated in diarrhea module 2 (D-M2). The non-diarrhea-associated Muribaculaceae (ASV28 and ASV44) and UBA1819 (ASV151) were aggregated in non-diarrhea module (ND-M). The non-diarrhea-associated Barnesiella (ASV497) and Ruminococcaceae_UCG-005 (ASV254) were aggregated in D-M1, and they formed negative correlations with the ASVs in D-M1. The ASVs in D-M1 and D-M2 had a higher (P < 0.05) total relative abundance in diarrheic calves than in non-diarrheic calves (Fig. 4B). The D-M1 was mainly occupied by Bacteroides, unclassified Barnesiellaceae, and Rikenellaceae_RC9_gut_group, with 1.49%, 1.53% and 0.75% relative abundance, respectively; D-M2 was occupied by Fusobacterium and Bacteroides, with 1.67% and 1.58% relative abundance, respectively, but the ND-M was mainly occupied by unclassified Muribaculaceae, Prevotellaceae_Ga6A1_group, Prevotellaceae_UCG-003, and Prevotella_9, with 1.90%, 0.69%, 0.57%, and 0.42% relative abundance, respectively (Fig. 4C).

Co-occurrent pattern of the fecal microbiota at stage 4 in trial 1. Pattern showing the diarrhea status-associated ASVs and modules (A). Mean relative abundance of diarrhea status-associated modules in diarrheic and non-diarrheic calves (B). ASVs relative abundance at genus level in different modules (C). ASV254 and ASV497 in module 2 were not included in plots (B) or (C)
Trial 2
Diarrhea characteristics of the study cohort
Over the course of the trial 2, all the calves had fecal score ≥ 3 at least 1 d. (Fig. 5A). In the 43 calves, 4, 20, 33, 31, 30, 18, 18, 16, 15, 4 and 5 calves had diarrhea on d 8 to 18 respectively. In which, 32 calves had one or more episodes of diarrhea 1 to 5 d after recovering from a previous episode (Fig. 5A). Except of d 9, 10 and 11, the microbial composition had no significant difference between diarrheic and non-diarrheic calves within the same age (Additional file 4: Fig. S3). At the same age, calves were in different days of diarrhea, so microbial changes were also out of synchronization (Fig. S3). So based on the temporal changes of the fecal scores, the fecal samples were divided into four phases: pre-diarrhea (when fecal score < 3), diarrhea (fecal scores ≥ 3 consecutively), post-diarrhea (fecal score falling below 3), and volatility (fecal score rising to ≥ 3 after 1 to 5 d below 3) (Fig. 5B). To track the microbial changes from the pre-diarrhea to post-diarrhea, samples of calves of different ages with or without diarrhea were combined into phases for analysis.

Transition phase division of the 43 calves based on fecal score in trial 2. Fecal score of the 43 calves from d 8 to 18 (A). Phase division based on samples fecal score (B). Principal coordinates analysis (PCoA) plot of fecal microbiotas among pre-diarrhea, diarrhea, and post-diarrhea phases (C)
Changes in fecal bacterial communities of calves from pre- to post-diarrhea
In total, 18,706,378 quality-filtered amplicon sequences were obtained from 340 fecal samples (the fecal samples of pre-diarrhea, diarrhea, and post-diarrhea phases) with an average of 55,019 ± 10,329 (mean ± SD) sequences per sample. The sequencing depth coverage reached > 99.97% on average (99.91% to 99.99%). The fecal microbiotas differed among pre-diarrhea, diarrhea, and post-diarrhea phases (P < 0.01, Fig. 5C).
The lowest number of observed species (on average 117 ASVs per sample) and Shannon index (3.2) were found in the fecal samples of the diarrhea phase (Fig. 6A). Both metrics increased (P < 0.05) in the post-diarrhea phase. Of the major phyla and genera (each with a relative abundance > 1% in > 60% of the fecal samples at any phase), the phylum Bacteroidota decreased (P < 0.05), while the phylum Fusobacteria increased (P < 0.05) from the pre-diarrhea to diarrhea phase (Fig. 6B), and the genera Bacteroides, Butyricicoccus, and Lachnoclostridium decreased (P < 0.05), while Clostridium_sensu_stricto_1 and Fusobacterium increased (P < 0.05) during the phase transition (Fig. 6C). In the post-diarrhea phase, the phyla Bacteroidota and Fusobacteria and the genus Fusobacterium tended to return their relative abundance to the pre-diarrhea level (P < 0.05), while the genus Clostridium_sensu_stricto_1 lost the relative abundance gained in the diarrhea phase (P > 0.05), and the genera Bacteroides, Butyricicoccus, and Lachnoclostridium maintained their relative abundance of the diarrhea phase. Although not having changed their relative abundance from the pre-diarrhea phase to the diarrhea phase, the genera Faecalibacterium, Prevotella_2, and Alloprevotella increased their relative abundance (P < 0.05), while the phylum Epsilonbacteraeota and the genus Campylobacter decreased their relative abundance (P < 0.05) in the post-diarrhea phase.

Fecal microbiota changes from pre-diarrhea, diarrhea, and post-diarrhea phases in trial 2. Two alpha diversity metrics of the fecal microbiota (A), Relative abundance of predominant bacterial phyla (B) and genera (C). Only the phyla and genera each with a relative abundance > 1% in at least 60% of samples in a single phase were shown. Significant differences between two phases were indicated with a * (P < 0.05)
ASVs and modules associated with the diarrhea phase
Comparison of the fecal microbiotas between pre-diarrhea and diarrhea phases revealed (based on fold change and analysis using LEfSe or DESeq2) 32 pre-diarrhea-associated ASVs and 29 diarrhea phase-associated ASVs (Additional file 5: Fig. S4). The pre-diarrhea phase-associated ASVs mainly consisted of Bacteroides (12 ASVs), Parabacteroides (4 ASVs), Butyricicoccus (3 ASVs), and Flavonifractor (3 ASVs). The diarrhea phase-associated ASVs mainly consisted of Clostridium_sensu_stricto_1 (8 ASVs), unclassified Fusobacteriaceae (7 ASVs), Fusobacterium (6 ASVs), and unclassified Enterobacteriaceae (3 ASVs). The co-occurrence pattern of the pre-diarrhea and diarrhea phases had 44 nodes, 78 edges, and 4 modules (Fig. 7A). The pre-diarrhea and diarrhea phase-associated modules (pre-D-M and D-P-M, respectively) were aggregated with six and 16 pre-diarrhea and diarrhea phase-associated ASVs, respectively, and both modules were independent. Module 3 and module 4 were aggregated with 14 and 5 ASVs, respectively. Both module 3 and module 4 had no diarrhea or pre-diarrhea phase-associated ASVs, but module 4 formed a negative correlation with the D-P-M through Prevotella_2 (ASV26) and Clostridium_sensu_stricto_1 (ASV48). The ASVs in the D-P-M had a higher (P < 0.05) total relative abundance in the diarrhea phase than in the pre-diarrhea phase, whereas the opposite was true for the ASVs in pre-D-M (Fig. 7B). The D-P-M was occupied by ASVs assigned to Clostridium_sensu_stricto_1 and Fusobacterium, with a relative abundance of 29.11% and 5.42%, respectively. The pre-D-M was only occupied by ASVs of Bacteroides, with a relative abundance about 11.77%. The ASVs of module 3 mainly consisted of Allprevotella, Bacteroides and Prevotella_9, with 2.94%, 1.65% and 0.55% relative abundance, respectively. The ASVs of module 4 were classified to Prevotella_2 and Lachnospiraceae_UCG-004, with 4.48% and 1.05% relative abundance, respectively (Fig. 7C).

Co-occurrent pattern of the fecal microbiota among three phases in trial 2. Pattern among pre-diarrhea and diarrhea phases (A). Mean relative abundance of phase-associated modules in pre-diarrhea and diarrhea (B). ASVs relative abundance at genus level in pre-diarrhea and diarrhea modules (C). Pattern among diarrhea and post-diarrhea phases (D). Mean relative abundance of phase-associated modules in diarrhea and post-diarrhea (E). ASVs relative abundance at genus level in diarrhea and post-diarrhea modules (F). ASV4 and ASV26 in diarrhea-M were divided into post-D-M1 in plot (E) and (F)
When compared the ASVs between diarrhea and post-diarrhea phases, 44 post-diarrhea phase-associated ASVs and 23 diarrhea phase-associated ASVs were identified based on fold change and analysis using LEfSe or DESeq2 (Additional file 4: Fig. S4). The post-diarrhea phase-associated ASVs mainly consisted of Prevotella_9 (13 ASVs), Alloprevotella (8 ASVs), Bacteroides (4 ASVs), and Collinsella (3 ASVs). The diarrhea phase-associated ASVs mainly consisted of Clostridium_sensu_stricto_1 (6 ASVs), unclassified Fusobacteriaceae (7 ASVs), and unclassified Enterobacteriaceae (4 ASVs). The co-occurrence pattern constructed of the fecal microbiotas at post-diarrhea and diarrhea phases had 57 nodes, 116 edges, and 5 modules (Fig. 7D). The D-P-M was aggregated with 11 diarrhea phase-associated ASVs and two post-diarrhea phase-associated ASVs (ASV4 and ASV26, both belonging to Prevotella_2), both of which had a negative relationship with Clostridium_sensu_stricto_1 (ASV48), a diarrhea phase-associated ASV and the keystone of D-P-M. Two post-diarrhea phase-associated modules (post-D-M1 and post-D-M2) were identified in the co-occurrence network, and post-D-M1 and post-D-M2 were aggregated with 11 and 5 post-diarrhea phase-associated ASVs, respectively. The composition of post-D-M1 was similar to that of module 3 in Fig. 7A, but post-D-M1 had more betweenness and close centrality than module 3 and formed a negative correlation with D-P-M through Alloprevotella (ASV14) and Prevotella_9 (ASV43), both of which negatively associated with Clostridium_sensu_stricto_1 (ASV48) in the D-P-M. The ASV composition of module 2 was the same as that of pre-D-M in Fig. 7A, and module 2 was standalone from the other four modules (Fig. 7D). Module 5 consisted of six ASVs. Although having no diarrhea and post-diarrhea phase-associated ASVs, module 5 positively connected with D-P-M through Tyzzerella_4 (ASV23) and Clostridium_sensu_stricto_1 (ASV48). The D-P-M was occupied by Fusobacterium, Prevotella_2 and Clostridium_sensu_stricto_1 with a relative abundance of 30.94%, 5.34% and 5.19%, respectively (Fig. 7F). The post-D-M1 was mainly occupied by Alloprevotella, Bacteroides and Prevotella_9, with a combined relative abundance of 4.80%, 2.32%, and 0.88%, respectively. The post-D-M2 was only occupied by Prevotella_9, with a relative abundance of 0.21%.
Discussion
A better understanding of the fecal microbiota in diarrheic and non-diarrheic calves can inform improved treatment and prevention strategies. Fecal microbiotas between diarrheic and non-diarrheic calves have been compared after interventions, such as trehalose supplementation [40], feeding waste milk containing antibiotic residues [41] or supplemented with sodium humate and glutamine combination [42], or single species [43] or multispecies probiotics [44]. These interventions affected the developing process of gut microbiota and increased the abundance of Bifidobacterium and Lactobacillus, which might conceal the natural development of resistance to pathogenic colonization in pre-weaned calves. Without these types of interventions, Kim et al. [20] described the ability of a fecal microbiota transplantation (inclusion was collected from the health calves of the similar age to diarrheic calves) could ameliorate diarrhea and restore gut microbial composition in pre-weaning calves. Most of these comparative studies focused on pre-weaning calves. In the present study, we first examined the dynamic development of the gut microbiota (represented by fecal microbiota) and diarrhea occurrence in dairy calves from birth to 15 d post-weaning with frequently fecal sampling in trial 1, which allowed us to identify two diarrhea peaks. Then we analyzed the fecal microbiota and diarrhea occurrence of another group of dairy calves over 11 d corresponding to the first diarrhea peak with fecal samples collected daily. Trial 1 helped test our hypothesis that some gut microbes might have resistance to dysbiotic process with calf diarrhea by dictating the microbial co-occurrence patterns during weaning, while trial 2 allowed us to examine in details as the calves transitioned from pre-diarrhea to diarrhea and then to post-diarrhea phases shortly after birth.
The development of the gut microbiota appeared to have three age periods with birth and weaning as the separatrices [45, 46]. As antibiotics had been used from d 1 to 5, the drastically decreasing of bacterial richness after birth (Fig. 1A) might be the effects of tylosin tartrate and sulfadimidine. However, in the recent study of neonatal dairy calves, without antibiotics, a similar decrease of bacterial richness was reported by Kim et al. [47] and Klein-Jöbstl et al. [48]. In the first two weeks after birth, the underdeveloped gut was colonized primarily by facultatively anaerobic microbes, especially Escherichia/Shigella, to render the intestinal environment suitable for anaerobic intestinal microbes to colonize, so the decrease of bacterial richness might be a result of bacterial adaptation. Escherichia/Shigella had a low relative abundance in the fecal samples at any day after birth to 15 d after weaning, but its relative abundance increased up to 45.36% and 31.51% on d 3 and 5, respectively, and then decreased to around 2% since d 15. The increased colonization by Escherichia/Shigella in young calves has been described previously [49], and it might explain the susceptibility of calves to diarrhea caused by E. coli. Klebsiella, which belongs to the same family, Enterobacteriaceae, as Escherichia/Shigella, also appeared to be an age-associated bacterial genus for this period. Klebsiella pneumoniae [50] and Klebsiella oxytoca [51] are opportunistic pathogenic in humans and are associated with increased infection mortality rate, particularly in immunocompromised individuals, neonates, and the elderly. However, infections in calves caused by Klebsiella have not been reported frequently. Glantz and Jacks reported that Klebsiella spp. occurred naturally in calves, and they might be responsible for some mortality [52]. Komatsu et al. reported fatal suppurative meningoencephalitis caused by K. pneumoniae in two calves [53]. Aslan et al. reported that K. pneumoniae could be isolated in calves suffering from respiratory tract infection, which was not cured by florfenicol [54]. In the present study, we observed changes of relative abundance of Klebsiella, decreasing from 1.07% and 2.09% on d 1 and 3, respectively, to less than 0.1% at d 5. Tylosin tartrate and sulfadimine are both broad spectrum antibiotics exerting their antimicrobial action by inhibiting the bacterial protein synthesis [55], and competing with para-aminobenzoic acid for dihydropteroate synthase [56], respectively. The decrease in Klebsiella might be a result of tylosin tartrate [57]. The peak of Klebsiella abundance did not correspond to a diarrhea peak, but it should be given special attention in calf industry because of its pathogenic significance.
Bacteroides was the most abundant genus through the whole period, but its relative abundance increased sharply to 41.96%, 49.66%, and 40.38% at d 5, 7, and 9, respectively, so it could be an age-associated bacterial genus for the first two weeks after birth. Its high abundance was attributed to its stronger saccharolytic ability than Prevotella in the gut of young calves [58, 59]. The acetate produced by Bacteroides could be consumed by other bacteria, such as Butyricicoccus and Megamonas, to produce butyrate and propionate [60], both of which are the main source of energy for intestinal epithelial cells, and butyrate can also inhibit the signaling pathways of pro-inflammatory cytokines [61], enhance intestinal barrier function by increasing mucin secretion and enhancing the tight-junctions [62]. Thus, the colonization by these microbes in the gut of young calves might facilitate the establishment of a functional gut. From the third week onward, weaning separated the microbiota characteristic of the two periods. Prior to weaning for more than 40 d, the microbiotas remained similar, suggesting that the gut might have established a stable functional microbiota over that period, with limited recruitment. However, with the transition from liquid to total solid feed, some of the gut bacteria displayed considerable changes, as exemplified by the replacement of Alloprevotella, Faecalibacterium, and Parabacteroides by Prevotellaceae_UCG-003, Rikenellaceae_RC9_gut_group, Ruminococcaceae_UCG-005, Ruminococcaceae_UGC-010, and Lachnospiraceae_FCS020_group. These five genera are within families that contain species capable of utilizing structural polysaccharides, and this replacement might facilitate the degradation of structural polysaccharides and the stability of the gut microbiota. Future research can help identify the species of these five genera and characterize their functions. Two diarrhea peaks were observed during the three developing periods of the gut microbiota, and less than half of the tested diarrheic fecal samples were pathogen positive, which suggests that microbiota homeostasis may be more important in preventing diarrhea than directly killing pathogens. Comparing fecal microbiota transplantation and antibiotic treatment for ameliorating calf diarrhea, Kim et al. [20] confirmed that gut microbial manipulation could offer another therapeutic paradigm, beyond antibiotic based therapies. Our data also suggest that prophylaxis/preventions with probiotics should be better administered in d 1 to 7 (stage 1, before the first diarrhea peak) and d 18 to 38 (stage 3, the stage after the first peak but before the second diarrhea peak). Supplementation of newborn calves with Lactobacillus and Bifidobacterium [63] or Faecalibacterium prausnitzii [64] within the first 7 d of life decreased diarrhea, but no study was found in the literature that had tested probiotic supplementation in stage 3. To promote the natural development of the gut microbiotas during the transition, the potential probiotics in the gut of calves should be identified.
Comparison of the fecal microbiota using LEfSe and the subsequent identification of the correlations between the differential bacterial genera and the core bacterial genera in the gut is a well-trodden path to reveal the effects of diarrhea on the microbiota and find potential probiotics in neonatal dairy calves for preventing diarrhea [65]. But many standard correlation analyses may lead to misleading results because 16S rRNA gene profiling data are sparse and compositional [37]. SparCC, which is tailored to the compositional and sparse features of genomic survey data and allows for inference of correlations between genes or species, has been used to elucidate the networks of interaction among microbial species living in or on the human body [37, 66]. Therefore, to reduce the incidence of false positive results, three levels of evaluation criteria including co-occurrence patterns examined using the SparCC algorithm (correlations with a P < 0.05 and a co-efficient R ≥ 0.5 or ≤ – 0.5 being considered positive and negative correlations, respectively), |log2 fold change|> 1, LDA score > 2 in LEfSe or adjusted P-value < 0.05 in DESeq2 were used in the present study. During the weaning, some ASVs and modules were associated with diarrhea, while some were associated with non-diarrhea (Fig. 4 and Fig. S2).
With the three levels of evaluation criteria, the identified diarrhea-associated ASVs were aggregated in Bacteroides (ASV39, ASV41, ASV145, ASV170, ASV206, and ASV259). These members might be biomarkers of diarrhea risk. Muribaculaceae (ASV28 and ASV44), UBA1819 (ASV151), Barnesiella (ASV497), and Ruminococcaceae_UCG-005 (ASV254) were non-diarrhea-associated ASVs in the co-occurrence pattern, and ASV28 and ASV497 had a direct inhibitory relationship with the members of D-M1 (Fig. 4A). Muribaculaceae, which was previously assigned as family S24-7 or Homeothermaceae, is a common and abundant family of symbiotic bacteria in the gut and specialized in fermenting complex carbohydrates [67]. It responded most positively to acarbose treatment for diabetes [68] and was linked to longevity [69]. Barnesiella belongs to the family Porphyromonadaceae within the phylum Bacteroidota. It was found to suppress the growth of intestinal vancomycin-resistant Enterococcus [70]. Members of Ruminococcaceae are mostly butyrate-producing bacteria. Weese et al. suggested that Firmicutes (particularly Lachnospiraceae and Ruminococcaceae)/Proteobacteria ratio might be used to potentially predict and prevent colic [71]. Although some members of Ruminococcacea were associated with diarrhea, both ASV151 and ASV254 (both assigned to Ruminococcaceae) were associated with non-diarrhea. So Muribaculaceae (ASV28 and ASV44), UBA1819 (ASV151), Barnesiella (ASV497), and Ruminococcaceae_UCG-005 (ASV254) might be used as potential probiotics for this specific commercial farm, with this specific microbial colonization patterns. The sequences of these ASVs were included in Table 2, and these ASVs can be verified in future studies. Fusobacteriaceae dominated D-M2 (Fig. 4C). Fusobacteriaceae was reported to have high relative abundances in dairy calves suffering from diarrhea, either infected [72] or uninfected [65], which indicates that module analysis can help identify bacteria associated with diarrhea or otherwise. With this module analysis, Muribaculaceae and Prevotella were identified as the core microbiota resisting diarrhea in weaning calves. Kim et al. reported that substituting fermented soybean meal (FSBM) for soybean meal (SBM) at 5% level in calf starter reduced the incidence of diarrhea and improved immunocompetence in neonatal calves after microbial infection [73]. The reason for the positive effects of FSBM on immunocompetence was not reported, but in a recent study, Feizi et al. found that FSBM increased the abundance of Prevotella ruminicola in the rumen of dairy calves [74]. Essential oils showed similar effects on dairy calves, increasing the Prevotellaceae abundance in the rumen [75] and decreasing the morbidity of neonatal diarrhea among pre-weaning calves [76]. Furthermore, a recent study reported that sodium humate and glutamine in combination also elevated the abundance of P. ruminicola in the rectum while reducing diarrhea incidence among dairy calves during the weaning period [42]. Tap et al. reported that Prevotellaceae enterotype was less susceptible to irritable bowel syndrome (IBS) compared with Bacteroidaceae enterotype [77]. Therefore, future research is warranted to investigate the relationship between calf diarrhea and Prevotella as a genus and its species. Prevotella may also be explored for its preventative ability to reduce calf diarrhea.
Consistent changes in relative abundance of Bacteroides, Butyricicoccus, Faecalibacterium, Alloprevotella, and Fusobacterium were observed in both trial 1 and trial 2 (Fig. 1, 2 and 6). When the fecal microbiota was examined in detail as the calves transitioned from pre-diarrhea to diarrhea and then to post-diarrhea phases in trial 2, the peak of both Clostridium_sensu_stricto_1 and Fusobacterium coincided with the peak of diarrhea. It has been reported that Clostridium_sensu_stricto_1 might cause epithelial inflammation in piglets [78] and stunting in infants (defined as height-for-age Z score equal to or lower than – 2, [79]). Therefore, research is needed to further investigate these two genera with respect to their role in calf diarrhea. Our co-occurrence analysis (Fig. 7) showed that the post-D-M1 might be a driver of diarrhea recovery because of the close interaction between its constituent members and the inhibitory relationship with D-P-M. Prevotella_2 and Alloprevotella increased their relative abundance in the post-diarrhea phase (Fig. 6C), and they dominated the post-D-M1 (Fig. 7F), which supports the importance of Prevotellaceae in resisting calf diarrhea. In particular, Prevotella_2 (ASV4 and ASV26), Alloprevotella (ASV14) and Prevotella_9 (ASV43), their sequences were included in Table 2, might be potential probiotics for preventing diarrhea in early stage. It should be noted that although most of the constituent members of post-D-M1 were detected before diarrhea (module 3 and 4 in Fig. 7A), their betweenness and close centrality increased in post-D-M1. This suggests that the interactions among different bacteria might play an important role in maintaining intestinal homeostasis in the gut. These potential probiotics may be supplemented in the first week after birth to prevent diarrhea, and fiber diets [80] or FSBM [71] may improve their efficacy. Clostridium_sensu_stricto_1 (ASV48, Fig. 7, Table 2), was negatively related with Prevotella_2 (ASV4 and ASV26), Alloprevotella (ASV14) and Prevotella_9 (ASV43). All of the four ASVs established the negative relationship between the post-D-M1 and D-P-M. Thus, Clostridium_sensu_stricto_1 (ASV48) might be a biomarker of diarrhea risk in the early stage.
Conclusions
In conclusion, microbial successions of the gut microbiome in dairy calves were rapid, and daily sampling is needed to capture the rapid dynamic gut microbial successions. Promoting indigenous Prevotella and Muribaculaceae might be a new strategy to reduce the incidence of diarrhea in neonatal calves and help calves to go through the weaning transition smoothly. Prevotella_2 (ASV4 and ASV26), Prevotella_9 (ASV43), Alloprevotella (AVS14), unclassified Muribaculaceae (ASV28 and ASV44), UBA1819 (ASV151), Ruminococcaceae_UCG-005 (ASV254), and Barnesiella (ASV497) might be used as probiotics to reduce or prevent calf diarrhea; Clostridium_sensu_stricto_1 (ASV48) might be a useful biomarker of diarrhea risk in this large-scale dairy farm locating in subtropical monsoon climate zone with automated milk feeder system.
Availability of data and material
The raw sequencing data generated in this study are publicly available in NCBI Sequence Read Archive (http://www.ncbi.nim.nih.gov/sra) under the accession number PRJNA716761.
Abbreviations
- E. coli K99+ :
-
Escherichia coli K99+
- BRV:
-
Bovine rotavirus
- BCoV:
-
Bovine coronavirus
- ASV:
-
Amplicon sequence variant
- Faith’s PD:
-
Faith’s phylogenetic diversity
- ANOSIM:
-
Aanalysis of similarity
- PCoA:
-
Principal coordinates analysis
- PERMANOVA:
-
Permutational multivariate analysis of variance
- LEfSe:
-
Linear discriminant analysis Effect Size
- D-M1:
-
Diarrhea module 1
- D-M2:
-
Diarrhea module 2
- ND-M:
-
Non-diarrhea module
- pre-D-M:
-
Pre-diarrhea phase-associated module
- D-P-M:
-
Diarrhea phase-associated module
- post-D-M1/2:
-
Post-diarrhea phase-associated modules
References
Cho YI, Yoon KJ. An overview of calf diarrhea - infectious etiology, diagnosis, and intervention. J Vet Sci. 2014;15(1):1–17. https://doi.org/10.4142/jvs.2014.15.1.1.
Meganck V, Hoflack G, Opsomer G. Advances in prevention and therapy of neonatal dairy calf diarrhoea: a systematical review with emphasis on colostrum management and fluid therapy. Acta Vet Scand. 2014;56(1):75. https://doi.org/10.1186/s13028-014-0075-x.
Castro JJ, Gomez A, White BA, Mangian HJ, Loften JR, Drackley JK. Changes in the intestinal bacterial community, short-chain fatty acid profile, and intestinal development of preweaned Holstein calves. 1. Effects of prebiotic supplementation depend on site and age. J Dairy Sci. 2016;99(12):9682–702. https://doi.org/10.3168/jds.2016-11006.
Windeyer MC, Leslie KE, Godden SM, Hodgins DC, Lissemore KD, LeBlanc SJ. Factors associated with morbidity, mortality, and growth of dairy heifer calves up to 3 months of age. Prev Vet Med. 2014;113(2):231–40. https://doi.org/10.1016/j.prevetmed.2013.10.019.
Aghakeshmiri F, Azizzadeh M, Farzaneh N, Gorjidooz M. Effects of neonatal diarrhea and other conditions on subsequent productive and reproductive performance of heifer calves. Vet Res Commun. 2017;41(2):107–12. https://doi.org/10.1007/s11259-017-9678-9.
Heinrichs AJ, Heinrichs BS, Harel O, Rogers GW, Place NT. A prospective study of calf factors affecting age, body size, and body condition score at first calving of holstein dairy heifers. J Dairy Sci. 2005;88(8):2828–35. https://doi.org/10.3168/jds.S0022-0302(05)72963-5.
Kargar S, Roshan M, Ghoreishi SM, Akhlaghi A, Kanani M, Abedi Shams-Abadi AR, et al. Extended colostrum feeding for 2 weeks improves growth performance and reduces the susceptibility to diarrhea and pneumonia in neonatal Holstein dairy calves. J Dairy Sci. 2020;103(9):8130–42. https://doi.org/10.3168/jds.2020-18355.
Khan MA, Lee HJ, Lee WS, Kim HS, Kim SB, Ki KS, et al. Pre- and postweaning performance of holstein female calves fed milk through step-down and conventional methods. J Dairy Sci. 2007;90(2):876–85. https://doi.org/10.3168/jds.S0022-0302(07)71571-0.
Klein-Jöbstl D, Iwersen M, Drillich M. Farm characteristics and calf management practices on dairy farms with and without diarrhea: a case-control study to investigate risk factors for calf diarrhea. J Dairy Sci. 2014;97(8):5110–9. https://doi.org/10.3168/jds.2013-7695.
Caballero S, Kim S, Carter RA, Leiner IM, Sušac B, Miller L, et al. Cooperating commensals restore colonization resistance to vancomycin-resistant Enterococcus faecium. Cell Host Microbe. 2017;21(5):592–602. https://doi.org/10.1016/j.chom.2017.04.002.
Jacobson A, Lam L, Rajendram M, Tamburini F, Honeycutt J, Pham T, et al. A gut commensal-produced metabolite mediates colonization resistance to Salmonella infection. Cell Host Microbe. 2018;24(2):296–307. https://doi.org/10.1016/j.chom.2018.07.002.
Kim YG, Sakamoto K, Seo SU, Pickard JM, Gillilland MG, Pudlo NA, et al. Neonatal acquisition of Clostridia species protects against colonization by bacterial pathogens. Science. 2017;356(6335):315–9. https://doi.org/10.1126/science.aag2029.
Wu H, Ma Y, Peng X, Qiu W, Kong L, Ren B, et al. Antibiotic-induced dysbiosis of the rat oral and gut microbiota and resistance to Salmonella. Arch Oral Biol. 2020;114:104730. https://doi.org/10.1016/j.archoralbio.2020.104730.
Jang JY, Kim S, Kwon MS, Lee J, Yu DH, Song RH, et al. Rotavirus-mediated alteration of gut microbiota and its correlation with physiological characteristics in neonatal calves. J Microbiol. 2019;57(2):113–21. https://doi.org/10.1007/s12275-019-8549-1.
Haag LM, Fischer A, Otto B, Plickert R, Kühl AA, Göbel UB, et al. Intestinal microbiota shifts towards elevated commensal Escherichia coli loads abrogate colonization resistance against Campylobacter jejuni in mice. PLoS ONE. 2012;7(5): e35988. https://doi.org/10.1371/journal.pone.0035988.
Kim YH, Nagata R, Ohtani N, Ichijo T, Ikuta K, Sato S. Effects of dietary forage and calf starter diet on ruminal pH and bacteria in Holstein calves during weaning transition. Front Microbiol. 2016;7:1575. https://doi.org/10.3389/fmicb.2016.01575.
Meale SJ, Chaucheyras-Durand F, Berends H, Guan LL, Steele MA. From pre- to postweaning: Transformation of the young calf’s gastrointestinal tract. J Dairy Sci. 2017;100(7):5984–95. https://doi.org/10.3168/jds.2016-12474.
Karasova D, Crhanova M, Babak V, Jerabek M, Brzobohaty L, Matesova Z, et al. Development of piglet gut microbiota at the time of weaning influences development of postweaning diarrhea - A field study. Res Vet Sci. 2021;135:59–65. https://doi.org/10.1016/j.rvsc.2020.12.022.
Ma T, Villot C, Renaud D, Skidmore A, Chevaux E, Steele M, et al. Linking perturbations to temporal changes in diversity, stability, and compositions of neonatal calf gut microbiota: prediction of diarrhea. ISME J. 2020;14(9):2223–35. https://doi.org/10.1038/s41396-020-0678-3.
Kim HS, Whon TW, Sung H, Jeong YS, Jung ES, Shin NR, et al. Longitudinal evaluation of fecal microbiota transplantation for ameliorating calf diarrhea and improving growth performance. Nat Commun. 2021;12(1):161. https://doi.org/10.1038/s41467-020-20389-5.
Lesmeister KE, Heinrichs AJ. Effects of corn processing on growth characteristics, rumen development, and rumen parameters in neonatal dairy calves. J Dairy Sci. 2004;87(10):3439–50. https://doi.org/10.3168/jds.S0022-0302(04)73479-7.
Zoetendal EG, Heilig HG, Klaassens ES, Booijink CC, Kleerebezem M, Smidt H, et al. Isolation of DNA from bacterial samples of the human gastrointestinal tract. Nat Protoc. 2006;1(2):870–3. https://doi.org/10.1038/nprot.2006.142.
Yu Y, Lee C, Kim J, Hwang S. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol Bioeng. 2005;89(6):670–9. https://doi.org/10.1002/bit.20347.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–3. https://doi.org/10.1038/nmeth.3869.
Strube ML. RibDif: can individual species be differentiated by 16S sequencing? Bioinform adv. 2021;1(1):vbab020. https://doi.org/10.1093/bioadv/vbab020.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6. https://doi.org/10.1093/nar/gks1219.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–7. https://doi.org/10.1128/AEM.00062-07.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8(4):e61217. https://doi.org/10.1371/journal.pone.0061217.
Nam HM, Srinivasan V, Gillespie BE, Murinda SE, Oliver SP. Application of SYBR green real-time PCR assay for specific detection of Salmonella spp. in dairy farm environmental samples. Int J Food Microbiol. 2005;102(2):161–71. https://doi.org/10.1016/j.ijfoodmicro.2004.12.020.
Pinheiro J, Bates D, DebRoy S, Sarkar D, Heisterkamp S, Van Willigen B, et al. nlme: Linear and nonlinear mixed effects models. R package version. 2022;3.1–155. Available from: https://cran.r-project.org/package=nlme.
Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biom J. 2008;50(3):346–63. https://doi.org/10.1002/bimj.200810425.
Oksanen J, Blanchet FG, Friendly M, Kindt R. vegan: Community ecology package 2019. Available from: https://cran.r-project.org/package=vegan.
Breiman L. Random Forests. Mach Learn. 2001;45(1):5–32. https://doi.org/10.1023/A:1010933404324.
Liaw A, Wiener M. Classification and regression by randomForest. R News. 2002;2:18–22. Available from: https://cran.r-project.org/doc/Rnews/Rnews_2002-3.pdf
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. https://doi.org/10.1186/gb-2011-12-6-r60.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. https://doi.org/10.1186/s13059-014-0550-8.
Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. PLoS Comput Biol. 2012;8(9):e1002687. https://doi.org/10.1371/journal.pcbi.1002687.
Csárdi G, Nepusz T. The igraph software package for complex network research. Int J Complex Syst. 2006;1695(5):1–9. Available from: https://igraph.org.
Pons P, Latapy M. Computing communities in large networks using random walks. In: Yolum P, Güngör T, Gürgen F, Özturan C, editors. Computer and Information Sciences - ISCIS 2005. Lecture Notes in Computer Science, vol 3733. Berlin Heidelberg: Springer; 2005. p. 284–93. https://doi.org/10.1007/11569596_31.
Miura H, Mukai K, Sudo K, Haga S, Suzuki Y, Kobayashi Y, et al. Effect of trehalose supplementation in milk replacer on the incidence of diarrhea and fecal microbiota in preweaned calves. J Anim Sci. 2021;99(1):skab012. https://doi.org/10.1093/jas/skab012.
Penati M, Sala G, Biscarini F, Boccardo A, Bronzo V, Castiglioni B, et al. Feeding pre-weaned calves with waste milk containing antibiotic residues is related to a higher incidence of diarrhea and alterations in the fecal microbiota. Front Vet Sci. 2021;8:650150. https://doi.org/10.3389/fvets.2021.650150.
Wang D, Du Y, Wang S, You Z, Liu Y. Effects of sodium humate and glutamine combined supplementation on growth performance, diarrhea incidence, blood parameters, and intestinal microflora of weaned calves. Anim Sci J. 2021;92(1): e13584. https://doi.org/10.1111/asj.13584.
Fernández-Ciganda S, Fraga M, Zunino P. Probiotic Lactobacilli administration induces changes in the fecal microbiota of preweaned dairy calves. Probiotics Antimicrob Proteins. 2021. https://doi.org/10.1007/s12602-021-09834-z.
Wu Y, Wang L, Luo R, Chen H, Nie C, Niu J, et al. Effect of a multispecies probiotic mixture on the growth and incidence of diarrhea, immune function, and fecal microbiota of pre-weaning dairy calves. Front Microbiol. 2021;12:681014. https://doi.org/10.3389/fmicb.2021.681014.
Meale SJ, Li SC, Azevedo P, Derakhshani H, DeVries TJ, Plaizier JC, et al. Weaning age influences the severity of gastrointestinal microbiome shifts in dairy calves. Sci Rep. 2017;7(1):198. https://doi.org/10.1038/s41598-017-00223-7.
Amin N, Schwarzkopf S, Kinoshita A, Tröscher-Mußotter J, Dänicke S, Camarinha-Silva A, et al. Evolution of rumen and oral microbiota in calves is influenced by age and time of weaning. Anim Microbiome. 2021;3(1):31. https://doi.org/10.1186/s42523-021-00095-3.
Kim ET, Lee SJ, Kim TY, Lee HG, Atikur RM, Gu BH, et al. Dynamic changes in fecal microbial communities of neonatal dairy calves by aging and diarrhea. Animals (Basel). 2021;11(4):1113. https://doi.org/10.3390/ani11041113.
Klein-Jöbstl D, Quijada NM, Dzieciol M, Feldbacher B, Wagner M, Drillich M, et al. Microbiota of newborn calves and their mothers reveals possible transfer routes for newborn calves’ gastrointestinal microbiota. PLoS ONE. 2019;14(8):e0220554. https://doi.org/10.1371/journal.pone.0220554.
Mayer M, Abenthum A, Matthes JM, Kleeberger D, Ege MJ, Holzel C, et al. Development and genetic influence of the rectal bacterial flora of newborn calves. Vet Microbiol. 2012;161(1–2):179–85. https://doi.org/10.1016/j.vetmic.2012.07.023.
Podschun R, Ullmann U. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin Microbiol Rev. 1998;11(4):589–603. https://doi.org/10.1128/CMR.11.4.589.
Broberg CA, Palacios M, Miller VL. Klebsiella: a long way to go towards understanding this enigmatic jet-setter. F1000Prime Rep. 2014;6:64. https://doi.org/10.12703/P6-64.
Glantz PJ, Jacks TM. A bacteriological and serological study of experimental escherichia coli infection of calves. Can J Comp Med. 1969;33(2):128–33.
Komatsu T, Yoshida E, Shigenaga A, Yasuie N, Uchiyama S, Takamura Y, et al. Fatal suppurative meningoencephalitis caused by Klebsiella pneumoniae in two calves. J Vet Med Sci. 2021;83(7):1113–9. https://doi.org/10.1292/jvms.21-0166.
Aslan V, Maden M, Erganis O, Birdane FM, Corlu M. Clinical efficacy of florfenicol in the treatment of calf respiratory tract infections. Vet Q. 2002;24(1):35–9. https://doi.org/10.1080/01652176.2002.9695122.
Brisson-Noël A, Trieu-Cuot P, Courvalin P. Mechanism of action of spiramycin and other macrolides. J Antimicrob Chemother. 1988;22(Suppl. B):13–23. https://doi.org/10.1093/jac/22.supplement_b.13.
Roland S, Ferone R, Harvey RJ, Styles VL, Morrison RW. The characteristics and significance of sulfonamides as substrates for Escherichia coli dihydropteroate synthase. J Biol Chem. 1979;254(20):10337–45. https://doi.org/10.1016/s0021-9258(19)86714-5.
Akhter S, Ansari MS, Andrabi S, Ullah N, Qayyum M. Effect of antibiotics in extender on bacterial and spermatozoal quality of cooled buffalo (Bubalus bubalis) bull semen. Reprod Domest Anim. 2008;43(3):272–8. https://doi.org/10.1111/j.1439-0531.2007.00890.x.
Shah HN, Collins MD. Proposal to restrict the genus Bacteroides (Castellani and Chalmers) to Bacteroides fragilis and closely related species. Int J Syst Bacteriol. 1989;39(1):85–7. https://doi.org/10.1099/00207713-39-1-85.
Willems A, Collins MD. 16S rRNA gene similarities indicate that Hallella seregens (Moore and Moore) and Mitsuokella dentalis (Haapsalo et al.) are genealogically highly related and are members of the genus Prevotella: emended description of the genus Prevotella (Shah and Collins) and description of Prevotella dentalis comb. nov. Int J Syst Bacteriol. 1995;45(4):832–6. https://doi.org/10.1099/00207713-45-4-832.
Trachsel J, Humphrey S, Allen HK. Butyricicoccus porcorum sp. nov., a butyrate-producing bacterium from swine intestinal tract. Int J Syst Evol Microbiol. 2018;68(5):1737–42. https://doi.org/10.1099/ijsem.0.002738.
Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13(9):R79. https://doi.org/10.1186/gb-2012-13-9-r79.
Jonkers D, Penders J, Masclee A, Pierik M. Probiotics in the management of inflammatory bowel disease: a systematic review of intervention studies in adult patients. Drugs. 2012;72(6):803–23. https://doi.org/10.2165/11632710-000000000-00000.
Abe F, Ishibashi N, Shimamura S. Effect of administration of bifidobacteria and lactic acid bacteria to newborn calves and piglets. J Dairy Sci. 1995;78(12):2838–46. https://doi.org/10.3168/jds.S0022-0302(95)76914-4.
Foditsch C, Pereira RV, Ganda EK, Gomez MS, Marques EC, Santin T, et al. Oral administration of Faecalibacterium prausnitzii decreased the incidence of severe diarrhea and related mortality rate and increased weight gain in preweaned dairy heifers. PLoS ONE. 2015;10(12):e0145485. https://doi.org/10.1371/journal.pone.0145485.
Fan P, Kim M, Liu G, Zhai Y, Liu T, Driver JD, et al. The gut microbiota of newborn calves and influence of potential probiotics on reducing diarrheic disease by inhibition of pathogen colonization. Front Microbiol. 2021;12:772863. https://doi.org/10.3389/fmicb.2021.772863.
Carr A, Diener C, Baliga NS, Gibbons SM. Use and abuse of correlation analyses in microbial ecology. ISME J. 2019;13(11):2647–55. https://doi.org/10.1038/s41396-019-0459-z.
Lagkouvardos I, Lesker TR, Hitch TCA, Galvez EJC, Smit N, Neuhaus K, et al. Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome. 2019;7(1):28. https://doi.org/10.1186/s40168-019-0637-2.
Smith BJ, Miller RA, Schmidt TM. Muribaculaceae genomes assembled from metagenomes suggest genetic drivers of differential response to acarbose treatment in mice. mSphere. 2021;6(6):e0085121. https://doi.org/10.1128/msphere.00851-21.
Sibai M, Altuntaş E, Yıldırım B, Öztürk G, Yıldırım S, Demircan T. Microbiome and longevity: High abundance of longevity-linked muribaculaceae in the gut of the long-living rodent Spalax leucodon. OMICS. 2020;24(10):592–601. https://doi.org/10.1089/omi.2020.0116.
Ubeda C, Bucci V, Caballero S, Djukovic A, Toussaint NC, Equinda M, et al. Intestinal microbiota containing Barnesiella species cures vancomycin-resistant Enterococcus faecium colonization. Infect Immun. 2013;81(3):965–73. https://doi.org/10.1128/IAI.01197-12.
Weese JS, Holcombe SJ, Embertson RM, Kurtz KA, Roessner HA, Jalali M, et al. Changes in the faecal microbiota of mares precede the development of post partum colic. Equine Vet J. 2015;47(6):641–9. https://doi.org/10.1111/evj.12361.
Ichikawa-Seki M, Motooka D, Kinami A, Murakoshi F, Takahashi Y, Aita J, et al. Specific increase of Fusobacterium in the faecal microbiota of neonatal calves infected with Cryptosporidium parvum. Sci Rep. 2019;9(1):12517. https://doi.org/10.1038/s41598-019-48969-6.
Kim MH, Yun CH, Kim HS, Kim JH, Kang SJ, Lee CH, et al. Effects of fermented soybean meal on growth performance, diarrheal incidence and immune-response of neonatal calves. Anim Sci J. 2010;81(4):475–81. https://doi.org/10.1111/j.1740-0929.2010.00760.x.
Feizi LK, Zad SS, Jalali SAH, Rafiee H, Jazi MB, Sadeghi K, et al. Fermented soybean meal affects the ruminal fermentation and the abundance of selected bacterial species in Holstein calves: a multilevel analysis. Sci Rep. 2020;10(1):12062. https://doi.org/10.1038/s41598-020-68778-6.
Poudel P, Froehlich K, Casper DP, St-Pierre B. Feeding essential oils to neonatal Holstein dairy calves results in increased ruminal Prevotellaceae abundance and propionate concentrations. Microorganisms. 2019;7(5):120. https://doi.org/10.3390/microorganisms7050120.
Campolina JP, Coelho SG, Belli AL, Machado FS, Pereira LGR, Tomich TR, et al. Effects of a blend of essential oils in milk replacer on performance, rumen fermentation, blood parameters, and health scores of dairy heifers. PLoS ONE. 2021;16(3):e0231068. https://doi.org/10.1371/journal.pone.0231068.
Tap J, Derrien M, Tornblom H, Brazeilles R, Cools-Portier S, Dore J, et al. Identification of an intestinal microbiota signature associated with severity of Irritable Bowel Syndrome. Gastroenterology. 2017;152(1):111-23.e8. https://doi.org/10.1053/j.gastro.2016.09.049.
Wang J, Ji H, Wang S, Liu H, Zhang W, Zhang D, et al. Probiotic Lactobacillus plantarum promotes intestinal barrier function by strengthening the epithelium and modulating gut microbiota. Front Microbiol. 2018;9:1953. https://doi.org/10.3389/fmicb.2018.01953.
Zambruni M, Ochoa TJ, Somasunderam A, Cabada MM, Morales ML, Mitreva M, et al. Stunting is preceded by intestinal mucosal damage and microbiome changes and is associated with systemic inflammation in a cohort of Peruvian infants. Am J Trop Med Hyg. 2019;101(5):1009–17. https://doi.org/10.4269/ajtmh.18-0975.
Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–8. https://doi.org/10.1126/science.1208344.
Acknowledgements
We thank the College Experimental Teaching Center, College of Animal Sciences, Zhejiang University for experimental equipment surport and assistance. We also thank our colleagues Guangyu Zhang, Lei Cao, Weibing Shi, Shaobo Yu, Zhibo Liu and Peng Wu at the Institute of Dairy Science, College of Animal Sciences, Zhejiang University for their assistance with sampling.
Funding
This work was supported by the National Key Research and Development Program of China (2017YFD0500502).
Author information
Authors and Affiliations
Contributions
JW and HC designed the study and drafted the manuscript. HC, YL and KH conducted the animal trials and collected the fecal samples. HC performed the DNA extraction and pathogen detection. HC and BY performed the data analyses and visualization. YZ provided experiment assistance. ZY provided critical guidance in the interpretation of results and revised the manuscript. JW supervised the study and revised the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental protocols used in the current study were approved by the Animal Care and Use Committee of Zhejiang University (Protocol number: ZJU-20262), and all experimental procedures were performed following the approved protocols.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Supplementary Information
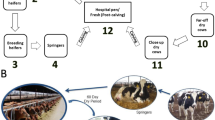
Additional file 1: Fig. S1.
A schematic showing the experimental design, milk feeding, and fecal sample collection of the two trials.
Additional file 2: Fig. S2.
Heatmap of the ASVs associated with diarrheic status in stage 4 of trial 1. The ASVs were identified based on fold change and analysis using LEfSe or DESeq2.
Additional file 3: Table S1.
The ASVs in co-occurrence pattern of trial 1 stage 4.
Additional file 4: Fig. S3.
Principal coordinates analysis (PCoA) plot of fecal microbiotas among ages in trial 2. The fecal microbiotas differences (P values) between diarrheic and non-diarrheic calves within the same age were compared.
Additional file 5: Fig. S4.
Heatmap of the ASVs that identified to be diarrheic status transition-associated in trial 2. The ASVs were identified based on fold change and analysis using LEfSe or DESeq2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, H., Liu, Y., Huang, K. et al. Fecal microbiota dynamics and its relationship to diarrhea and health in dairy calves. J Animal Sci Biotechnol 13, 132 (2022). https://doi.org/10.1186/s40104-022-00758-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40104-022-00758-4