
Abstract
Background
Improving the feed efficiency would increase profitability for producers while also reducing the environmental footprint of livestock production. This study was conducted to investigate the relationships among feed efficiency traits and metabolizable efficiency traits in 180 male broilers. Significant loci and genes affecting the metabolizable efficiency traits were explored with an imputation-based genome-wide association study. The traits measured or calculated comprised three growth traits, five feed efficiency related traits, and nine metabolizable efficiency traits.
Results
The residual feed intake (RFI) showed moderate to high and positive phenotypic correlations with eight other traits measured, including average daily feed intake (ADFI), dry excreta weight (DEW), gross energy excretion (GEE), crude protein excretion (CPE), metabolizable dry matter (MDM), nitrogen corrected apparent metabolizable energy (AMEn), abdominal fat weight (AbF), and percentage of abdominal fat (AbP). Greater correlations were observed between growth traits and the feed conversion ratio (FCR) than RFI. In addition, the RFI, FCR, ADFI, DEW, GEE, CPE, MDM, AMEn, AbF, and AbP were lower in low-RFI birds than high-RFI birds (P < 0.01 or P < 0.05), whereas the coefficients of MDM and MCP of low-RFI birds were greater than those of high-RFI birds (P < 0.01). Five narrow QTLs for metabolizable efficiency traits were detected, including one 82.46-kb region for DEW and GEE on Gallus gallus chromosome (GGA) 26, one 120.13-kb region for MDM and AMEn on GGA1, one 691.25-kb region for the coefficients of MDM and AMEn on GGA5, one region for the coefficients of MDM and MCP on GGA2 (103.45–103.53 Mb), and one 690.50-kb region for the coefficient of MCP on GGA14. Linkage disequilibrium (LD) analysis indicated that the five regions contained high LD blocks, as well as the genes chromosome 26 C6orf106 homolog (C26H6orf106), LOC396098, SH3 and multiple ankyrin repeat domains 2 (SHANK2), ETS homologous factor (EHF), and histamine receptor H3-like (HRH3L), which are known to be involved in the regulation of neurodevelopment, cell proliferation and differentiation, and food intake.
Conclusions
Selection for low RFI significantly decreased chicken feed intake, excreta output, and abdominal fat deposition, and increased nutrient digestibility without changing the weight gain. Five novel QTL regions involved in the control of metabolizable efficiency in chickens were identified. These results, combined through nutritional and genetic approaches, should facilitate novel insights into improving feed efficiency in poultry and other species.
Similar content being viewed by others

Introduction
Feed efficiency is the most important trait in the poultry industry because feed accounts for approximately 70% of the total production cost [1]. In poultry production, feed efficiency is generally defined as the relative ability of an animal to convert feed to product. The most widely used indexes for evaluating feed efficiency are the feed conversion ratio (FCR) and residual feed intake (RFI). FCR is the ratio between feed intake and body weight gain during the measurement period. RFI, which was first used by Koch et al. [2] for cattle, is generally defined as the difference between actual and expected feed intake, the latter of which is based on an animal’s requirements for maintaining body weight and for production [3]. Genetic selection for RFI has been reported to lead to reductions in true metabolizable energy intake and diet-induced thermogenesis in chickens [4]. Digestive efficiency is defined as the proportion of dietary intake minus feces, and metabolizable efficiency is defined as the proportion of dietary intake minus feces and urine [5]. In cattle, digestive efficiency is one of the five major physiological processes controlling RFI, and it conservatively explains 10% of the variation in RFI [6]. In poultry, metabolizable efficiency is easier to determine and is a more practical measure than digestive efficiency, because feces and urine are voided together via the single channel of the cloaca [5]. The heritability values of metabolizable efficiency, such as nitrogen corrected apparent metabolizable energy (AMEn), are moderate to high, ranging from 0.33 to 0.47 [7, 8]. Selection for metabolizable efficiency is accompanied by improved feed efficiency and reduced environmental impact [7, 9].
Quantitative trait loci (QTLs) for economically important traits in animals have been studied for more than 20 years. In chickens, 587 and 40 QTLs associated with FCR and RFI, respectively, have been detected in the Animal QTL Database (Animal QTLdb; https://www.animalgenome.org/cgi-bin/QTLdb/GG/index, 10/24/2019). To our knowledge, only three studies have reported 22 QTLs significantly associated with metabolizable efficiency traits, such as AMEn, dry excreta weight (DEW), and crude protein excretion (CPE) [8, 10, 11]. Few studies have been conducted in other chicken populations and most previous studies have been performed on two broiler lines divergently selected for low or high AMEn on a wheat-based diet.
A genome-wide association study (GWAS) is a powerful tool that can be used to explore the genomic variation associated with complex traits in farm animals. GWAS studies for RFI have been performed in broilers [12] and layers [13]. To date no GWAS for metabolizable efficiency traits has been reported in chickens. This study was conducted to clarify the relationships among feed efficiency traits and metabolizable efficiency traits, and identify significant loci and genes affecting metabolizable efficiency traits in fast-growing white-feathered broiler chickens.
Materials and methods
Experimental birds
In the present study, the workflow for the experiment is illustrated in Fig. 1, and all chickens were obtained from the fast-growing white-feathered pure line B. Line B is a synthetic line produced by Foshan Gaoming Xinguang Agricultural and Animal Industrials Co., Ltd. (Foshan, China), and it has been selected for high body weight and growth rate traits for seven generations. In generation 6, a total of 189 male broiler breeders at 24 d of age, which were produced in the same hatch from 68 sires and 127 dams in generation 5, were randomly selected. They were housed in identical individual cages and provided with water and feed ad libitum. Each day, the amount of fresh feed provided was recorded individually, and residual feed was recorded daily and removed for an intervening period at 28 d of age. The broilers were fed a common corn-soybean meal diet until the end of the trial (42 d of age). The diet contained 2,900 kcal/kg metabolic energy and 183 g/kg crude protein, and detailed information about the diet is summarized in Table 1. The birds were slaughtered at 43 d of age after a 12-h overnight fast to obtain records of carcass traits, including abdominal fat weight (AbF).

The workflow for the experiment in the current study
Metabolism trial and chemical analysis
A metabolism trial was conducted on 189 male broilers subjected to an adaptation period of 4 d followed by a subsequent collection period from 28 to 42 d of age. A total excreta collection method was used. During the collection period, the individual body weight and total feed intake were measured, and the daily (24 h) excreta was collected in white enamel trays. Contaminating materials (e.g., feathers, scales, and debris) were carefully removed. Then a small amount of 10% hydrochloric acid solution was sprayed to prevent nitrogen loss [14]. Excreta were collected daily for a fixed period of time and immediately placed in forced-air ventilation ovens at 65 °C for 24 h.
At the end of the collection period, dried excreta were weighted, ground through a 40-mesh screen, and stored in airtight plastic bags at − 20 °C for dry matter (DM), gross energy (GE), and crude protein (CP) analyses. The moisture content of the feed and excreta was removed by drying in forced-ventilation ovens at 105 °C until a constant weight was reached (AOAC method 934.01) [15]. GE values were determined through bomb calorimetry with a Parr 6400 adiabatic calorimeter (Parr Instrument Company, Illinois, USA), with benzoic acid used as the standard. CP content in samples was determined with the Kjeldahl method (AOAC method 984.13) using a Foss Kjeltec 2300 semiautomatic analyzer (Foss Tecator AB, Höganäs, Sweden) [15].
Phenotypic measurements
Seventeen traits were measured or calculated: body weight at 28 d (BW28), BW42, average daily feed intake (ADFI), average daily gain (ADG), FCR, RFI, DEW, gross energy excretion (GEE), CPE, metabolizable dry matter (MDM), AMEn, metabolizable crude protein (MCP), coefficients of MDM, AMEn, and MCP, AbF, and percentage of abdominal fat (AbP). The total feed intake for each broiler was calculated by summing the feed consumption during the test period, which was then used to calculate the ADFI. The individual body weight gain was calculated on the basis of BW28 and BW42, and was then used to derive the ADG. The FCR was obtained by using the total feed intake divided by the total weight gain. For AMEn, 8.22 kcal/g of nitrogen was used as the correction factor [16]. The coefficients of MDM, AMEn, and MCP were calculated as input in feed minus output in excreta, divided by input in feed and multiplied by 100. The AbP was calculated as the ratio of AbF to BW42. The metabolic body weight at mid-test (MWT) was calculated as the average of BW28 and BW42 (MBW), raised to the power of 0.75 (MBW0.75) for each bird. RFI was computed as the difference between the observed and predicted ADFI. The predicted ADFI was calculated as:
where μ represents the intercept, MWT represents the metabolic body weight at mid-test. ADG represents the average daily gain, β1 and β2 represent partial regression coefficients, and e represents the residual. The estimated e was taken as the measure of RFI.
Quality control of phenotypes was applied to RFI, FCR, and the coefficients of MDM, AMEn, and MCP, and data that deviated more than three standard deviations from the mean were removed. Nine birds were excluded from the analysis, and the remaining 180 records were available for further analyses.
Genotyping, imputation, and quality control
Genomic DNA was extracted from blood samples with the phenol-chloroform method. The 189 broilers were genotyped with the customized chicken 55 K SNP array from Beijing Compass Biotechnology Co., Ltd. (Beijing, China) [17]. To improve the accuracy of imputation, in the following procedure we introduced a larger genotyping data for 3,449 broilers (1,926 males and 1,523 females) from three generations (generation 5, 6, and 7) of line B using 55 K SNP arrays previously acquired by our group.
For the target panel, a total of 3,638 broilers (2,115 males and 1,523 females) were used for genotype imputation. Quality control criteria were applied to the target panel: individual call rate ≥ 90%, SNP call rate ≥ 90%, and minor allele frequency (MAF) ≥ 0.01. In addition, SNPs located on the sex chromosomes were removed. Ultimately, 41,856 autosome variants and 3,607 broilers (2,097 males and 1,510 females) remained for further analyses.
For the reference panel, 230 broilers (101 males and 129 females) in generation 7 were randomly selected from 3,449 birds and re-sequenced with 150 bp paired-end reads on an Illumina NovaSeq 6000 platform with an average depth of approximately 10×1 L coverage. The sequencing was performed by Zhejiang Annoroad Biotechnology Co., Ltd. (Zhejiang, China). Variant calling was performed according to a standardized bioinformatics pipeline for all samples [18, 19]. Specifically, clean sequencing data were aligned to the chicken reference genome (GRCg6a/galGal6; ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/002/315/GCF_000002315.6_GRCg6a/) with the Burrows-Wheeler Aligner (BWA)-MEM algorithm [20]. Then, PCR duplicates were removed and local indel realignment and base-quality score recalibration were performed with the Genome Analysis Toolkit (GATK version 3.5) [21]. Variant calling was then performed via the HaplotypeCaller in GVCF mode with joint genotyping on all samples. Finally, SNPs were filtered with the GATK VariantFiltration protocol. The filtering settings were as follows: variant confidence score (QUAL) < 30.0, QualByDepth (QD) < 2.0, ReadPosRankSum < − 8.0, total depth of coverage (DP) < 4.0, FisherStrand (FS) > 60.0. SNPs on Gallus gallus chromosome (GGA) 16 were omitted because there were fewer common SNPs between the target panel and the reference panel. The sex chromosomes were also removed. In addition, quality control of the reference panel was conducted with the criteria of individual call rate ≥ 90%, SNP call rate ≥ 90%, and MAF ≥ 0.01. After filtering, a total of 12,377,431 autosome variants remained for the 230 sequenced birds.
Genotype imputation of the 55 K genotypes of the broilers to the imputed whole-genome sequence (WGS) level was performed with Beagle 5.0 [22]. Before imputation, inconsistencies between the target panel and the reference panel were checked with conform-gt software (http://faculty.washington.edu/browning/conform-gt.html). Subsequently, 40,342 autosomal SNPs from the target panel were retained. One factor, the effective population size [23], affects the accuracy of genotype imputation, because it is much smaller in livestock than in humans [24]. The effect of this factor on imputation from 55 K SNP chip data to WGS data was investigated to achieve higher accuracy. The microchromosome GGA28 was selected to improve computational efficiency.
The reference panel was pre-phased with Beagle 5.0 (default settings) [22]. Then the imputation from 55 K to WGS level was also executed in Beagle 5.0 with default parameters, except for setting the effective population size to 61,500 instead of the default of 1 million (Additional file 1: Fig. S1). To assess the accuracy of imputation from the target panel to the reference panel, we assessed the genotype concordance rate and allelic R2 measures for each variant. The genotype concordance rate was calculated by comparing the imputed and real genotypes for the 230 birds analyzed with both panels. The allelic R2 was calculated as the estimated squared correlation of the imputed sequence genotype on the true sequence, which was given by Beagle 5.0. We applied strict post-imputation filtering criteria per SNP: allelic R2 ≥ 0.9 and MAF ≥ 0.05. Finally, 1,279,346 autosomal variants and 180 samples remained for the GWAS analyses (Additional file 2: Table S1).
Estimation of phenotypic correlation and genome-wide association study
Pairwise phenotypic correlations of two feed efficiency traits (RFI and FCR) with metabolizable efficiency traits were analyzed in the Hmisc R package. To better understand the relationships between RFI and metabolizable efficiency traits, the lowest and highest 20% of the RFI (low-RFI and high-RFI) birds were selected from 180 chickens and compared with Tukey’s test in SAS 9.4 (SAS Institute, NC) [25]. P < 0.05 (*) or P < 0.01 (**) was considered significant.
The GWAS for metabolizable efficiency traits was performed using the univariate linear mixed model (LMM) implemented in GEMMA version 0.98.1 software (https://github.com/genetics-statistics/GEMMA/releases) [26]. The genotype was set as the fixed factor and the additive polygenic effect as the random effect. Except for the coefficients of MDM, AMEn, and MCP, BW28 was considered as a covariate in the LMM for other traits. The statistical model was as follows:
where y represents the vector of phenotypic values; W represents the vector of covariates, including a column of 1 s; α represents the vector of the corresponding coefficients including the intercept; x represents the vector of marker genotypes; β represents the effect size of the marker; u represents the vector of random polygenic effects; ϵ represents the vector of errors; τ−1 represents the variance of the residual errors; λ represents the ratio between the two variance components; K represents the centered relatedness matrix estimated from 1,279,346 variants and In represents the identity matrix. MVNn represents the n-dimensional multivariate normal distribution. The Wald test was used as a criterion to select SNPs associated with metabolizable efficiency traits.
The genome-wide significance was assessed using the simple M method [27], to infer effective independent tests. A total of 85,247 independent tests over the entire chromosomal SNPs were obtained, and then genome-wide significant and suggestive thresholds were set to 5.87e-7 (0.05/85,247) and 1.17e-5(1/85,247), respectively. Manhattan and Q-Q plots were constructed for each trait by the qqman package (https://cran.r-project.org/web/packages/ qqman/) in R (version 3.6.0). Linkage disequilibrium (LD) blocks of target regions were performed using the Haploview version 4.2 software [28]. SNP positions were updated according to the newest release from UCSC (GRCg6a/galGal6 genome version). Identification of the closest genes to genome-wide significant and suggestive variants was obtained using UCSC annotation of the GRCg6a/galGal6 genome version (http://genome-asia.ucsc.edu/cgi-bin/hgGateway?hgsid=472768848_otkBtCHKhHMTV1xrxHuq737iivJ1). Boxplots were produced by the ggplot2 package in R (version 3.6.0).
Results
Descriptive statistics of traits
We determined the descriptive statistics for the traits associated with growth, feed efficiency, and metabolizable efficiency (Table 2). In this study, the ranges of RFI were − 16.72 g/d to 17.03 g/d, and the average FCR was 1.89 during the growing period from 28 to 42 d of age. The average BW28 was 1,254.67 g and reached 2,499.08 g of BW42. Birds digested an average of 167.95 g/d of feed, absorbed approximately 103.35 g/d of MDM, produced approximately 88.89 g/d of ADG, and excreted nearly 40.65 g/d of DEW. The coefficients of MDM, AMEn, and MCP averaged 71.80%, 71.42%, and 60.81%, respectively. The coefficients of variation of these traits in the population ranged from 1.36% to 33.96%.
Phenotypic correlation analysis
Pearson correlation coefficients for feed efficiency traits (RFI and FCR) with other traits are shown in Table 3 and Additional file 3: Table S2. The positive phenotypic correlations of RFI with FCR, ADFI, DEW, GEE, CPE, MDM, AMEn, AbF, and AbP were moderate to high, ranging from 0.29 to 0.76 (P < 0.01). RFI showed a low correlation with MCP (r = 0.18; P < 0.05) and the coefficient of MDM (r = − 0.17; P < 0.05). RFI was phenotypically independent of BW28, BW42, and ADG (r = 0.00; P > 0.05). FCR was positively correlated with BW28 (r = 0.28; P < 0.01) and CPE (r = 0.18; P < 0.05), and negatively correlated with BW42, ADG, and MCP, ranging from − 0.49 to − 0.20 (P < 0.01). The relationships of FCR with ADFI, DEW, GEE, MDM, AMEn, AbF, and AbP were close to zero (P > 0.05). The same negative relationships existed between the coefficient of MCP and the two feed efficiency traits (RFI and FCR). Poor phenotypic correlations were found between the coefficient of AMEn and the two traits (RFI and FCR) (P > 0.05).
Phenotypic differences between the low- and high-RFI broilers
Descriptive statistics for growth, feed efficiency, and metabolizable efficiency traits compared between the high- and the low-RFI broiler chickens are presented in Table 4. No significant difference was observed between high-RFI birds and low-RFI birds for BW28, BW42, ADG, MCP, and the coefficient of AMEn (P > 0.05). RFI and FCR were significantly lower for low-RFI birds than for high-RFI birds (P < 0.01). ADFI, DEW, GEE, CPE, MDM, AMEn, and AbP were 7.79%, 9.70%, 8.59%, 14.09%, 7.03%, 7.73%, and 20.90% lower, respectively, in low-RFI birds than high-RFI birds (P < 0.01). AbF was 18.97% lower in low-RFI chickens than high-RFI chickens (P < 0.05). Interestingly, the coefficients of MDM and MCP were greater for low-RFI birds than for high-RFI birds (P < 0.01).
Imputation accuracy
The average imputation accuracies of different effective population sizes on GGA28 are shown in Additional file 1: Fig. S1. As the effective population size increased from 50 to the default of 1 million, the genotype concordance rate slightly increased first and then decreased. The effective population size was set as 61,500 to obtain the highest accuracy of imputation.
The number of SNPs in different MAF classes for different datasets are shown in Fig. 2a. In general, the MAF distribution from the four datasets showed the same trend, in which the number of SNPs per class slightly declined with increasing of MAF. Consistency was observed in the distribution of SNPs between 55 K array data and imputed WGS data after post-imputation filtering, and between WGS data and imputed WGS data after imputation (MAF ≥ 0.05). In addition, the MAF distribution based on 55 K array data was not significantly different from that based on WGS data (χ2-test, P = 0.15).

Distribution of MAF and imputation accuracy. (a) Percentage of SNPs in each MAF class for 55 K array data for 3607 birds, WGS data for 230 birds, and imputed WGS data for 3607 birds after imputation and post-imputation filtering. Post-imputation SNP filtering criteria: allelic R2 ≥ 0.9 and MAF ≥ 0.05. The imputation accuracy for imputed WGS data for 3607 birds after imputation according to MAF (b) and per chromosome (c) is also shown. MAF represents minor allele frequency
To evaluate the imputation accuracy for imputed WGS data for 3,607 birds after imputation in detail, we determined the average genotype concordance rate and R2 according to the MAF and chromosome, as shown in Fig. 2b, c. With increasing MAF, the genotype concordance rate decreased from 0.97 to approximately 0.69, whereas R2 increased from 0.12 to 0.75. At the chromosome level, the genotype concordance rate slightly fluctuated between 0.81 and 0.90, whereas R2 fluctuated between 0.20 and 0.69. The distribution of SNPs used in the GWAS analyses after post-imputation filtering is summarized in Additional file 2: Table S1. The genotypic concordance rate and R2 reached an average of 0.924 and 0.937, respectively.
Genome-wide association study of metabolizable efficiency traits
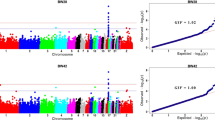
We focused on analysis of metabolizable efficiency traits in the current study. The Manhattan and quantile-quantile (Q-Q) plots are presented in Figs. 3, 4 and 5, and Table 5.

Manhattan and quantile-quantile plots of GWAS for excreta traits. Each dot represents a SNP in the dataset. The horizontal red and blue lines indicate the thresholds for genome-wide significance (P value = 5.87e-7) and suggestive significance (P value = 1.17e-5), respectively. DEW, GEE, and CPE represent dry excreta weight, gross energy excretion, and crude protein excretion, respectively

Manhattan and quantile-quantile plots of GWAS for metabolizable traits. Each dot represents a SNP in the dataset. The horizontal red and blue lines indicate the thresholds for genome-wide significance (P value = 5.87e-7) and suggestive significance (P value = 1.17e-5), respectively. MDM, AMEn, and MCP represent metabolizable dry matter, nitrogen corrected apparent metabolizable energy, and metabolizable crude protein, respectively

Manhattan and quantile-quantile plots of GWAS for the coefficients of metabolizable traits. Each dot represents a SNP in the dataset. The horizontal red and blue lines indicate the thresholds for genome-wide significance (P value = 5.87e-7) and suggestive significance (P value = 1.17e-5), respectively. The coefficients of MDM, AMEn, and MCP represent the coefficients of metabolizable dry matter, nitrogen corrected apparent metabolizable energy, and metabolizable crude protein, respectively
For DEW and GEE, the same 95.17-kb region in GGA26 (4.26–4.36 Mb) was identified, which contained 61 significant SNPs. LD analysis showed that one high LD block was detected in this region (Fig. 6a). The most significant SNP in this region, rs14300817, had a negative effect (β < 0) of DEW and GEE, respectively. These SNPs on GGA26 were located either within or near the nearest genes, including chromosome 26 C6orf106 homolog (C26H6orf106).

Association results of the candidate region on GGA26 (4.26–4.36 Mb) for DEW and GEE. (a) Linkage disequilibrium (LD) analysis of the 61 significant SNPs on GGA26. (b) Box plot for the effect of the SNP rs14300817 on DEW. (c) Box plot for the effect of the SNP rs14300817 on GEE. DEW and GEE represent dry excreta weight and gross energy excretion, respectively
For MDM and AMEn, four significant SNPs were clustered within a 120.13-kb region (GGA1: 93.26–93.38 Mb). The region with one strong block contained four significant SNPs (Additional file 4: Fig. S2a). The top SNP in this region, rs732655996, was located near a novel gene, LOC396098.
For the coefficients of MDM and AMEn, a common 691.25-kb region (GGA5: 18.31–19.00 Mb) was detected, which contained 34 and 53 significant SNPs, respectively. LD analysis revealed one strong block in this significant region (Fig. 7a). The top SNP for the coefficient of MDM, rs741135348, was located in the 17th intron of SH3 and multiple ankyrin repeat domains 2 (SHANK2). The SNP rs315854959 was located near the ETS homologous factor (EHF). In addition, four significant SNPs associated with the coefficient of MDM were detected, which clustered within a 0.38-kb region in GGA2 (103.48 Mb). These SNPs were located near histamine receptor H3-like (HRH3L).

Association results of the candidate region on GGA5 (18.31–19.00 Mb) with the coefficients of MDM and AMEn. (a) Linkage disequilibrium (LD) analysis of the 53 significant SNPs on GGA5. (b) Box plot for the effect of the SNP rs741135348 on the coefficient of MDM. (c) Box plot for the effect of the SNP rs315854959 on the coefficient of AMEn. The coefficients of MDM and AMEn represent the coefficients of metabolizable dry matter and nitrogen corrected apparent metabolizable energy, respectively
For the coefficient of MCP, 251 significant SNPs were located in GGA2 and GGA14. The 45 significant SNPs were clustered within a 74.62-kb region (GGA2: 103.45–103.53 Mb), and four of these SNPs were the same as those found for the coefficient of MDM; 206 SNPs were clustered within a 690.50-kb region (GGA14: 10.02–10.71 Mb). Extremely strong LD status was found in the two regions (Fig. 8a and Additional file 5: Fig. S3a). The top variant on GGA2, rs15137100, was located near the HRH3L gene.

Association results of the candidate region on GGA2 (103.45–103.53 Mb) with the coefficients of MDM and MCP. (a) Linkage disequilibrium (LD) analysis of the 45 significant SNPs on GGA2. (b) Box plot for the effect of the SNP rs740788104 on the coefficient of MDM. (c) Box plot for the effect of the SNP rs15137100 on the coefficient of MCP. The coefficients of MDM and MCP represent the coefficients of metabolizable dry matter and metabolizable crude protein, respectively
The effects of the most significant SNPs resulted in observed differences in the DEW, GEE, MDM, AMEn, and coefficients of MDM, AMEn, and MCP, as shown in Figs. 6, 7 and 8, and S2–S3. These results indicate that the lowest and highest phenotypic values belonged to homozygotes, whereas the intermediate values belonged to heterozygotes. Broilers with homozygous GG (rs14300817) excreted more DEW and GEE than did those with homozygous AA (Fig. 6b, c). MDM and AMEn were higher in the homozygous CC (rs732655996) than the homozygous TT broilers (Additional file 4: Fig. S2b–c). The coefficients of MDM, AMEn, and MCP were higher for broilers with homozygous CC (rs741135348), AA (rs315854959), GG (rs740788104), TT (rs15137100), and GG (rs738484580) than AA, GG, AA, CC, and AA (Figs. 7 and 8b–c, and Additional file 5: Fig. S3b).
Discussion
Phenotypic analysis
Metabolizable efficiency in classical animal nutrition can be used not only to evaluate the nutrient values of diets but also to directly account for individuals’ abilities to digest and absorb nutrients. For poultry, the index and total collection methods have been widely used for the determination of metabolizable efficiency. Although these methods involve laborious quantitative records of feed intake and output, the total collection method was chosen in the current study because it is more accurate, precise, and reproducible than the index method [5, 29]. To date there is no ideal inert indicator that has a uniform distribution in the diet and can be easily chemically determined [5]. In our study, feed intake and excreta were recorded for a 14-d test period consistent with poultry production, a period generally longer than those in previous studies, which have used a 2-d to 5-d period [10, 29, 30]. The coefficients of AMEn and MCP ranged from 68.34% to 74.57% (mean 71.42%) and from 53.15% to 67.12% (mean 60.81%), respectively, in agreement with findings by Wu et al. [31], who found that the coefficients of AMEn and MCP averaged 73% and 62% with ranges of 66–80% and 51–71%, respectively, in Ross 308 broilers fed 19 diets formulated with varying nutrient composition. Our results showed significant phenotypic correlations between growth traits (BW28, BW42, and ADG) and FCR in juvenile broilers, but not RFI, which have also been observed in slow-growing chickens [32] and ducks [33]. Therefore, selection for lower FCR focused on improving growth performance without decreasing feed intake.
Poultry excreta has always been of concern because it is associated with environmental pollution. Nutritional techniques such as reduction of dietary CP content have been used to decrease pollution, but these techniques have some undesirable effects on performance and appetite [34]. Therefore, genetic solutions should be determined. The heritability values of excretion traits from 17 to 23 d of age were low to moderate and ranged from 0.09 to 0.30 [9]. Tran et al. [10] detected nine QTLs significantly associated with metabolizable efficiency traits and selection for these traits should decrease excreta. In the current study, RFI presented a favorable phenotypic correlation with ADFI, DEW, GEE, CPE, MDM, and AMEn with moderate or high values, whereas weak correlations (almost zero) were observed between FCR and these traits. Different results for correlations between AMEn and the two traits (RFI and FCR) have been reported by Mignon-Grasteau et al. [7] and de Verdal et al. [30]; these studies used birds of the D+ and D- lines and found strongly negative genetic correlations between AMEn (kcal/kg) with FCR and RFI. Mignon-Grasteau et al. [35] estimated the heritability for AMEn on a corn-based diet and AMEn on a wheat-based diet to be 0.15 and 0.32, respectively, thereby indicating that the variability in the heritability of AMEn is largely influenced by diet composition. Thus, a reasonable explanation for the differences is that AMEn is an extremely complex trait affected by multiple factors including diet composition, test period, and calculation methods.
In addition, low-RFI birds had significantly lower ADFI, DEW, GEE, CPE, MDM, and AMEn, and higher the coefficients of MDM and MCP than the high-RFI birds. Similar results for low-RFI and high-RFI animals have been observed by Metzler-Zebeli et al. [36], who reported that low-RFI male broilers have lower DEW and CPE than high-RFI male broilers. Furthermore, Harris et al. [37] reported that low-RFI pigs have higher digestibility values for DM (87.3% vs. 85.9%), nitrogen (88.3% vs. 86.1%), and GE (86.9% vs. 85.4%) than high-RFI pigs. Mauch et al. [38] used divergent RFI lines and found that under a low-energy, high-fiber diet, low RFI pigs have greater digestibility of DM, GE, nitrogen, and neutral detergent fiber (7%, 7%, 10%, and 32%) than high RFI pigs (P < 0.05).
Excess AbF deposition is undesirable in the poultry industry because AbF is considered a waste product [39]. In the current study, AbF and AbP had moderate, positive phenotypic correlations with RFI but not with FCR. This is in agreement with findings from Wen et al. [32], in which abdominal fat had a moderate positive phenotypic correlation with RFI and not with FCR in slower growing chickens. In addition, we found that the low-RFI birds had significantly lower AbF and AbP than the high-RFI birds. This result suggests that selection for lower RFI animals could decrease fat deposition in growing animals.
Genotype imputation
Genotype imputation has been widely used in GWAS to boost power [40]. This method can aid in identifying many novel SNPs and QTLs associated with phenotypes of interest. In previous GWAS-based studies, imputation from low density SNP chip genotypes to the WGS level had been implemented in chickens [41], pigs [42], and cattle [43]. Imputed genotypes with sufficiently high imputation accuracy are necessary for reliable results in follow-up analyses such as GWAS. In the current study, the genotypic concordance rate and R2 between imputed and true genotypes reached an average of 0.924 and 0.937, respectively, values that were higher than those reported by Huang et al. [41], who used sequence data imputed from a SNP array for GWAS in chickens and achieved an average imputation accuracy of 0.914. Hayes et al. [44] reported that the accuracy of mimic imputation from the 50 K panel to WGS was 83–93% in Beagle (version 3) for sheep breeds. Ni et al. [45] reported that the post-imputation filtering criterion for imputation accuracy should be 0.80 to ensure the high quality of the imputed WGS data.
Genome-wide association study of metabolizable efficiency traits
Loci and genes for DEW, GEE, MDM, and AMEn
The genomic region of 95.17 kb on GGA26 (4.26–4.36 Mb) was detected to be associated with DEW and GEE. Previous studies have reported QTLs on GGA26 for DEW (2.4–3.2 Mb) and excreta nitrogen to phosphorus ratio (3.2–4.2 Mb) in an F2 resource population of medium-growth broilers [11]. The most significant SNP associated with DEW and GEE was located in the fifth exon of the C26H6orf106 gene. Its homolog, C6orf106, has been investigated for activation of extracellular-signal-regulated kinase signaling pathways to accelerate cell proliferation [46].
A genomic region (GGA1: 93.26–93.38 Mb) was found to be associated with MDM and AMEn, and has also been found to be included in a feed efficiency QTL (GGA1: 90.35–123.03 Mb) in a meat-type × egg-type resource population by Hansen et al. [47]. The highly significant SNP is located in the unannotated gene LOC396098, which has no known function in chickens.
Loci and genes associated with the coefficients of MDM, AMEn, and MCP
We detected one important QTL region (GGA5: 18.31–19.00 Mb) associated with the coefficients of MDM and AMEn, which was not previously reported. The top SNPs associated with DEW and GEE were within the SHANK2 and EHF genes. SHANK2 (also known as ProSAP1) is the second member of the Shank protein family and is involved in neurodevelopmental and psychiatric disorders [48]. SHANK2 plays a key role in regulating transepithelial salt and water transport by modulating Na+/H+ exchanger 3 (NHE3) expression and activity in epithelial cells, including those in the gastrointestinal tract [49]. SHANK2-knockout mice show hyperactivity and repetitive behaviors [50]. EHF is a member of the epithelium-specific ETS transcription factor family, which is highly expressed in multiple epithelial cell types including intestinal epithelium [51]. EHF plays an important role in the regulation of epidermal proliferation and differentiation [52].
Two novel regions (GGA2: 103.45–103.53 Mb and GGA14: 10.02–10.71 Mb) associated with the coefficient of MCP were detected and the top variant was located near the HRH3L gene. Very few studies on HRH3L are available in the literature. HRH3 is an autoreceptor on numerous neurons that inhibits the synthesis and release of histamine [53, 54]. Previous studies have shown that HRH3 negatively regulates food intake in rodents, in a manner independent of its histaminergic tone modulation [55].
Conclusions
In summary, selection for low RFI significantly decreased chicken feed intake, excreta output, and abdominal fat deposition, and increased nutrient digestibility without changing weight gain. In addition, five novel QTL regions involved in the control of metabolizable efficiency in chickens were identified. These results, obtained from both nutritional and genetic approaches, should facilitate novel insights into improving feed efficiency in poultry and other species.
Availability of data and materials
The raw whole genome sequencing data reported in this paper have been deposited in the Genome Sequence Archive [56] in BIG Data Center [57] under accession number CRA002454 that can be publicly accessed at https://bigd.big.ac.cn/gsa. The genotype and phenotype data of the 180 samples used in GWAS are available at the figshare repository (https://figshare.com/s/8c882576e0bc014fe382).
Abbreviations
- AbF:
-
Weight of abdominal fat
- AbP:
-
Percentage of abdominal fat
- ADFI:
-
Average daily feed intake
- ADG:
-
Average daily gain
- AMEn:
-
Nitrogen corrected apparent metabolizable energy
- BW28:
-
Body weight at 28 d of age
- BW42:
-
Body weight at 42 d of age
- C26H6orf106:
-
Chromosome 26 C6orf106 homolog
- CP:
-
Crude protein
- CPE:
-
Crude protein excretion
- CV:
-
Coefficient of variation
- d:
-
Day
- DEW:
-
Dry excreta weight
- DM:
-
Dry matter
- EHF:
-
ETS homologous factor
- FCR:
-
Feed conversion ratio
- GE:
-
Gross energy
- GEE:
-
Gross energy excretion
- GGA:
-
Gallus gallus chromosome
- GWAS:
-
Genome-wide association study
- HRH3L:
-
Histamine receptor H3-like
- LD:
-
Linkage disequilibrium
- MCP:
-
Metabolizable crude protein
- MDM:
-
Metabolizable dry matter
- MWT:
-
Metabolic body weight at mid-test
- QTL:
-
Quantitative trait locus
- Animal QTLdb:
-
Animal QTL Database
- SHANK2:
-
SH3 and multiple ankyrin repeat domains 2
- SNP:
-
Single nucleotide polymorphism
- WGS:
-
Whole-genome sequence
- RFI:
-
Residual feed intake
References
Zampiga M, Flees J, Meluzzi A, Dridi S, Sirri F. Application of omics technologies for a deeper insight into quali-quantitative production traits in broiler chickens: a review. J Anim Sci Biotechno. 2018;9:61.
Koch RM, Swiger LA, Chambers D, Gregory KE. Efficiency of feed use in beef cattle. J Anim Sci. 1963;22:486–94.
Archer JA, Arthur PF, Herd RM, Parnell PF, Pitchford WS. Optimum postweaning test for measurement of growth rate, feed intake, and feed efficiency in British breed cattle. J Anim Sci. 1997;75:2024–32.
Gabarrou JF, Geraert PA, Picard M, Bordas A. Diet-induced thermogenesis in cockerels is modulated by genetic selection for high or low residual feed intake. J Nutr. 1997;127:2371–6.
Vohra P. Evaluation of metabolizable energy for poultry. Worlds Poul Sci J. 1972;28:204–14.
Herd RM, Arthur PF. Physiological basis for residual feed intake. J Anim Sci. 2009;87:E64–71.
Mignon-Grasteau S, Muley N, Bastianelli D, Gomez J, Peron A, Sellier N, et al. Heritability of digestibilities and divergent selection for digestion ability in growing chicks fed a wheat diet. Poult Sci. 2004;83:860–7.
Van Goor A, Bolek KJ, Ashwell CM, Persia ME, Rothschild MF, Schmidt CJ, et al. Identification of quantitative trait loci for body temperature, body weight, breast yield, and digestibility in an advanced intercross line of chickens under heat stress. Genet Select Evol. 2015;47:96.
de Verdal H, Narcy A, Bastianelli D, Chapuis H, Meme N, Urvoix S, et al. Improving the efficiency of feed utilization in poultry by selection. 2. Genetic parameters of excretion traits and correlations with anatomy of the gastro-intestinal tract and digestive efficiency. BMC Genet. 2011;12:71.
Tran TS, Narcy A, Carre B, Gabriel I, Rideau N, Gilbert H, et al. Detection of QTL controlling digestive efficiency and anatomy of the digestive tract in chicken fed a wheat-based diet. Genet Select Evol. 2014;46:25.
Mignon-Grasteau S, Rideau N, Gabriel I, Chantry-Darmon C, Boscher MY, Sellier N, et al. Detection of QTL controlling feed efficiency and excretion in chickens fed a wheat-based diet. Genet Select Evol. 2015;47:74.
Xu Z, Ji C, Zhang Y, Zhang Z, Nie Q, Xu J, et al. Combination analysis of genome-wide association and transcriptome sequencing of residual feed intake in quality chickens. BMC Genomics. 2016;17:594.
Yuan J, Wang K, Yi G, Ma M, Dou T, Sun C, et al. Genome-wide association studies for feed intake and efficiency in two laying periods of chickens. Genet Sel Evol. 2015;47:82.
Ribeiro AML, Penz AM Jr, Belay TK, Teeter RG. Comparison of different drying techniques for nitrogen analysis of poultry excreta, feces, and tissue. J Appl Poult Res. 2001;10:21–3.
Horwitz W, Chichilo P, Reynolds H. Official methods of analysis of the Association of Official Analytical Chemists. Washington: Association of Official Analytical Chemists; 1970.
Hill FW, Anderson DL. Comparison of metabolizable energy and productive energy determinations with growing chicks. J Nutr. 1958;64:587–603.
Liu R, Xing S, Wang J, Zheng M, Cui H, Crooijmans R, et al. A new chicken 55K SNP genotyping array. BMC Genomics. 2019;20:410.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Prot Bioinformatics. 2013;43:11 10 11–33.
Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v2 [q-bio.GN]. 2013.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Browning BL, Zhou Y, Browning SR. A one-penny imputed genome from next-generation reference panels. Am J Hum Genet. 2018;103:338–48.
van den Berg S, Vandenplas J, van Eeuwijk FA, Bouwman AC, Lopes MS, Veerkamp RF. Imputation to whole-genome sequence using multiple pig populations and its use in genome-wide association studies. Genet Sel Evol. 2019;51:2.
Hall SJ. Effective population sizes in cattle, sheep, horses, pigs and goats estimated from census and herdbook data. Animal. 2016;10:1778–85.
SAS. User’s guide: statistics. version 9.4. NC, USA: SAS Institute Cary; 2016.
Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44:821–4.
Gao X. Multiple testing corrections for imputed SNPs. Genet Epidemiol. 2011;35:154–8.
Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5.
Kong C, Adeola O. Evaluation of amino acid and energy utilization in feedstuff for swine and poultry diets. Asian-Australas J Anim Sci. 2014;27:917–25.
de Verdal H, Narcy A, Bastianelli D, Chapuis H, Meme N, Urvoix S, et al. Improving the efficiency of feed utilization in poultry by selection. 1. Genetic parameters of anatomy of the gastro-intestinal tract and digestive efficiency. BMC Genet. 2011;12:59.
Wu SB, Swick RA, Noblet J, Rodgers N, Cadogan D, Choct M. Net energy prediction and energy efficiency of feed for broiler chickens. Poult Sci. 2019;98:1222–34.
Wen C, Yan W, Zheng J, Ji C, Zhang D, Sun C, et al. Feed efficiency measures and their relationships with production and meat quality traits in slower growing broilers. Poult Sci. 2018;97:2356–64.
Zhang Y, Guo ZB, Xie M, Zhang Z, Hou S. Genetic parameters for residual feed intake in a random population of Pekin duck. Asian-Australas J Anim Sci. 2017;30:167–70.
Namroud NF, Shivazad M, Zaghari M. Effects of fortifying low crude protein diet with crystalline amino acids on performance, blood ammonia level, and excreta characteristics of broiler chicks. Poult Sci. 2008;87:2250–8.
Mignon-Grasteau S, Juin H, Sellier N, Bastianelli D, Gomez J, Carré B. Genetic parameters of digestibility of wheat-or corn-based diets in chickens. In: Proceedings of the 9th World Congress on Genetics Applied to Livestock Production: 1-6 August 2010. Leipzig, German. 2010;0249.
Metzler-Zebeli BU, Molnar A, Hollmann M, Magowan E, Hawken RJ, Lawlor PG, et al. Comparison of growth performance and excreta composition in broiler chickens when ranked according to various feed efficiency metrics. J Anim Sci. 2016;94:2890–9.
Harris AJ, Patience JF, Lonergan SM, JMD C, Gabler NK. Improved nutrient digestibility and retention partially explains feed efficiency gains in pigs selected for low residual feed intake. J Anim Sci. 2012;90(Suppl 4):164–6.
Mauch ED, Young JM, Serao NVL, Hsu WL, Patience JF, Kerr BJ, et al. Effect of lower-energy, higher-fiber diets on pigs divergently selected for residual feed intake when fed higher-energy, lower-fiber diets. J Anim Sci. 2018;96:1221–36.
Emamgholi Begli H, Vaez Torshizi R, Akbar Masoudi A, Ehsani A, Jensen J. Relationship between residual feed intake and carcass composition, meat quality and size of small intestine in a population of F 2 chickens. Livest Sci. 2017;205:10–5.
Spencer CC, Su Z, Donnelly P, Marchini J. Designing genome-wide association studies: sample size, power, imputation, and the choice of genotyping chip. PLoS Genet. 2009;5:e1000477.
Huang S, He Y, Ye S, Wang J, Yuan X, Zhang H, et al. Genome-wide association study on chicken carcass traits using sequence data imputed from SNP array. J Appl Genet. 2018;59:335–44.
Yan G, Guo T, Xiao S, Zhang F, Xin W, Huang T, et al. Imputation-based whole-genome sequence association study reveals constant and novel loci for hematological traits in a large-scale swine F2 resource population. Front Genet. 2018;9:401.
Hoglund JK, Buitenhuis B, Guldbrandtsen B, Lund MS, Sahana G. Genome-wide association study for female fertility in Nordic red cattle. BMC Genet. 2015;16:110.
Hayes BJ, Bowman PJ, Daetwyler HD, Kijas JW, van der Werf JH. Accuracy of genotype imputation in sheep breeds. Anim Genet. 2012;43:72–80.
Ni G, Cavero D, Fangmann A, Erbe M, Simianer H. Whole-genome sequence-based genomic prediction in laying chickens with different genomic relationship matrices to account for genetic architecture. Genet Select Evol. 2017;49:8.
Li X, Dong M, Zhou JP, Zhu DH, Zhao JB, Sheng WW. C6orf106 accelerates pancreatic cancer cell invasion and proliferation via activating ERK signaling pathway. Mol Cell Biochem. 2019;454:87–95.
Hansen C, Yi N, Zhang YM, Xu S, Gavora J, Cheng HH. Identification of QTL for production traits in chickens. Anim Biotechnol. 2005;16:67–79.
Lee S, Lee E, Kim R, Kim J, Lee S, Park H, et al. Shank2 deletion in Parvalbumin neurons leads to moderate hyperactivity, Enhanced Self-Grooming and Suppressed Seizure Susceptibility in Mice. Front Mol Neurosci. 2018;11:209.
Han W, Kim KH, Jo MJ, Lee JH, Yang J, Doctor RB, et al. Shank2 associates with and regulates Na+/H+ exchanger 3. J Biol Chem. 2006;281:1461–9.
Schmeisser MJ, Ey E, Wegener S, Bockmann J, Stempel AV, Kuebler A, et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature. 2012;486:256–60.
Luk IY, Reehorst CM, Mariadason JM. ELF3, ELF5, EHF and SPDEF transcription factors in tissue homeostasis and Cancer. Molecules. 2018;23.
Rubin AJ, Barajas BC, Furlan-Magaril M, Lopez-Pajares V, Mumbach MR, Howard I, et al. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nat Genet. 2017;49:1522–8.
Arrang JM, Garbarg M, Schwartz JC. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature. 1983;302:832–7.
Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev. 2008;88:1183–241.
Yoshimoto R, Miyamoto Y, Shimamura K, Ishihara A, Takahashi K, Kotani H, et al. Therapeutic potential of histamine H3 receptor agonist for the treatment of obesity and diabetes mellitus. Proc Natl Acad Sci U S A. 2006;103:13866–71.
Wang Y, Song F, Zhu J, Zhang S, Yang Y, Chen T, et al. GSA: genome sequence archive<sup/>. Genomics Proteomics Bioinformatics. 2017;15:14–8.
National Genomics Data Center M, Partners. Database Resources of the National Genomics Data Center in 2020. Nucleic Acids Res. 2020;48:D24–33.
Acknowledgements
The authors thank Professor Zhou Zhengkui (Chinese Academy of Agricultural Sciences, CAAS) for helpful suggestions when performing the genotype imputation analysis.
Funding
The research was supported by grants from the National Nonprofit Institute Research Grant (Y2020PT02), the earmarked fund for the modern agro-industry technology research system (CARS-41), Agricultural Science and Technology Innovation Program (ASTIP-IAS04; ASTIP-IAS-TS-15).
Author information
Authors and Affiliations
Contributions
WL contributed to performing of the study and the analysis of data. RRL contributed to analysis, interpretation of data, and writing of the manuscript. MQZ, FRF & DWL contributed to the sample and data collection. YMG contributed to interpretation of data and reviewing of the manuscript. GPZ & JW contributed to the design of the study, interpretation of data, and reviewing of the manuscript. All authors submitted comments on the draft, read, and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All experimental procedures with broilers were performed according to the Guidelines for Experimental Animals established by the Ministry of Science and Technology (Beijing, China). Ethical approval on animal survival was given by the animal welfare and ethics committee of the Institute of Animal Sciences (IAS), Chinese Academy of Agricultural Sciences (CAAS, Beijing, China) with the following reference number: IAS2019–44.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Supplementary information
Additional file 1: Figure S1.
Average imputation accuracies of different effective population sizes on GGA28.
Additional file 2: Table S1.
Distribution of SNPs used in the GWAS analyses after post-imputation filtering.
Additional file 3: Table S2.
Pearson correlation coefficients for growth and metabolizable traits in broilers.
Additional file 4: Figure S2.
Association results of the candidate region on GGA1 (93.26–93.38 Mb) for MDM and AMEn. (a) Linkage disequilibrium (LD) analysis of the 4 significant SNPs on GGA1. (b) Box plot for the effect of the SNP rs732655996 on MDM. (c) Box plot for the effect of the SNP rs732655996 on AMEn. MDM and AMEn represent metabolizable dry matter and nitrogen corrected apparent metabolizable energy, respectively.
Additional file 5: Figure S3.
Association results of the candidate region on GGA14 (10.02–10.71 Mb) for coefficient of MCP. (a) Linkage disequilibrium (LD) analysis of the 206 significant SNPs on GGA14. (b) Box plot for the effect of the SNP rs738484580 on the coefficient of MCP. Coefficient of MCP represents the coefficient of metabolizable crude protein.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, W., Liu, R., Zheng, M. et al. New insights into the associations among feed efficiency, metabolizable efficiency traits and related QTL regions in broiler chickens. J Animal Sci Biotechnol 11, 65 (2020). https://doi.org/10.1186/s40104-020-00469-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40104-020-00469-8