
Abstract
Background
Beta-blockade is contraindicated in severe aortic regurgitation (AR) due to the fear of prolonging diastole and thus aggravate regurgitation. However, this has never been scientifically proven and positive effects of targeting the sympathetic system in AR has been demonstrated in several studies.
Method
Thirty-nine Sprague–Dawley rats with AR were randomized to ten weeks of medical treatment with carvedilol or no treatment. Treatment was initiated either early or late after AR induction. The effect of carvedilol was assessed by serial echocardiography and invasive hemodynamic measurements.
Results
AR resulted in eccentric hypertrophy and left ventricular (LV) dysfunction. LV remodeling and function as measured by echocardiography was unaffected by treatment. LV dimensions were similar between treated and untreated groups and measures of LV performance (including strain and strain rate) were also unaltered. This result was confirmed by invasive measurements showing maximal and minimal pressure–time development, LV volumes, and LV pressures, to be unaltered by treatment. On the contrary, despite relative bradycardia carvedilol did not reflect any negative impact on the heart.
Conclusion
Carvedilol did not improve left ventricular remodeling or function in rats with surgically induced AR. Despite relative bradycardia, we did not find carvedilol to negatively impact the heart, either when treatment was initiated early or late in the course of disease.
Similar content being viewed by others

Background
Chronic aortic regurgitation (AR) is a common heart valve disease with high morbidity and mortality rates once symptoms occur (Dujardin et al. 1999). However, disease–progression is often gradual and patients with even severe degree of regurgitation may be asymptomatic for decades without any need of surgical intervention (Bonow et al. 1991; Goldbarg and Halperin 2008). Ultimately, left ventricular (LV) compensatory mechanisms are exhausted, which results in LV dilation and dysfunction and consequently heart failure and eventually death (Maurer 2006). The time of surgical intervention (aortic valve replacement/repair (AVR)) relies on when patients develop symptoms or when significant alterations in LV size or function occurs (Bonow et al. 2008; Vahanian et al. 2012). Medical treatment of AR has been debated for decades and results from clinical studies have been equivocal (Levine and Gaasch 1996; Evangelista et al. 2005; Lin and Stewart 2011). Thus, medical management of AR is not routinely recommended.
Long–term cardiac overload, as seen in congestive heart failure, leads to over–activation of the sympathetic and renin–angiotensin–aldosterone system (Schrier and Abraham 1999). This results in various deleterious effects (Levine et al. 1982; Bristow 1984) and reversal of this state has demonstrated beneficial effects on the heart (Packer et al. 1996). Beta-blockers are thus first-line treatment in congestive heart failure (Hunt et al. 2009; McMurray et al. 2012). The use of beta-blockers in AR is however controversial, mostly due to the risk of prolonging diastole, which could aggravate regurgitation. Only a few studies have addressed the effects of beta–blockade in volume overload, but the rationale for targeting the sympathetic system in AR has been shown by Champetier et al. (Champetier et al. 2009). They found several pathophysiological similarities between hearts from rats with AR and hearts exposed to other types of cardiac disease. Furthermore, in a study by Plante et al. (Plante et al. 2004) it was shown that treating AR for 24 weeks with metoprolol had a global impact on the heart, resulting in improved LV function and reduction in LV remodeling. Additionally, it has been shown in experimental and clinical studies that beta-blockers reduce mortality in chronic AR (Plante et al. 2008; Sampat et al. 2009). However, experimental studies testing the effects of beta–blockade in AR have only tested the effects of treatment started early in the course of disease. In a clinical setting, patients may have AR for years before the condition is discovered, and at this time–point LV changes may be too advanced to exploit the positive effects of beta–blockade. Thus starting treatment late may induce an increase in regurgitation–volume that could disturb the balance between load and compensatory mechanisms.
We therefore undertook this study to investigate the effects of beta-blockade in a setting reproducing a clinical situation where treatment is initiated at different time points along the course of disease.
Results
Clinical data
Five rats died during the study–period; one from the AR (early) group, two from the AR + CAR (early) group, one from the AR (late) group, and one from the AR + CAR (late) group. All rats dying were found dead in the morning with no preceding signs of heart failure. At sacrifice heart weight in sham (early) operated rats was significantly lower than in rats with AR (early), while no significant difference was found between AR and AR + CAR in either the early or the late treated groups, Table 1. Furthermore we did not find any differences in lung tissue weights among groups.
Echocardiographic data
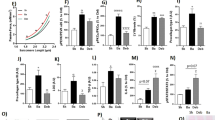
At baseline no echocardiographic differences were observed between groups. Rats with AR developed eccentric hypertrophy with increased LV dimensions (LVEDD and LVESD) and wall thickness, Table 2. Additionally, AR resulted in LV dysfunction. Carvedilol had no effect on this, and echocardiographic measures of LV remodelling was unaffected by treatment in both the early and late treated group, Figure 1. Also the rate of LV systolic dysfunction–development measured by conventional or speckle-tracking echocardiography was unaltered by treatment in both groups, Figure 2.

LV dimensions. LV dimensions measured by serial echocardiography in sham-operated (sham), treated (Carvedilol), and untreated (AR) in the early (left) and late (right) treated groups.

LV function. LV function measured by different echocardiographic techniques in sham-operated (sham), treated (Carvedilol), and untreated (AR) in the early (left) and late (right) treated groups. Strain rate (SR).
Thus, we did not find carvedilol to alter the development of eccentric remodeling or LV dysfunction as measured by echocardiography, regardless of treatment–start.
Hemodynamic measurements
Invasive hemodynamic measurements were performed at baseline and repeated immediately before sacrifice. Although only borderline significant in the late treated group we observed a lower heart rate in carvedilol–treated compared to untreated rats.
Carvedilol did not result in any significant improvement in LV maximum or minimum pressure–time development (dp/dtmax and dp/dtmin) or LV volumes (relative end diastolic or end systolic volume) compared to untreated rats. Furthermore, end–diastolic and end–systolic LV pressures were unaltered by treatment in both study-legs, Table 3. We found tau to be lower in treated compared to untreated rats in the late treated group, while other measures of LV diastolic function was unchanged by treatment. It may reflect less ECM disturbance in the treated group, but this is an isolated finding and should be interpreted in the scope of a low sample size.
Overall, we did not observe carvedilol to consistently or significantly change LV hemodynamic conditions, either when treatment was started early or late in the course of disease.
Discussion
In this study of rats with surgically induced AR we demonstrate the cardiac effects of carvedilol when treatment is initiated early and late in the course of disease. Rats with AR clearly developed eccentric remodeling. Treatment with carvedilol did not change this. Thus, we found LV–function and remodeling, as measured by echocardiography and invasive hemodynamic catheterization to be unaltered by ten weeks of carvedilol treatment. Carvedilol induced relative bradycardia without revealing negative impact on the heart.
In a clinical setting AVR is indicated in patients with chronic severe AR once symptoms or significant alterations in LV size or function occurs (Vahanian et al. 2007; Bonow et al. 2008). AVR is an effective treatment with good long-term prognosis if performed at the correct time (Klodas et al. 1997). However, open chest surgery and anticoagulant treatment carries a considerable peri–operative risk. Consequently, medical therapy to alleviate LV load and ultimately postpone the time of surgery has long been sought (Lin and Stewart 2011).
Blockade of the sympathetic system
In heart failure it is known that activation of neurohormonal systems exerts deleterious effects on the heart (Bristow 1984). Additionally, when chronically increased, high concentrations of circulating catecholamines are cardiotoxic and the extra inotropic and chronotropic drive results in excessive oxygen expenditure (Mann et al. 1992). Blocking this over–activation can reverse these negative effects. Nevertheless, due to the risk of bradycardia and the following increase in regurgitation and afterload, beta-blockers are contraindicated in severe AR. This relies on little scientific proof and the time is right to investigate this further. Recently it was shown that sustained volume overload activate neurohormonal systems, similar to other types of cardiac disease, and genetic studies has proven beta–blockade to reverse up–regulated genes related to this over–activation (Suzuki et al. 1997; Champetier et al. 2009; Zendaoui et al. 2011). In our study the aim of treatment was to only bluntly inhibit the sympathetic system, as to reduce the risk of negative consequences on the heart while still exploiting the possible positive biological properties of carvedilol. This was achieved by a small dose of carvedilol to only mildly reduced HR. In previous studies of AR a similar small dose of beta-blockers has been used, but has mainly investigating the effects of early-started treatment (Suzuki et al. 1997; Plante et al. 2004; Zendaoui et al. 2011). Treatment was well tolerated in both (early and late started) groups and we did not observe any reflections of negative consequences on hemodynamic and morphometric measurements, as would be expected if regurgitation was significantly increased (e.g. cardiac incompensation).
LV remodeling and function
Severe AR is initially characterized by a long compensated phase, where hemodynamic consequences of sustained volume overload causes an adaptive increase in LV volumes without reduction in LV performance. Eccentric hypertrophy develops to compensate the initial overload–burden, but once compensatory mechanisms are exhausted, more ventricular load results in further remodeling and increased afterload. In this process, loss of elastin and alterations in collagen fiber organization results in decreased LV compliance and increased stiffness. Gradually filling pressures and cardiac work increases that ultimately result in decreased cardiac performance.
Similar to the natural history in man, we observed AR rats to develop LV remodeling. This process started early after AR-induction and continued throughout the course of disease. This confirms the findings of Plante et al. (Champetier et al. 2009) who found in a model of experimental AR that sarcomeric alpha–actin and smooth muscle alpha–actin protein were significantly increased along with increased levels of mRNA for several ECM component, including collagen I and III. This was reflected on LV dilation and increased relative wall thickness measured by echocardiography. In their study early started metoprolol reduced LV end systolic diameter but did not change wall thickness. This was explained by a change in several ECM components rather than reversal of myocyte proteins. Similarly, in a study by Zendaoui et al. (Zendaoui et al. 2011), a high–dose beta–blocker initiated early has shown comparable effects. Thus, they found six months of treatment to reduce end–systolic diameter and expression of mRNA for collagen I, while echocardiographic measures of hypertrophy was unaffected. We found a similar result of unchanged hypertrophy in both early and late treated groups, although we found no difference in LV diameters. This could be explained by differences in AR severity and heart rate response amongst studies but more likely due to long-term versus short-term treatment regimes. As an isolated result we found (tau) to be decreased in the late treated group, which could reflect less ECM disturbance. In conclusion these results indicate that beta–blockade may positively influence LV dimensions but has not shown to be able to consistently reduce myocyte hypertrophy.
We did not find any difference in positive or negative dp/dt and LV filling pressures were similar between sham–operated and rats with AR in both study–legs. Treatment did not affect this. These results partly confirm the finding of others.
Previous studies have shown inconsistent effects on LV performance. In one study, long–term treatment with metoprolol did not change pressure development or LVEF after 6 and 12 months (Plante et al. 2008), while another study showed dp/dtmin to be significantly increased by disease and dp/dtmax to be unaffected, while high–dose carvedilol did not change this. In the same study LVEF measured by echocardiography decreased marginally in rats with AR (LVEF 77 ± 0.8% in sham vs. 66 ± 0.5% in AR) and carvedilol slightly reversed this mild decrease (71 ± 1.4% in treated vs. 66 ± 0.5% in untreated rats). In another study invasive hemodynamic measurements were not performed but echocardiography showed improved systolic and diastolic function in rats treated early with metoprolol for twenty–four weeks. However, in this study LV systolic function was also only marginally affected by treatment. We did not find carvedilol to improve LV systolic function. In the studies showing positive effects systolic function, LVEF was within the limits of reference and did not decrease significantly during the course of disease. This may reflect a more benign type of AR where six months of severe volume overload does not impair systolic function. Our model somehow resulted in a reduction in LV function measured by FS and speckle tracking echocardiography and may represent a more progressive type of disease less susceptible to the positive effects of beta–blockade.
Although we were unable to show carvedilol to improve LV remodeling and performance, regardless of treatment start, we did not find beta–blokade to negatively impact the heart.
Limitations
For several reasons (e.g. great biological differences) animal studies cannot and should never be extrapolated to humans.
Using a higher dose of carvedilol or a different beta-blocker (e.g. a selective) may show more pronounced results. However, we sought to only bluntly inhibit the sympathetic system in fear of increasing regurgitation. The dose of carvedilol chosen resulted in a heart rate drop of 5-10% compared to non-treated AR groups. This is similar to what has previously shown significant effects on the heart on other experimental studies. Although selective and non-selective beta-blockers have pharmacological actions in common they differ greatly, and cannot be directly compared. Thus, we interpret our results in the scope hereof.
Due to a small sample size it is hard to conclude definitively on our results and thus we suggest a larger scale study to investigated this interesting field. However, we believe that we have interpreted our findings accordingly.
Conclusion
Carvedilol did not improve left ventricular remodeling or performance in rats with surgically induced severe AR. Despite relative bradycardia, we did not find carvedilol to negatively impact the heart, either when treatment was initiated early or late in the course of disease.
Methods
Animal model
AR was created in rats by echocardiography–guided closed–chest operation, as described elsewhere (Arsenault et al. 2002). Briefly, two aortic valve leaflets were punctured in a retrograde manner by a right–sided carotid arteriotomy. Isoflurane (1.5-2.0% mixed with oxygen) sedation was followed by an intra peritoneal cocktail (ketamine 90 mg kg−1 and Xylazine 10 mg kg−1) injection. AR perforation and severity was confirmed by echocardiography during the procedure. Treatment was performed by mixing carvedilol (10 mg/kg/d) in transgenic dough–diet (Bioserv: Frenchtown, NJ, U.S.A.), which was fed to rats daily for the duration of medical treatment. All animal experiments were approved by the Johns Hopkins University Institutional Animal Care and Use Committee.
Early treatment protocol
The twenty-five male Sprague Dawley rats (age 19–20 weeks: Charles River: Wilmington, MA, U.S.A.) included in this part of the study were divided into three groups: Sham–operated rats receiving no treatment (Sham (early), n = 8), rats with AR receiving no medical treatment (AR (early), n = 8), and rats treated with carvedilol beginning two weeks after AR induction (AR + CAR (early), n = 9). In sham–operated rats all procedures were performed, except perforation of aortic valves. All rats were sacrificed twelve weeks after induction of AR.
Late treatment protocol
Fourteen male Sprague Dawley rats (age 9–10 weeks) were subjected to severe AR and divided into two groups: Controls with severe AR receiving no medical treatment (AR (late), n = 6) and rats with severe AR treated with carvedilol beginning at twelve weeks after induction of AR (AR + CAR (late), n = 8). All rats were sacrificed twenty–two weeks after induction of AR.
Echocardiography
Echocardiography was performed on a Vivid 7 machine (GE Healthcare: Horton, Norway) with a 14 MHZ linear vascular probe in unconscious rats anesthetized with gas (Isoflurane 1.5–2.0%). Examinations were performed at following time points: Early study: 0 (baseline), 2, 6, 9 and 12 weeks after AR induction. Late study: 0 (baseline), 2, 6, 9, 10, 12, 16, 19, 22 weeks after AR induction. Regurgitation was confirmed by a color-Doppler ratio of regurgitant jet >50% of the LV outflow tract diameter. LV measurements were assessed by conventional M-mode echocardiography; end diastolic diameter (LVEDD), end systolic diameter (LVESD), shortening fraction (FS) and wall thickness (WT). Measurements were averaged from four beats, two parasternal long–axis (PLAX) and two short–axis (SAX) planes. FS was calculated as [LVEDD–LVESD]/ [LVEDD]. As a measure of WT, septum and LV posterior wall were averaged. Speckle tracking was performed by acquiring 2D cine loops from at least six cardiac cycles, in mid–ventricular SAX plane at frame rates >70 s−1. Tracking was performed by using at least three consecutive cardiac cycles in EchoPAC’s strain modality analysis software (v. 7.0, GE Healthcare, Waukesha, WI, U.S.A.). Averaged circumferential deformation represented by the global trace was used. For strain–rate measurements the global traces were used.
Hemodynamic measurements
A micro–tip conductance pressure–volume catheter (Millar Instruments, Inc.: Houston, TX, U.S.A.) was inserted into the heart by right–sided carotid arteriotomy using a classic closed–chest approach (Pacher et al. 2008). Pressure-volume (PV) loops were obtained under resting conditions. Measurements were performed at baseline and immediately before sacrifice.
Statistical analysis
By comparing sham and untreated rats the overload caused by AR was documented. The effect of medical treatment was documented by comparing untreated AR-rats with carvedilol-treated AR-rats. The difference between multiple groups was analyzed with ANOVA and Tukey’s test for multiple comparisons. Serial measurements were compared by repeated measures. All p–values are two–tailed and a significance level of 0.05 was used. Statistics are given as mean ± SEM, unless stated otherwise. All analysis were performed using SAS® software (SAS for windows, release 9.1, SAS Institute Inc., Cary, NC, U.S.A.) and PASW statistics 18.0 (SPSS IBM for Macintosh, release 18.0.3, Armonk, USA).
Abbreviations
- AR:
-
Aortic regurgitation
- AVR:
-
Aortic valve replacement/repair
- DBP:
-
Diastolic blood pressure
- ECM:
-
Extra cellular matrix
- EDD:
-
End diastolic diameter
- EDP:
-
End-diastolic pressure
- EDV:
-
End-diastolic volume
- ESD:
-
End systolic diameter
- ESP:
-
End-systolic pressure
- ESV:
-
End-systolic volume
- HR:
-
Heart rate
- LV:
-
Left ventricle
- PLAX:
-
Parasternal long–axis
- PV:
-
Pressure-volume
- dp/dt:
-
Pressure-volume time development
- RV:
-
Right ventricle
- SAX:
-
Short–axis
- FS:
-
Shortening fraction
- SR:
-
Strain rate
- SBP:
-
Systolic blood pressure
- WT:
-
Wall thickness
References
Arsenault M, Plante E, Drolet M-C, Couet J (2002) Experimental aortic regurgitation in rats under echocardiographic guidance. J Heart Valve Dis 11:128–134
Bonow RO, Lakatos E, Maron BJ, Epstein SE (1991) Serial long-term assessment of the natural history of asymptomatic patients with chronic aortic regurgitation and normal left ventricular systolic function. Circulation 84:1625–1635, doi:10.1161/01.CIR.84.4.1625
Bonow RO, Carabello BA, Chatterjee K, de Leon AC Jr, Faxon DP, Freed MD, Gaasch WH, Lytle BW, Nishimura RA, O’Gara PT, O’Rourke RA, Otto CM, Shah PM, Shanewise JS, American College of Cardiology/American Heart Association Task Force on Practice Guidelines (2008) 2008 focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to revise the 1998 guidelines for the management of patients with valvular heart disease). Endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol 52:e1–e142, doi:10.1016/j.jacc.2008.05.007
Bristow MR (1984) The adrenergic nervous system in heart failure. N Engl J Med 311:850–851, doi:10.1056/NEJM198409273111310
Champetier S, Bojmehrani A, Beaudoin J, Lachance D, Plante E, Roussel E, Couet J, Arsenault M (2009) Gene profiling of left ventricle eccentric hypertrophy in aortic regurgitation in rats: rationale for targeting the β-adrenergic and renin-angiotensin systems. Am J Physiol Heart Circ Physiol 296:H669–H677, doi:10.1152/ajpheart.01046.2008
Dujardin KS, Enriquez-Sarano M, Schaff HV, Bailey KR, Seward JB, Tajik AJ (1999) Mortality and morbidity of aortic regurgitation in clinical practice. A long-term follow-up study. Circulation 99:1851–1857
Evangelista A, Tornos P, Sambola A, Permanyer-Miralda G, Soler-Soler J (2005) Long-term vasodilator therapy in patients with severe aortic regurgitation. N Engl J Med 353:1342–1349, doi:10.1056/NEJMoa050666
Goldbarg SH, Halperin JL (2008) Aortic regurgitation: disease progression and management. Nat Clin Pract Cardiovasc Med 5:269–279, doi:10.1038/ncpcardio1179
Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW, Yancy CW, American College of Cardiology Foundation; American Heart Association (2009) 2009 Focused Update Incorporated Into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the International Society for Heart and Lung Transplantation. J Am Coll Cardiol 53:e1–e90, doi:10.1016/j.jacc.2008.11.013
Klodas E, Enriquez-Sarano M, Tajik AJ, Mullany CJ, Bailey KR, Seward JB (1997) Optimizing timing of surgical correction in patients with severe aortic regurgitation: role of symptoms. J Am Coll Cardiol 30:746–752, doi:10.1016/S0735-1097(97)00205-2
Levine HJ, Gaasch WH (1996) Vasoactive drugs in chronic regurgitant lesions of the mitral and aortic valves. J Am Coll Cardiol 28:1083–1091, doi:10.1016/S0735-1097(96)00288-4
Levine TB, Francis GS, Goldsmith SR, Simon AB, Cohn JN (1982) Activity of the sympathetic nervous system and renin-angiotensin system assessed by plasma hormone levels and their relation to hemodynamic abnormalities in congestive heart failure. Am J Cardiol 49:1659–1666
Lin A, Stewart R (2011) Medical treatment of asymptomatic chronic aortic regurgitation. Expert Rev Cardiovasc Ther 9:1249–1254, doi:10.1586/erc.11.97
Mann DL, Kent RL, Parsons B, Cooper G (1992) Adrenergic effects on the biology of the adult mammalian cardiocyte. Circulation 85:790–804, doi:10.1161/01.CIR.85.2.790
Maurer G (2006) Aortic regurgitation. Heart 92:994–1000, doi:10.1136/hrt.2004.042614
McMurray JJ, Adamopoulos S, Anker SD, Auricchio A, Böhm M, Dickstein K, Falk V, Filippatos G, Fonseca C, Gomez-Sanchez MA, Jaarsma T, Køber L, Lip GY, Maggioni AP, Parkhomenko A, Pieske BM, Popescu BA, Rønnevik PK, Rutten FH, Schwitter J, Seferovic P, Stepinska J, Trindade PT, Voors AA, Zannad F, Zeiher A, Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology, Bax JJ, Baumgartner H, Ceconi C et al (2012) ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012 The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 14:803–869, doi:10.1093/eurjhf/hfs105
Pacher P, Nagayama T, Mukhopadhyay P, Bátkai S, Kass DA (2008) Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat Protoc 3:1422–1434, doi:10.1038/nprot.2008.138
Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM, Shusterman NH (1996) The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N Engl J Med 334:1349–1355, doi:10.1056/NEJM199605233342101
Plante E, Lachance D, Gaudreau M, Drolet MC, Roussel E, Arsenault M, Couet J (2004) Effectiveness of beta-blockade in experimental chronic aortic regurgitation. Circulation 110:1477–1483, doi:10.1161/01.CIR.0000141733.55236.9D
Plante E, Lachance D, Champetier S, Drolet MC, Roussel E, Arsenault M, Couet J (2008) Benefits of long-term beta-blockade in experimental chronic aortic regurgitation. Am J Physiol Heart Circ Physiol 294:H1888–H1895, doi:10.1152/ajpheart.01286.2007
Sampat U, Varadarajan P, Turk R, Kamath A, Khandhar S, Pai RG (2009) Effect of beta-blocker therapy on survival in patients with severe aortic regurgitation results from a cohort of 756 patients. J Am Coll Cardiol 54:452–457, doi:10.1016/j.jacc.2009.02.077
Schrier RW, Abraham WT (1999) Hormones and hemodynamics in heart failure. N Engl J Med 341:577–585, doi:10.1056/NEJM199908193410806
Suzuki M, Yoshikawa T, Wainai Y, Nagami K, Handa S (1997) Beta-blockade prevents ventricular failure following aortic regurgitation in rabbits. Cardiology 88:418–424
Vahanian A, Baumgartner H, Bax J, Butchart E, Dion R, Filippatos G, Flachskampf F, Hall R, Iung B, Kasprzak J, Nataf P, Tornos P, Torracca L, Wenink A, Task Force on the Management of Valvular Hearth Disease of the European Society of Cardiology, ESC Committee for Practice Guidelines (2007) Guidelines on the management of valvular heart disease: the task force on the management of valvular heart disease of the European society of cardiology. Eur Heart J 28:230–268, doi:10.1093/eurheartj/ehl428
Vahanian A, Alfieri O, Andreotti F, Antunes MJ, Barón-Esquivias G, Baumgartner H, Borger MA, Carrel TP, De Bonis M, Evangelista A, Falk V, Lung B, Lancellotti P, Pierard L, Price S, Schäfers HJ, Schuler G, Stepinska J, Swedberg K, Takkenberg J, Von Oppell UO, Windecker S, Zamorano JL, Zembala M, ESC Committee for Practice Guidelines (CPG), Joint Task Force on the Management of Valvular Heart Disease of the European Society of Cardiology (ESC), European Association for Cardio-Thoracic Surgery (EACTS) (2012) Guidelines on the management of valvular heart disease (version 2012) The Joint Task Force on the Management of Valvular Heart Disease of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS). Eur Heart J 33:2451–2496, doi:10.1093/eurheartj/ehs109
Zendaoui A, Lachance D, Roussel E, Couet J, Arsenault M (2011) Usefulness of carvedilol in the treatment of chronic aortic valve regurgitation. Circ Heart Fail 4:207–213, doi:10.1161/CIRCHEARTFAILURE.110.958512
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KE has made substantial contributions to conception and design, acquisition of data, and analysis and interpretation of data. KE drafted the manuscript. NTO has made substantial contributions to conception, design, and analysis and interpretation of data. NTO has been involved in drafting and revising the manuscript critically for important intellectual content; VLD has made substantial contributions to analysis and interpretation of data. TFH has been involved in revising the manuscript critically for important intellectual content. PS has been involved in revising the manuscript critically for important intellectual content. TPA has made substantial contributions to analysis and interpretation of data. TPA has been involved in revising the manuscript critically for important intellectual content. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eskesen, K., Olsen, N.T., Dimaano, V.L. et al. Effects of early and late-onset treatment with carvedilol in an experimental model of aortic regurgitation. SpringerPlus 4, 52 (2015). https://doi.org/10.1186/s40064-015-0829-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-015-0829-6