
Abstract
Enantiopure N(Boc)-β3-amino nitriles, valuable synthetic intermediates in the multistep homologation of α-amino acids, were alkylated using n-BuLi as base. Alkylations afforded easily separable, almost equimolecular mixtures of diastereomeric N(Boc)-protected syn and anti β2,3-amino nitriles. Suitable manipulations of both cyano and amino groups eventually led to enantiopure N- and/or C-protected β2,3-amino acids. For example, methanolysis using conc. HCl gas in MeOH, provides C-protected β2,3 amino acids in excellent yields. This methodology is applied to the synthesis of a series N(Boc)-β2,3-dialkyl amino nitriles derived from l-phenylalanine, d-phenylalanine, l-valine and one C-protected β2,3 amino acid. We demonstrate an efficient procedure for the preparation of anti and syn β2,3-amino acids with alkyl side chains, from α-amino acids in reasonable yields.
Similar content being viewed by others

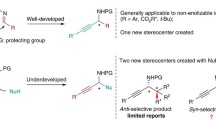

Background
Beta amino acids have shown great potential for a wide range of applications in many fields of organic chemistry in recent years (Juaristi and Soloshonok 2005). The growing interest in this class of compounds, which are widely used in medicinal chemistry forming new secondary structures and as valuable synthetic building blocks, can be better appreciated by database searching (e.g. Sci-Finder). Almost all β3-amino acids with proteinogenic side chains are now commercially available, but are quite expensive. Several synthetic procedures have been reported for the preparation of β-amino acids, and the field has been extensively reviewed (Cole 1994; Liu and Sibi 2002; Weiner et al. 2010).
β2,3-Amino acids have two substituent at the α (C2) and β (C3) positions (Fig. 1). They are relatively rare in nature, although occur as substructures in several bioactive compounds and important metabolites (Juaristi and Soloshonok 2005). These β-amino acids bearing two side chains are of particular interest for the synthesis of β-peptides (oligomers of β-amino acids) in view of their conformation-inducing ability and thereby the ability to afford new foldamers (Seebach et al. 1999; Cheng et al. 2001; Martinek and Fülöp 2012).

Anti (1) and syn (2) β2,3-amino acids
β2,3-Amino acids may be either homochiral (anti- or like-β2,3-amino acids) or heterochiral (syn- or unlike-β2,3-amino acids) and it is noteworthy that, when included as building blocks in peptides, the former (1) afford predominantly helical structures, with all substituents in lateral positions, whereas their syn diastereomers (2) adopt an extended conformation, with formation of pleated sheets (Seebach et al. 1999; Balamurugan and Muraleedharan 2015).
A number of synthetic procedure have been proposed for the preparation of β2,3-amino acids and many of them have been reported recently in an excellent review (Kiss et al. 2015), but very few have been originate from amino acid precursors (Burgess et al. 1993) which have the obvious advantage of transferring pre-existing structural information, such as the nature of side chain and the chirality, into the final products. Among these, the Arndt-Eistert homologation is the most commonly used procedure (Podlech and Seebach 1995). The process involves the conversion of N-protected α-amino acid mixed anhydrides into the corresponding α-diazoketones using diazomethane, followed by the Wolff rearrangement. It has seen a resurgence in popularity in recent years due to the work carried out at ETH in Zurich, which has also identified new secondary structures by inserting several β3 residues in homologous peptide sequences (Seebach et al. 2004). However, this procedure is aimed at obtaining β3-amino acids and only involves intermediate α-diazoketones.
These intermediates have also been used in the preparation of α-methyl β3-residues via KHMDS/HMPA methylation, in modest yields (Yang et al. 2000). Anti-β2,3-amino acids can be prepared by the alkylation of β3-amino esters (Estermann and Seebach 1988; Cardillo et al. 1996; Capone et al. 2007). An alternative approach to the asymmetric synthesis of syn and anti β2,3-amino acids from non-amino acid precursors, exploits the conjugate addition of a chiral N-benzyl-N-(α-methylbenzyl) lithium amide to a specially prepared α,β-unsaturated ester, already bearing the side chains needed in the final product (Davies et al. 1994; Langenhan and Gellman 2003].
A remarkable, unified approach to both syn and anti β2,3-amino acids in enantiomerically pure form with natural or unnatural side chain, was reported (Yu et al. 2010). The strategy is based on asymmetric cycloaddition of a nitrone derived from d-gulose to Z or E acrylates, followed by a facile N–O cleaving fragmentation reaction. The availability of convenient synthetic routes to monomers predisposed to form helical structures has accelerated the pace of discoveries involving β-peptide helices. In contrast, a general, scalable synthesis of syn-β2,3-amino acids from α-amino acids has not been reported so far. Our interests towards non-natural amino acids synthesis began some time ago, when we proposed a procedure for converting α-amino acids into their β3-homologues (Caputo et al. 1995). The methodology reported generated valuable synthetic intermediates, such as β-amino alcohols 3, β-amino iodides 4 and β-amino nitriles 5 (Scheme 1). This homologation procedure has since been enhanced, and the β3-amino acids, as well as all homologation intermediates, can be also prepared labeled with isotopic 2H, using NaBD4 in D2O in the reduction step [Caputo and Longobardo 2007].

Multistep homologation of α-amino acids to N- and/or C-protected β3-amino acids
The homologation intermediate N-protected β-amino iodides 4 are extremely useful starting materials for the preparation of new classes of unnatural amino acids (Bolognese et al. 2006; Sureshbabu et al. 2011; Longobardo et al. 2013].
Furthermore, N-protected β3-amino nitriles 5, with different amine Pgs, are substrates in biotransformation reactions to give the corresponding amides and/or amino acids catalyzed by nitrilases and nitrile hydratases (Liljeblad and Kanerva 2006; Veitía et al. 2009).
We therefore considered it timely to report a practical synthetic application of intermediates 5 in a novel approach to the simultaneous synthesis of syn and anti-dialkyl β2,3-amino acids (including deuterium labeled compounds) from α-amino acids.
Methods
Solvents, inorganic salts and organic reagents were purchased from commercial resources and used without further purification unless otherwise noted. All of the compounds for which analytical and spectroscopic data are quoted were homogeneous by TLC. TLC analyses were performed using silica gel plates (E. Merck silica gel 60 F-254) and components were visualized by the following methods: ultraviolet light absorbance, iodine adsorbed on silica gel, and ninhydrin spray. Melting points were measured with a Kofler apparatus and are uncorrected. Column chromatography was carried out on silica gel (E. Merck, 70–230 mesh). THF was dried over Na in presence of benzophenone under an Ar atmosphere. All the compounds were characterized by 1H and 13C NMR spectroscopy. NMR spectra were recorded using Varian Inova 500 and Bruker DRX-400 spectrometers: chemical shifts are in ppm and J coupling constants in Hz. High-resolution ES mass spectra were obtained with a Micromass Q-TOF UltimaTM API. Optical rotations were measured with a Jasco 1010 polarimeter (k = 589 nm). One suitable crystal was mounted at room temperature on a Bruker–Nonius Kappa-CCD diffractometer.
General procedure for Alkylation of N(Boc)-β3-amino nitriles
To a magnetically stirred solution of N(Boc)-β3-amino nitrile 5 (R1 = CH2Ph, 1.0 mmol) in anhydrous THF (5 mL), at −78 °C under an argon atmosphere, was added n-BuLi (1.6 M in hexane, 2.2 mmol) dropwise over a few minutes. After 15 min, a solution of benzyl bromide (1.5 mmol) in anhydrous THF (1 mL) was added in one portion, and the mixture was allowed to warm to room temperature over 1 h, before diluting with aq. 0.1 M HCl (10 mL) and extracting with EtOAc (3 × 20 mL). The combined organic layers were washed with water until neutral, and dried (Na2SO4). Evaporation of the solvents under reduced pressure afforded a crude residue consisting of two main products that were readily separated by column chromatography (silica gel, EtOAc-hexane, 1:9). The more mobile product (49 % yield) turned out to be N(Boc)-(2S,3S)-β2,3-dibenzylamino propionitrile (8, Fig. 2). m.p. = 127–128 °C (from Et2O), [α] 25D = −8.1 (c 0.5, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 1.48 (s, 9H, H-Boc), 2.84–3.05 (non resolvable m, 5H, H-2, H-4, H-5), 4.19 (m, 1H, H-3), 4.79 (d, 1H, J = 7.8 Hz, H-NBoc), 7.18-7.34 (m, 10H, H-Ar). 13C NMR (125 MHz, CDCl3) δ: 28.2 (×3), 35.8, 38.8, 40.6, 52.2, 80.2, 119.5, 127.0, 127.2, 128.7 (×4), 128.9 (×4), 136.3, 136.7, 155.3. Anal. Calcd for C22H26N2O2 (350.45): C, 75.40; H, 7.48; N, 7.99. Found: C, 75.37; H, 7.50; N, 8.03. HRMS (ESI), exact mass calcd. for [C22H27N2O2, 351.2067 (M + H)+], found 351.2075. Single crystal of 8 (from Et2O solution); data collection at room temperature (MoKα radiation, graphite monocromator, φ and ω scan mode). Crystal data: C22H26N2O2, M 350.45, hexagonal, P65, Z 6, a (Å) 11.454(5), b (Å) 11.454(5), c (Å) 27.594(4), V (Å3) 3135(2), γ 120.0°, ρc (g/cm3) 1.114, μ (mm−1) 0.071. Semiempirical absorption correction (sadabs) applied. No. of rflns 4817, collected up to θmax 27.52° [No. of independent rflns 2448, No. of rflns with I > 2σ(I) 1068]. The structure was solved by direct methods and anisotropically refined by full matrix least-squares method on F2 against all the independent measured reflections. H atoms were geometrically positioned, except for the carbamic H atom that was located in a difference Fourier map, and were refined in a riding model with Uiso thermal parameters equal to Ueq of the carrier atoms. The final R-factors for 238 refined parameters were R1 0.0717 [on reflections with I > 2σ(I)] and wR2 0.1120 on all the independent reflections. Max and min residual electron densities (eÅ−3) +0.117 and −0.120. The absolute configuration (S) at C2 was assigned by comparison with the chirality at C3, whose absolute configuration (S) is that of the starting l-Phe used for the synthesis of 8. All crystallographic data were deposited with the Cambridge Crystallographic Data Centre, CCDC No. 632994. The second reaction product (45 % yield) was N(Boc)-(2R,3S)-β2,3-dibenzylamino propionitrile (9):): m.p. = 132–133 °C (from Et2O); [α] 25D = −43.2 (c 0.7, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 1.38 (s, 9H, H-Boc), 2.88 (dd, 1H, J = 10.0 and 14.0 Hz, Ha-4), 2.94 (dd, 1H, J = 9.1 and 13.4 Hz, Ha-5), 2.97 (dd, 1H, J = 4.7 and 13.4 Hz, Hb-5), 3.18 (dd, 1H, J = 4.0 and 14.0 Hz, Hb-4), 3.27 (m, 1H, H-2), 4.05 (m, 1H, H-3), 4.60 (d, 1H, J = 7.8 Hz, H-NBoc), 7.21–7.39 (m, 10H, H-Ar). 13C NMR (125 MHz, CDCl3) δ: 28.2 (×3), 35.8, 37.4, 39.6, 51.2, 79.9, 119.9, 127.0, 127.4, 128.9 (×4), 129.2 (×4), 136.3, 136.6, 155.0. The NMR assignments were made using COSY and HSQC experiments. HRMS (ESI), exact mass calcd. for [C22H27N2O2, 351.2067 (M + H)+], found 351.2059. Under the same conditions, the enantiomer of N(Boc)-β3-amino nitrile 5 (R1 = CH2Ph) gave in order: compound 10 (48 %) and compound 11 (43 %). The NMR spectra of 10 and 11 were super imposable to those of compounds 8 and 9, respectively.

N(Boc)-β2,3-amino nitriles prepared from l-Phe, d-Phe and l-Val
Under the same conditions, from the N(Boc)-β3-amino nitrile 5 [R1 = CH(CH3)2] and benzyl bromide we obtained:
12. (2S,3S)-3-(tert-butoxycarbonylamino)-2-benzyl-4-methylpentanenitrile anti 12, 60 %: Amorphous solid; [α] 25D = +2° (c = 1.1, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 0.96 and 1.00 (d, 6H, J = 6.8 Hz, H-5 and H-6), 1.49 (s, 9H, H-Boc), 1.81 (m, 1H, H-4), 2.93–2.99 (m, 2H, H-7) 2.98-3.04 (m, 1H, H-2), 3.55–3.62 (m, 1H, H-3), 4.70 (d, 1H, J = 10.7 Hz, H-NBoc), 7.26–7.38 (m, 5H, H-Ph). 13C-NMR (125 MHz, CDCl3) δ: 19.1, 19.7, 28.3 (×3), 32.4, 36.5, 38.3, 56.8, 79.9, 119.7, 127.2, 128.7 (×2), 128.9 (×2), 137.1, 155.9. Anal. Calcd for C18H26N2O2 (302.41): C, 71.49; H, 8.67; N, 9.26. Found: C, 71.37; H, 8.50; N, 9.23. HRMS (ESI), exact mass calcd. for [C18H27N2O2, 303.2067 (M + H)+], found 303.2074.
13. (2R,3S)-3-(tert-butoxycarbonylamino)-2-benzyl-4-methylpentanenitrile syn 13, 30 %: m.p. = 102–103° C (Et2O); [α] 25D = −67° (c = 0.4, CHCl3,); 1H-NMR (CDCl3, 500 MHz) δ: 0.95 and 1.00 (d, 6H, J = 6.8 Hz, H-5 and H-6), 1.47 (s, 9H, H-Boc), 2.20–2.30 (m, 1H, H-4), 2.80–2.90 (m, 2H, H-7), 2.88–3.05 (m, 1H, H-2), 3.80–3.90 (m, 1H, H-3), 4.54 (d, J = 10.8, 1H, H-NBoc), 7.26–7.36 (m, 5H, H-Ph). 13C-NMR (125 MHz, CDCl3) δ: 15.5, 20.3, 28.3 (×3), 29.6, 35.4, 38.8, 55.6, 80.1, 119.9, 127.2, 128.8 (×2), 128.9 (×2), 136.9, 155.8. HRMS (ESI), exact mass calcd. for C18H27N2O2 303.2067 [(M + H)+], found 303.2052.
Under the same conditions, the alkylation of N(Boc)-β3-amino nitrile 5 (R1 = CH2Ph) with isopropyl iodide furnished:
14. (2S,3S)-3-(tert-butoxycarbonylamino)-2-isopropyl-4-phenylbutanenitrile anti 14, 51 %: m.p. = 91-93 °C (Et2O); [α] 25D = −12,6° (c = 0.2, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 0.97 and 1.09 (d, 6H, J = 6.8 Hz, H-6 and H-7), 1.40 (s, 9H, H-Boc), 1.88–2.00 (m, 1H, H-5), 2.25 (dd, 1H, J = 9.0 and 3.2 Hz, H-2), 2.81 (dd, 1H, J = 13.7 and 8.8 Hz, H4a), 3.03 (dd, 1H, J = 13.7 and 6.8 Hz, H-4b), 4.18–4.27 (m, 1H, H-3), 4.71 (d, 1H, J = 10.3 Hz, H-NBoc), 7.20–7.35 (m, 5H, H-Ph). 13C NMR (125 MHz, CDCl3) δ: 20.5, 20.9, 28.1(×3), 29.6, 41.1, 43.8, 50.4, 79.9, 119.3, 126.9, 128.7 (×2), 129.0 (×2), 136.6, 155.1. Anal. Calcd for C18H26N2O2 (302.41): C, 71.49; H, 8.67; N, 9.26. Found: C, 71.37; H, 8.50; N, 9.23. HRMS (ESI), exact mass calcd. for [C18H27N2O2, 303.2067 (M + H)+], found 303.2049.
15. (2R,3S)-3-(tert-butoxycarbonylamino)-2-isopropyl-4-phenylbutanenitrile syn 15, 36 %: m.p. = 113–115 °C (Et2O); [α] 25D = −19,7° (c = 0.5, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 1.10 and 1.16 (d, 6H, J = 6.8 Hz, H-6 and H-7), 1.35 (s, 9H, H-Boc), 1.90–2.04 (m, 1H, H-5), 2.65–2.70 (m, 1H, H-4a), 2.77–2.86 (m, 1H, H-4b), 3.13 (dd, J = 14.2 and 3.4 Hz, 1H, H-2), 4.12 (m, 1H, H-3), 4.46 (d, 1H, J = 8.8 Hz, H-NBoc), 7.20–7.36 (m, 5H, H-Ph). 13C NMR (125 MHz, CDCl3) δ: 19.7, 20.6, 28.1 (×3), 29.6, 37.4, 44.7, 50.3, 79.9, 119.6, 126.8, 128.6 (×2), 129.2 (×2), 136.4, 154.9. HRMS (ESI), exact mass calcd. for [C18H27N2O2, 303.2067 (M + H)+], found 303.2083.
Under the same conditions, from the di-deuterated enantiomer of N(Boc)-β3-amino nitrile 5 (R1 = CH2Ph) (Caputo and Longobardo 2007), alkylation with 4-iodochlorobutane provided:
16. (2R)-2-[(1R)-1-(tert-butoxycarbonylamino)-2-phenylethyl]-6-chloro(2-2H)hexanenitrile anti 16, 56 %: m.p = 75–77 °C; [α] 25D = −20.5° (c = 0.5, CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 1.41 (s, 9H, H-Boc), 1.58 (m, 4H, H-6 and H-7), 1.72 (m, 2H, H-5), 2.82 (dd, 1H, J = 13.7 and 8.8 Hz, H-4a), 3.02 (dd, 1H, J = 13.7 and 6.8, H-4b), 3.49 (t, 2H, J = 6.3, H-8), 4.09 (m, 1H, H-3), 4.68 (d, 1H, J = 10.3 Hz, H-NBoc), 7.20–7.38 (m, 5H, H-Ph). 13C-NMR (125 MHz, CDCl3) δ: 24.9, 28.5 (×3), 29.1, 32.1 (×2), 40.9, 44.5, 52.5, 80.4, 119.9, 127.3, 129.1 (×2), 129.2 (×2), 136.7, 155.3. Anal. Calcd for C19H26DClN2O2 (351.89): C, 64.85; H, 7.45; D, 0.57; Cl, 10.08; N, 7.96. Found: C, 64.67; H, 8.06; N, 7.88. HRMS (ESI), exact mass calcd. for [C19H27DClN2O2, 352.1896 (M + H)+], found 352.1883.
17. (2S)-2-[(1R)-1-(tert-butoxycarbonylamino)-2-phenylethyl]-6-chloro(2-2H)hexanenitrile syn 17, 28 %, m.p. = 83–85 °C; [α] 25D = −21.6° (c = 0.65 CHCl3); 1H-NMR (500 MHz, CDCl3) δ: 1.36 (s, 9H, H-Boc), 1.61 (m, 4H, H-6 and H-7), 1.81 (m, 2H, H-5), 2.80–2.90 (m, 1H, H-4a), 3.10 (dd, 1H, J = 13.7 and 6.8 Hz, H-4b), 3.54 (t, 2H, J = 6.3 Hz, H-8), 3.98–4.02 (m, 1H, H-3), 4.55 (d, 1H, J = 10.3 Hz, H-NBoc), 7.22–7.33 (m, 5H, H-Ph). 13C-NMR (125 MHz, CDCl3) δ: 24.9, 28.4 (×3), 28.5, 28.8, 32.1, 37.3, 44.5, 52.5, 80.4, 120.4, 127.2 (×2), 128.9 (×2), 129.4 136.5, 155.3. HRMS (ESI), exact mass calcd. for [C19H27DClN2O2, 352.1896 (M + H)+], found 352.1890.
18. (2R,3R)-Methyl 3-Amino-2-benzyl-4-phenylbutanoate hydrochloride, 81 %. Methanolysis of compound 10 in the conditions previously described (Caputo et al. 1995; Caputo and Longobardo 2007] yielded the hydrochloride salt (81 %) as colorless crystals: m.p. 205–207 °C from Et2O; [α] 25D = −21.6 (c 0.2, MeOH). HRMS (ESI), exact mass calcd. for [C18H22NO2, 284.1645 (M + H)+], found 284.1658. The 1H NMR and 13C NMR spectra matched those reported in the literature (Seki et al. 2000) m.p. 208–210 °C; [α] 25D = −22.5 (c 0.17, MeOH).
Results and discussion
LDA and metallated-HMDS which are commonly used to produce α-carbanions from N(Boc)-β3-amino esters, turned out to be completely ineffective toward N(Boc)-protected nitriles 5. We found that chiral N(Boc)-β3-amino nitriles are smoothly alkylated at the α-position. The reaction is not at all diastereoselective, hence leads to essentially equimolecular mixtures of syn and anti di-substituted β-amino nitriles. The diastereomers can be easily separated and individually converted into the corresponding N- and/or C-protected β2,3-amino acids. The alkylations were carried out at −78 °C in anhydrous THF, using 2.2 equivalents of n-BuLi per mole of the starting N(Boc)-β3-amino nitrile 5. Under such conditions the resulting lithium dianion is rapidly formed and precipitates as a white solid from THF. These dianions are quite stable below −65 °C, and rapid quenching with alkyl halides afforded almost equimolecular mixtures of both diastereomeric anti and syn N(Boc)-α,β-dialkyl β-amino nitriles (e.g. 6 and 7 in Scheme 2). One example of moderate selectivity (from 60:40 to 95:5 anti:syn) observed in the alkylation of β3-amino nitriles has been reported, but the starting nitriles were fully protected with two bulky benzyl groups at the amino nitrogen atom in that case, therefore cannot be directly compared to our work (Reetz et al. 1994).

Anti (6) and syn (7) N(Boc)-α,β-dialkyl β-amino nitriles from N(Boc)-β3-amino nitriles
Various N(Boc)-β2,3-amino nitriles with simple alkyl side chains were prepared under the same conditions. The overall yields of the alkylated products were higher than 80 % in all cases. Details of the synthesis of a series N(Boc)-β2,3-dialkyl amino nitriles derived from l-Phe, d-Phe and l-Val are shown in Fig. 2 and Table 1.
In particular, N(Boc)-β3-amino nitriles 5 (R1 = CH2Ph) was alkylated with benzyl bromide to afford N(Boc)-β2,3-dibenzylamino nitriles anti 8 and syn 9 in 94 % yield. The same alkylation, performed on ent-5, (R1 = CH2Ph from d-Phe) gave 91 % yield of anti 10 (ent-8) and syn 11 (ent-9). Alkylation of 5 with isopropyl iodide (R1 = CH2Ph) gave the corresponding C2-alkylated derivatives 14 and 15 in 87 % yield. Alkylation of 5 [R1 = CH(CH3)2 ] with benzyl bromide provide the N(Boc)-β2,3-dialkylamino nitriles anti 12 and syn 13 in 90 % yield, that are structural isomers of 14 and 15, which have the same side chains but installed on different C2 and C3 carbons.
Finally, a specially prepared C2-dideuterated N(Boc)-β3-alkyl amino nitrile 5, derived from d-Phe, was alkylated with 4-iodochlorobutane yielding diastereomers 16 and 17 (containing one deuterium atom) in 84 % yield. These derivatives contain a side chain useful for further synthetic elaboration, for example in the preparation of β2,3-amino acids with a lysine side chain (Langenhan and Gellman 2003).
In all the alkylations studied, single enantiomers were obtained in very good yields after a simple chromatographic separation, as shown in Table 1. The calculation of the dipole moment for compounds 8–17, using the module Chem3D Ultra of ChemDraw software, suggested that for each pair of diastereomers, the anti-stereoisomer was the least polar, and we found that they were eluted first from the silica gel chromatographic columns (EtOAc-Hex 1:9).
Two of the four N(Boc)-β2,3-dibenzylamino nitriles, namely 10 and 11 (prepared from d-Phe) could be readily converted into already known amino acids (Seki et al. 2000), which enabled the assignment of the absolute stereochemistry to the chiral centers of compounds 8–11. Nitrile 10 was converted by methanolysis (14 M HCl in MeOH, 0 °C to room temperature, 12 h) into the corresponding methyl ester hydrochloride 18 in 81 % yield (Scheme 3). The need for a high HCl concentration is necessary for both the transformation of the cyano group and the removal of the amino Pg. The remaining diastereomeric pair, N(Boc)-β2,3-amino nitriles 8 and 9 (prepared from l-Phe) were enantiomers of 10 and 11, respectively, and thus could be assigned the opposite configurations at the chiral center. All the stereochemical assignments were eventually confirmed by X-ray analysis of N(Boc)-β2,3-dibenzylamino nitrile 8 (Fig. 3). The conversions above, along with our existing experience, emphasize the reliability of N(Boc)-β2,3-amino nitriles as intermediates to afford N- and/or C-protected β2,3-amino acids efficiently, preserving the integrity of the chiral centers.

Methanolysis of nitrile 10 to the corresponding methyl ester hydrochloride 18

Structure of anti N(Boc)-β2,3-dibenzylamino nitrile 8 from X-ray structural analysis data
Conclusions
The procedure we report here is suitable for the simultaneous preparation of both syn and anti β2,3-amino acids from α-amino acids. The straightforward preparation of the starting chiral N(Boc)-protected β3-amino nitriles (including deuterium-labeled derivatives) from commercial N(Boc)-protected proteinogenic α-amino acids is a noteworthy feature of the whole synthetic scheme. The easy preparation of β2,3-amino acids in enantiomeric pure form, with natural or unnatural side chains in position 2 and with the option to introduce other substituents than alkyl groups in this position, starting from low-cost materials, represents a valued synthetic methodology which may find a wide number of applications in the chemistry of amino acids and peptides. In light of the present example, the already mentioned homologation of α-amino acids via β-amino iodides appears to be an effective alternative to the classical Arndt-Eistert homologation procedure, due to the possibility of isolating valuable intermediates, which can be exploited in novel amino acid syntheses.
Abbreviations
- Boc:
-
tert-butoxycarbonyl
- Cbz:
-
benzyloxycarbonyl
- Fmoc:
-
fluorenylmethyloxycarbonyl
- ImH:
-
imidazole
- HMDS:
-
hexamethyldisilylazane
- HMPA:
-
hexamethylphosphoramide
- LDA:
-
lithium diisopropylamide
- Moc:
-
methyloxycarbonyl
- NMM:
-
N-methylmorpholine
- Pg:
-
protecting group
- THF:
-
tetrahydrofuran
- TPP:
-
triphenylphosphine
References
Balamurugan D, Muraleedharan KM (2015) Can helical peptides unwind one turn at a time?—controlled conformational transitions in α, β2,3-hybrid peptides. Chem Eur J 21(26):9332–9338
Bolognese A, Fierro O, Guarino D, Longobardo L, Caputo R (2006) One-pot synthesis of orthogonally protected enantiopure S-(Aminoalkyl)-cysteine derivatives. Eur J Org Chem 1:169–173
Burgess K, Liu LT, Pal B (1993) Asymmetric syntheses of optically active α, β-disubstituted β-amino acids. J Org Chem 58(17):4158–4763
Capone S, Pedatella S, Guaragna A, De Nisco M, Palumbo G (2007) Highly diastereoselective preparation of anti-α, β-dialkyl β-amino acids containing natural α-amino acid side chains. Tetrahedron 63(49):12202–12206
Caputo R, Longobardo L (2007) Enantiopure β3-amino acids-2,2-d2 via homologation of proteinogenic α-amino acids. Amino Acids 32(3):401–404
Caputo R, Cassano E, Longobardo L, Palumbo G (1995) Synthesis of enantiopure N-and C-protected homo-β-Amino acids by direct homologation of α-amino acids. Tetrahedron 51(45):12337–12350
Cardillo G, Tolomelli A, Tomasini C (1996) Enzymatic resolution of α-alkyl β-amino acids using immobilized penicillin G acylase. J Org Chem 61(24):8651–8654
Cheng R, Gellman SH, DeGrado WF (2001) β-Peptides: from structure to function. Chem Rev 101(10):3219–3232
Cole DC (1994) Recent stereoselective synthetic approaches to β-amino acids. Tetrahedron 50(32):9517–9582
Davies SG, Walters IAS (1994) Asymmetric synthesis of anti-α-alkyl-β-amino Acids. J Chem Soc Perkin Trans I 9:1129–1139
Estermann H, Seebach D (1988) Diastereoselective alkylations of 3-amino butanoic acid in the 2-position. Helv Chim Acta 71(7):1824–1839
Juaristi E, Soloshonok V (eds) (2005) Enantioselective synthesis of β-amino acids, 2nd edn. Wiley-Interscience, New York
Kiss L, Cherepanova M, Fülöp F (2015) Recent advances in the stereoselective syntheses of acyclic disubstituted β2,3-amino acids. Tetrahedron 71(14):2049–2069
Langenhan JM, Gellman SH (2003) Preparation of protected syn-α, β-dialkyl β-amino acids that contain polar side chain functionality. J Org Chem 68(16):6440–6443
Liljeblad A, Kanerva LT (2006) Biocatalysis as a profound tool in the preparation of highly enantiopure β-amino acids. Tetrahedron 62(25):5831–5854
Liu M, Sibi MP (2002) Recent advances in the stereoselective synthesis of β-amino acids. Tetrahedron 58(40):7991–8035
Longobardo L, Cecere N, DellaGreca M, de Paola I (2013) Novel thiol- and thioether-containing amino acids: cystathionine and homocysteine families. Amino Acids 44(2):443–448
Martinek TA, Fülöp F (2012) Peptidic foldamers: ramping up diversity. Chem Soc Rev 41(2):687–702
Podlech J, Seebach D (1995) On the preparation of β-amino acids from α-amino acids using the Arndt-Eistert reaction: scope, limitations and stereoselectivity. Application to carbohydrate peptidation. Stereoselective α-alkylations of some β-amino acids. Liebigs Annalen 7:1217–1228
Reetz MT, Kayser F, Harms K (1994) Stereoselective synthesis of β-amino nitriles and 1,3-diamines. Tetrahedron Lett 35(47):8769–8772
Seebach D, Abele S, Gademann K, Jaun B (1999) Pleated sheets and turns of β-peptides with proteinogenic side chains. Angew Chem Int Ed Engl 38(11):1595–1597
Seebach D, Beck AK, Bierbaum DJ (2004) The world of β- and γ-peptides comprised of homologated proteinogenic amino acids and other components. Chem Biodiv 1(8):1111–1239
Seki M, Shimizu T, Matsumoto K (2000) Stereoselective synthesis of β-benzyl-α-alkyl-β-amino acids from L-aspartic acid. J Org Chem 65(5):1298–1304
Sureshbabu VV, Vasantha B, Hemantha HP (2011) Synthesis of N-Fmoc-protected amino alkyl thiocyanates/selenocyanates and their application in the preparation of 5-substitutedS/Se-linked tetrazoles. Synthesis 9:1447–1455
Veitía MSI, Brun PL, Jorda P, Falguières A, Ferroud C (2009) Synthesis of novel N-protected β3-amino nitriles: study of their hydrolysis involving a nitrilase-catalyzed step. Tetrahedron Asymmetry 20(18):2077–2089
Weiner B, Szymanski W, Janssen DB, Minnaard AJ, Feringa BL (2010) Recent advances in the catalytic asymmetric synthesis of β-amino acids. Chem Soc Rev 39(5):1656–1691
Yang H, Foster K, Stephenson CRJ, Brown W, Roberts E (2000) Asymmetric wolff rearrangement reactions with α-Alkylated-α-diazoketones: stereoselective synthesis of α-substituted-β-amino acid derivatives. Org Lett 2(14):2177–2179
Yu S, Ishida H, Juarez-Garcia ME, Bode JW (2010) Unified synthesis of enantiopure β2h, β3h and β2,3-amino acids. Chem Sci 1:637–641
Authors’ contributions
LL design and performed the series of experiments and wrote the manuscript. MDG performed the NMR analysis, including 2D experiments. IdP performed the series of experiments, including purification and characterization of novel molecules. All authors read and approved the final manuscript.
Acknowledgements
The authors are gratefully indebted with Prof. A. Tuzi for her skillful help with X-ray analysis.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Longobardo, L., DellaGreca, M. & de Paola, I. A practical route to β2,3-amino acids with alkyl side chains. SpringerPlus 4, 553 (2015). https://doi.org/10.1186/s40064-015-1351-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-015-1351-6