
Abstract
The transportation of solutes across the inner membrane of the mitochondria is catalyzed by a nuclear-coded family of transport proteins called mitochondrial carriers (MCs). Sequences from dictyostelid genome projects have facilitated analysis of the evolution of the dictyostelid mitochondrial carrier family (MCF). The average evolutionary distances between various regions in the MCF shows that the transmembrane region (TR) and conical pit region (CPR) are the only two conserved structural regions. A phylogenetic tree built using the concatenated orthologous TR and CPR sequences of 7 MCs showed that dictyostelids are similar to metazoans in this way. A close evolutionary relationship was observed between dictyostelids and metazoans in 4 MCs known to be related to ADP/ATP transport (MAA). This was further evidenced by the fact that dictyostelids have undergone gene expansion similar to that of metazoans during the evolution of MAA. Sequence logo analysis of CPR in MAA showed that dictyostelids have motifs similar to those of Metazoa. Combined with the conserved substrate binding site of 7 MCs in eukaryotes, it is postulated that dictyostelids are closely related to Metazoa with respect to the evolution of MAA.
Similar content being viewed by others

Background
Social Amoebae, or Dictyostelia, are eukaryotic microbes with a unique life cycle consisting of both uni- and multicellular stages (Basu et al. 2013). Each organism starts its life as a unicellular amoeba, but when starved it aggregates with others to form a multicellular fruiting body. This process has been best described in the model organism Dictyostelium discoideum (Schaap et al. 2006; Romeralo et al. 2011). The phylogeny of D. discoideum constructed on a proteome scale indicated divergence along the branch diverged from the metazoan–fungal lineage (Eichinger et al. 2005), which means that the phylogenetic status of D. discoideum is distinct from that of Metazoa as the whole. However, D. discoideum has achieved multicellularity using strategies similar to those of Metazoa, indicating their evolutionary relationship (Eichinger et al. 2005).
Previous studies have shown the evolutionary correlation of D. discoideum and Metazoa in signaling systems. The D. discoideum genome encodes numerous G-protein-coupled frizzled/smooth receptors, metabotropic glutamate, SH2 domain based phosphoprotein signaling, and secretin families that were previously thought to be specific to animals (Sucgang et al. 2011). Molecular channels facilitate cell–cell translocation of solutes and signaling molecules and are widely distributed in almost all complex multicellular organisms (Knoll 2011). It is necessary to further study the evolutionary relationship between Dictyostelia and Metazoa in solute channel proteins.
Mitochondrial carriers (MCs) are widespread in eukaryotes and are important solute transport channels that provide a link between metabolic reactions occurring in the cytosol and those that occur in the mitochondrial matrix by catalyzing the translocation of numerous solutes across the membrane (Palmieri 2013; Palmieri and Pierri 2010; Kunji and Robinson 2010; Arco and Satrústegui 2005; Palmieri 2004). MCs are involved in many important metabolic pathways, such as oxidative phosphorylation, the citric acid cycle, fatty acid oxidation, amino acid degradation, and Ca2+ cell signaling (Palmieri 2004; Arco and Satrústegui 2005; Palmieri 2013). MCs have several sequence features in common: a tripartite structure, a threefold repeated signature motif, and 6 transmembrane α-helices. These structural features are different from those of any other solute carrier family and allow to unequivocally recognize MCs (Palmieri 2013). Together with the fact that MCs are the largest of solute carriers and widespread in eukaryotes (Palmieri 2013), the metabolic importance and specific structural features of MCs give them a competitive advantage in the study of the evolutionary relationship between Dictyostelia and Metazoa in solute channel proteins. Dictyostelid genome projects, which were performed on the 4 representative social amoeba dictyostelids genomes (Heidel et al. 2011; Sucgang et al. 2011), have facilitated analysis of the evolution of the mitochondrial carrier family (MCF) in Dictyostelia.
Methods
Detection of conserved structural region
The structure of the MCF consists of three segments: NCLOOP24, which faces the cytosol; the transmembrane region (TR), which is located in the inner membrane; and LOOP135, which faces the matrix. The conical pit region (CPR) in LOOP135 is composed of three groups with the PX[D/E]XX[K/R]X[K/R] motif and five amino acids downstream near the C-terminus. The classification of different structural region has been reported previously (Gong et al. 2010).
Average evolutionary distance (AED) was used to estimate the degree of conservation of each structural region: the smaller the mean value during evolution, the higher the conservation. To characterize the conservation of different structural regions, the AED of the corresponding structural region sequence from each orthologous subfamily of human MCF (SLC25) was calculated using MEGA 5.0 (model = Dayhoff matrix; gamma parameter = 2.0). The details of the processes by which each orthologous subfamily were determined can be found in a previous work (Gong et al. 2010).
Phylogenetic analysis
Eukaryotic orthologous sequences of SLC25 were obtained from the InParanoid eukaryotic ortholog database (Ostlund et al. 2010). SLC25 served as text query in the search of human sequences in the InParanoid 8 database to obtain the eukaryotic orthologous sequences of SLC25. The sequences supported by the best bootstrap values in InParanoid database were selected as the orthologous sequences. Members of SLC25 with a high coverage of taxa (>90 %) in the inParanoid database were selected for phylogenetic analysis. The tandem concatenated sequences consisting of the orthologous TR and CPR sequences in these eukaryotic MCs were used to construct a phylogenetic tree. The accession numbers of these orthologous sequences can be found in Additional file 2: Table S1.
Protein sequences were aligned using Muscle with default parameters (Edgar 2004). Maximum likelihood trees were inferred using PhyML v3.0 (Guindon et al. 2010) with the LG model (Le and Gascuel 2008). Clade support was calculated using SH-like approximate likelihood ratio tests (aLRT) (Anisimova et al. 2011). PhyML analyses were performed using NNI tree topology searches with estimated Gamma shape parameters.
Gene family size analysis of MAA
The protein sequences of 18 representative eukaryotic species were used for the comparative genomic analysis of MCs. An E-value of 10−5 was used for the BLASTp search to obtain the homologous sequences of SLC25A4, 5, 6, and 24 in the genomes of 18 representative eukaryotic species. Multigene families were generated from all of the obtained MC proteins of selected genomes using SCPS tools at default settings (BLASTp, cut-off e-value ≤10−35) (Paccanaro et al. 2006). The multigene families were then analyzed for evolutionary changes in gene family size using the CAFÉ program with the input phylogenetic tree (De Bie et al. 2006).
Because alpha tubulin provides materials for the phylogenetic analysis of species, alpha tubulin protein sequences were used to building the input phylogenetic tree for analysis of the size of the MAA gene family (Parfrey et al. 2010). The orthologous sequences of human alpha tubulin in 18 representative eukaryotic species were obtained from the InParanoid 8 database. Protein sequences were aligned using Muscle with default parameters (Edgar 2004). Maximum likelihood trees were produced using PhyML v3.0, with the LG model (Guindon et al. 2010; Le and Gascuel 2008).
Results and discussion
Conserved structural regions in MCF
Although MCs display a wide variety of transported solutes (i.e., a large variety of metabolites, nucleotides and coenzymes), all MCs have common sequence features: a tripartite structure, a threefold repeated signature motif PX[D/E]XX[K/R]X[K/R], and six transmembrane a-helices (Palmieri 2013). These common structural features allow the program to treat MCs as a single unit for study of the evolution of MCF.
The 3D structure of ADP/ATP has been used as a template for the study of various MCs (Palmieri 2013; Pebay-Peyroula et al. 2003). For this reason, the spatial distribution of TR and CPR was here depicted using the 3D structure of the ADP/ATP (PDB code: 1OKC). A six-transmembrane a-helix bundle (H1–H6) consisted of TR (Fig. 1a). The CPR 1, 3, and 5 consisting of the threefold repeated signature motifs PX[D/E]XX[K/R]X[K/R] and their C-terminal 13 sites are located at the C-terminus of the three odd-numbered transmembrane α-helices, respectively (Fig. 1a). The average evolutionary distances of various regions in the MCF showed that the TR and CPR were the only two regions conserved during evolution (Fig. 1b). Combined with the TR and CPR, which constitute the core of solute carrier transport (Gong et al. 2010), these observations indicated that they can represent MCF.

The conserved regions of MCs. a The three-dimensional structure of the ADP/ATP carrier in Bos taurus (PDB code: 1OKC). The region marked in purple represents the CPR. The protein structure was drawn using VMD (Humphrey et al. 1996). b Average evolutionary distances of different regions in the MCF
Evolutionary similarity of dictyostelids and metazoans with respect to MCs
The distribution of orthologous sequences of human MCs (SLC25) showed SLC25A3, 4, 5, 6, 24, 28, and 37 to have the most coverage (>90 %) in the Inparanoid eukaryotic ortholog database (Additional file 2: Table S2), indicating the orthologous sequences of 7 SLC25s that were found to be widely distributed in eukaryotes. Detailed information regarding these 7 SLC25s is given in Table 1. These 7 SLC25s and their orthologous sequences (7 MCs) were to represent the MCF. Because the common substrate binding site has already been identified in the three even α-helices (TR246) of MCs, attention was shifted to variation in substrates transported by these 7 MCs in eukaryotic evolution (Robinson and Kunji 2006). The TR246 sequences were concatenated according to the order of the even transmembrane α-helices (H2, 4, and 6) from the N-terminal to C-terminal (Fig. 1a). Logo analysis of the concatenated TR246 showed that the residues in substrate binding site of 7 MCs were basically conserved in eukaryotic evolution (Fig. 2), which provided evidence for the similar types of substrates transported by 7 MCs in eukaryotic evolution. Together with many conserved signature motifs in the CPR of 7 MCs (Table 1), these observations indicate that the phylogenetic tree constructed using the concatenated TR and CPR of 7 MCs may reflect solute transport capabilities.

Logo analysis of TR246 in the orthologous sequences of 7 SLC25s in eukaryotes. Different color shading from left to right indicates TR2, 4, and 6, respectively. The orthologous sequences of 7 SLC25s from 48 eukaryotic taxa in Additional file 2: Table S3 were selected for logo analysis of TR246. Contact point I, II, and III indicate substrate contact sites. WebLogo 3 was used for the logo analysis (Crooks et al. 2004)
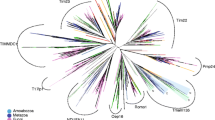
The TR and CPR sequences were concatenated according to the order of the 6 transmembrane α-helices (H1–H6) and CPR (CPR1, 3, and 5) from the N-terminal to C-terminal (Fig. 1a). Here, 7 MCs were concatenated in the order of the SLC25 classification number, from low to high (SLC25A3, 4, 5, 6, 24, 28, and 37). The tree constructed using the concatenated orthologous TR and CPR of 7 MCs with the average sequence length (1040) clearly showed five clusters, the Metazoa, dictyostelids, fungi, plantae, and SAR (Fig. 3). Phylogenetic tree showed that four dictyostelids were clustered close to Metazoa (Fig. 3). These observations indicate dictyostelids may have solute transport mechanisms similar to those of Metazoa. This is partially consistent with the fact that D. discoideum amoebae display distinct characteristics of true multicellularity, such as cell–cell signaling, cellular specialization, and coherent cell movement (Raper 1984; Kessin 2001). As the only solitary free-living representative Amoebozoa, Acanthamoeba castellanii has been used a reference for studying the evolution of dictyostelid MCF (Clarke et al. 2013). In contrast, the Amoebozoa A. castellanii was not found near Metazoa or the dictyostelid cluster (Fig. 3). The MC-based phylogenetic tree produced here showed Amphimedon queenslandica and Nematostella vectensis belonging to basal metazoans not to be nested within any metazoan group. This suggests that they cannot transport solutes any more effectively than other complex metazoans to satisfy the energy needs of complex metabolic activities. This was evidenced by the fact that no molecular functions related to metabolism were found to be enriched in an enrichment analysis of molecular functions of basal metazoans (Srivastava et al. 2010). To some extent, these observations suggest that dictyostelids have a more developed capacity to transport solutes, which would satisfy the energy needs of true multicellularity.

Maximum likelihood tree of eukaryotes reconstructed with the tandem concatenated orthologous TR and CPR sequences of 7 SLC25s in 48 taxa. Branch support was given as SH-like aLRT scores. Only SH-like aLRT scores above 70 % are shown. Bold red branches represent the dictyostelid cluster. SAR is a clade that includes stramenopiles, alveolates, and Rhizaria. The first letter of each group provides the “SAR” in the name. The details regarding the distribution of orthologous sequences of SLC25 in InParanoid database can be found in Additional file 2: Table S3
Evolutionary similarity of dictyostelids and metazoans with respect to MAA evolution
Further phylogenetic analysis using the orthologous TR and CPR sequences of 7 SLC25A showed the close evolutionary relationship of the orthologous sequences of SLC25A4-5-6 and SLC25A24 between dictyostelids and metazoans (Additional file 1: Figure S1). The 4 MCs, are mainly responsible for transporting ADP/ATP (Palmieri 2013). This indicated the close evolutionary relationship between dictyostelids and metazoans with respect to these 4 MCs, which are known to be related to ADP/ATP transport (MAA).
Attention was shifted to the evolutionary changes in subfamilies of the MAA in eukaryotes. The phylogenetic tree built using alpha tubulin protein sequences showed that fungi (Coprinopsis cinerea and Neurospora crassa) and invertebrate chordates (Ciona intestinalis and Branchiostoma floridae) consisted of a group instead of dictyostelids. This was consistent with the phylogeny of D. discoideum constructed on a proteome scale (Eichinger et al. 2005). Then, the phylogenetic tree using alpha tubulin protein sequences was used for the analysis of evolutionary changes in the size of the MAA gene family. However, although gene expansion has been observed in the nodes connected to the MAA of most model invertebrate chordates, gene expansion was not observed in the nodes leading to fungi MAA, only in the nodes leading to dictyostelids MAA (Fig. 4). Together with the close phylogenetic relationship between dictyostelids and metazoans in the MAA (Additional file 1: Figure S1), these observations indicated that dictyostelids were closely related to Metazoa in the evolution of MAA.

Evolutionary changes in the eukaryotic MAA. The figure represents the total number of MAA subfamilies in each species or node. The numerals on branches show the numbers of expanded (left, red), unchanged (middle, black), and contracted (right, blue) MAA subfamilies along the lineages. The red box contains the four dictyostelids
Common motifs shared by dictyostelid and metazoan MAA
After the phylogenetic analysis of MCs in Fig. 3, sequence logo analysis of the most conserved CPR region (Fig. 1b) was conducted in the MAA of the three groups (Dictystelids, Metazoa, and Fungi). Results showed that dictyostelids had a motif more similar to that of Metazoa rather than to that of fungi (Additional file 1: Figure S2), at such sites as 1, 11, 12, and 13. Combined with the conserved substrate binding site in the TR246 of 7 MCs (Fig. 2), these observations indicated that dictyostelids shared the common motif with Metazoa in the conserved TR and CPR of MAA.
Because the charged residues in the conserved motif of CPR formed a salt bridge network (Robinson et al. 2008; Robinson and Kunji 2006), attention was shifted to the polar residues in the CPR motif. The proportion of polar glutamine (59.3 %) in the twelfth position (Q12) of CPR near the C-terminus in dictyostelids is similar to that (59.6 %) of Metazoa, which is markedly higher than that of fungi (33.3 %). The polar NH2 group on the long-chain branch of Q was found to strengthen the salt bridge network through participation in the hydrogen bonding reaction. Because the salt bridge network in CPR is involved in substrate transport, this indicates the enriched Q12 in CPR may be associated closely with substrate transport (Robinson et al. 2008). The common motif shared by dictyostelids and metazoans in MAA indicate that dictyostelid MAA may be capable of ADP/ATP transport similar to that performed by Metazoa MAA.
Conclusions
The MC-based phylogenetic tree described here showed that dictyostelids were similar to metazoans in the evolution of the MAA. Dictyostelid MAA exhibited gene expansion similar to that of metazoan MAA. Further sequence logo analysis of the conserved TR and CPR in MAA showed that dictyostelids had a motif similar to that observed in Metazoa. These findings may aid understanding of the evolutionary correlation between dictyostelids and Metazoa in MAA.
References
Anisimova M, Gil M, Dufayard JF, Dessimoz C, Gascuel O (2011) Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst Biol 60(5):685–699. doi:10.1093/sysbio/syr041
Arco AD, Satrústegui J (2005) New mitochondrial carriers: an overview. Cell Mol Life Sci 62(19–20):2204–2227. doi:10.1007/s00018-005-5197-x
Basu S, Fey P, Pandit Y, Dodson R, Kibbe WA, Chisholm RL (2013) DictyBase 2013: integrating multiple Dictyostelid species. Nucleic Acids Res 41 (Database issue):D676–D683. doi:10.1093/nar/gks1064
Clarke M, Lohan AJ, Liu B, Lagkouvardos I, Roy S, Zafar N, Bertelli C, Schilde C, Kianianmomeni A, Burglin TR, Frech C, Turcotte B, Kopec KO, Synnott JM, Choo C, Paponov I, Finkler A, Heng Tan CS, Hutchins AP, Weinmeier T, Rattei T, Chu JS, Gimenez G, Irimia M, Rigden DJ, Fitzpatrick DA, Lorenzo-Morales J, Bateman A, Chiu CH, Tang P, Hegemann P, Fromm H, Raoult D, Greub G, Miranda-Saavedra D, Chen N, Nash P, Ginger ML, Horn M, Schaap P, Caler L, Loftus BJ (2013) Genome of Acanthamoeba castellanii highlights extensive lateral gene transfer and early evolution of tyrosine kinase signaling. Genome Biol 14(2):R11. doi:10.1186/gb-2013-14-2-r11
Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14(6):1188–1190. doi:10.1101/gr.849004
De Bie T, Cristianini N, Demuth JP, Hahn MW (2006) CAFE: a computational tool for the study of gene family evolution. Bioinformatics (Oxford, England) 22(10):1269–1271. doi:10.1093/bioinformatics/btl097
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797. doi:10.1093/nar/gkh340
Eichinger L, Pachebat JA, Glockner G, Rajandream MA, Sucgang R, Berriman M, Song J, Olsen R, Szafranski K, Xu Q, Tunggal B, Kummerfeld S, Madera M, Konfortov BA, Rivero F, Bankier AT, Lehmann R, Hamlin N, Davies R, Gaudet P, Fey P, Pilcher K, Chen G, Saunders D, Sodergren E, Davis P, Kerhornou A, Nie X, Hall N, Anjard C, Hemphill L, Bason N, Farbrother P, Desany B, Just E, Morio T, Rost R, Churcher C, Cooper J, Haydock S, van Driessche N, Cronin A, Goodhead I, Muzny D, Mourier T, Pain A, Lu M, Harper D, Lindsay R, Hauser H, James K, Quiles M, Madan Babu M, Saito T, Buchrieser C, Wardroper A, Felder M, Thangavelu M, Johnson D, Knights A, Loulseged H, Mungall K, Oliver K, Price C, Quail MA, Urushihara H, Hernandez J, Rabbinowitsch E, Steffen D, Sanders M, Ma J, Kohara Y, Sharp S, Simmonds M, Spiegler S, Tivey A, Sugano S, White B, Walker D, Woodward J, Winckler T, Tanaka Y, Shaulsky G, Schleicher M, Weinstock G, Rosenthal A, Cox EC, Chisholm RL, Gibbs R, Loomis WF, Platzer M, Kay RR, Williams J, Dear PH, Noegel AA, Barrell B, Kuspa A (2005) The genome of the social amoeba Dictyostelium discoideum. Nature 435(7038):43–57. doi:10.1038/nature03481
Gong M, Li J, Wang M, Wang J, Zen K, Zhang CY (2010) The evolutionary trajectory of mitochondrial carrier family during metazoan evolution. BMC Evol Biol 10(1):282. doi:10.1186/1471-2148-10-282
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59(3):307–321. doi:10.1093/sysbio/syq010
Heidel AJ, Lawal HM, Felder M, Schilde C, Helps NR, Tunggal B, Rivero F, John U, Schleicher M, Eichinger L, Platzer M, Noegel AA, Schaap P, Glockner G (2011) Phylogeny-wide analysis of social amoeba genomes highlights ancient origins for complex intercellular communication. Genome Res 21(11):1882–1891. doi:10.1101/gr.121137.111
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14(1):33–38, 27–38
Kessin RH (2001) Dictyostelium: evolution, cell biology and the development of multicellularity. Cambridge University Press, Cambridge
Knoll AH (2011) The multiple origins of complex multicellularity. Annu Rev Earth Planet Sci 39:217–239
Kunji ER, Robinson AJ (2010) Coupling of proton and substrate translocation in the transport cycle of mitochondrial carriers. Curr Opin Struct Biol 20(4):440–447. doi:10.1016/j.sbi.2010.06.004
Le SQ, Gascuel O (2008) An improved general amino acid replacement matrix. Mol Biol Evol 25(7):1307–1320. doi:10.1093/molbev/msn067
nature02056
Ostlund G, Schmitt T, Forslund K, Kostler T, Messina DN, Roopra S, Frings O, Sonnhammer EL (2010) InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic acids research 38 (Database issue):D196–D203. doi:10.1093/nar/gkp931
Paccanaro A, Casbon JA, Saqi MA (2006) Spectral clustering of protein sequences. Nucleic Acids Res 34(5):1571–1580. doi:10.1093/nar/gkj515
Palmieri F (2004) The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch 447(5):689–709. doi:10.1007/s00424-003-1099-7
Palmieri F (2013) The mitochondrial transporter family SLC25: identification, properties and physiopathology. Mol Aspects Med 34(2–3):465–484. doi:10.1016/j.mam.2012.05.005
Palmieri F, Pierri CL (2010) Structure and function of mitochondrial carriers—role of the transmembrane helix P and G residues in the gating and transport mechanism. FEBS Lett 584(9):1931–1939. doi:10.1016/j.febslet.2009.10.063
Parfrey LW, Grant J, Tekle YI, Lasek-Nesselquist E, Morrison HG, Sogin ML, Patterson DJ, Katz LA (2010) Broadly sampled multigene analyses yield a well-resolved eukaryotic tree of life. Syst Biol 59(5):518–533. doi:10.1093/sysbio/syq037
Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trezeguet V, Lauquin GJ, Brandolin G (2003) Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 426(6962):39–44. doi:10.1038/nature02056
Raper KB (1984) The dictyostelids. Princeton university Press, Princeton
Robinson AJ, Kunji ER (2006) Mitochondrial carriers in the cytoplasmic state have a common substrate binding site. Proc Natl Acad Sci USA 103(8):2617–2622. doi:10.1073/pnas.0509994103
Robinson AJ, Overy C, Kunji ER (2008) The mechanism of transport by mitochondrial carriers based on analysis of symmetry. Proc Natl Acad Sci USA 105(46):17766–17771. doi:10.1073/pnas.0809580105
Romeralo M, Cavender JC, Landolt JC, Stephenson SL, Baldauf SL (2011) An expanded phylogeny of social amoebas (Dictyostelia) shows increasing diversity and new morphological patterns. BMC Evol Biol 11:84. doi:10.1186/1471-2148-11-84
Schaap P, Winckler T, Nelson M, Alvarez-Curto E, Elgie B, Hagiwara H, Cavender J, Milano-Curto A, Rozen DE, Dingermann T, Mutzel R, Baldauf SL (2006) Molecular phylogeny and evolution of morphology in the social amoebas. Science 314(5799):661–663. doi:10.1126/science.1130670
Srivastava M, Simakov O, Chapman J, Fahey B, Gauthier ME, Mitros T, Richards GS, Conaco C, Dacre M, Hellsten U, Larroux C, Putnam NH, Stanke M, Adamska M, Darling A, Degnan SM, Oakley TH, Plachetzki DC, Zhai Y, Adamski M, Calcino A, Cummins SF, Goodstein DM, Harris C, Jackson DJ, Leys SP, Shu S, Woodcroft BJ, Vervoort M, Kosik KS, Manning G, Degnan BM, Rokhsar DS (2010) The Amphimedon queenslandica genome and the evolution of animal complexity. Nature 466(7307):720–726. doi:10.1038/nature09201
Sucgang R, Kuo A, Tian X, Salerno W, Parikh A, Feasley CL, Dalin E, Tu H, Huang E, Barry K, Lindquist E, Shapiro H, Bruce D, Schmutz J, Salamov A, Fey P, Gaudet P, Anjard C, Babu MM, Basu S, Bushmanova Y, van der Wel H, Katoh-Kurasawa M, Dinh C, Coutinho PM, Saito T, Elias M, Schaap P, Kay RR, Henrissat B, Eichinger L, Rivero F, Putnam NH, West CM, Loomis WF, Chisholm RL, Shaulsky G, Strassmann JE, Queller DC, Kuspa A, Grigoriev IV (2011) Comparative genomics of the social amoebae Dictyostelium discoideum and Dictyostelium purpureum. Genome Biol 12(2):R20. doi:10.1186/gb-2011-12-2-r20
Authors’ contributions
The manuscript was conceived and designed by QT and QMZ. MG QMZ and QT performed acquisition and analysis of data. All authors were involved in revising the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by the Grants from Shanghai Agricultural Science Development 2013(10), Shanghai Agriculture Committee (HuNongKeGongZi2011D-1-2), and the Shanghai Municipal Science and Technology Commission [10DZ2212400].
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Additional files
40064_2016_3146_MOESM1_ESM.doc
Additional file 1. Figure S1. Maximum likelihood tree of eukaryotes reconstructed with the orthologous TR and CPR sequences of 7 SLC25. Branch support was given in the form of SH-like aLRT scores. Red branches represent the metazoans cluster. Blue branches represent the dictyostelid cluster. SLC25A4-5-6 represents the orthologous TR and CPR sequences of SLC25A4, 5, and 6 in 48 taxa. Figure S2. Logo analysis of CPR in the MAA. The MAA sequences from the species in the three groups based on the phylogenetic tree were used for the logo analysis (Figure 3).
40064_2016_3146_MOESM2_ESM.xlsx
Additional file 2. Table S1. Accession numbers of the orthologous sequences of 7 SLC25 used in the MCs-based phylogenetic tree. Table S2. Orthologous sequences distribution of SLC25 in 230 taxa in the InParanoid database. High coverage of SLC25 was marked by brown shading. Table S3. Distribution of orthologous sequences of SLC25 in 41 taxa of InParanoid database. Orthologous sequences of 7 SLC25 (blue shading) with 0 hits (gray shading) were obtained using reciprocal best hits BLAST in NCBI non-redundant database. The orthologous sequences in 6 other taxa were obtained using reciprocal best hits BLAST in NCBI non-redundant database.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gong, M., Zhu, Q. & Tan, Q. Evolutionary analyses of mitochondrial carrier family of dictyostelids. SpringerPlus 5, 1465 (2016). https://doi.org/10.1186/s40064-016-3146-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-016-3146-9