
Abstract
A growing amount of evidence has indicated contributions of variants in causative genes of Parkinson’s disease (PD) to the development of sleep disturbance in PD and prodromal PD stages. In this article, we aimed to investigate the role of genetics in sleep disorders in PD patients and asymptomatic carriers at prodromal stage of PD. A systematic review and meta-analysis of observational studies was conducted based on the MEDLINE, EMBASE and PsychINFO databases. A pooled effect size was calculated by odds ratio (OR) and standard mean difference (SMD). Forty studies were selected for quantitative analysis, including 17 studies on glucocerebrosidase (GBA), 25 studies on Leucine-rich repeat kinase 2 (LRRK2) and 7 on parkin (PRKN) genes, and 3 studies on alpha-synuclein gene (SNCA) were used for qualitative analysis. Patients with PD carrying GBA variants had a significantly higher risk for rapid-eye-movement behavior disorders (RBD) (OR, 1.82) and higher RBD Screening Questionnaire scores (SMD, 0.33). Asymptomatic carriers of GBA variants had higher severity of RBD during follow-up. Patients with PD carrying the LRRK2 G2019S variant had lower risk and severity of RBD compared with those without LRRK2 G2019S. Variants of GBA, LRRK2 and PRKN did not increase or decrease the risk and severity of excessive daytime sleepiness and restless legs syndrome in PD. Our findings suggest that the genetic heterogeneity plays a role in the development of sleep disorders, mainly RBD, in PD and the prodromal stage of PD.
Similar content being viewed by others


Background
Parkinson’s disease (PD) is one of the most common neurodegenerative disorders, manifesting mainly as bradykinesia, resting tremor and various non-motor symptoms. Increasing evidence has witnessed that the non-motor symptoms have a significant impact on the quality of life of patients with PD. The non-motor symptoms can occur either after motor onset or before motor symptoms at a prodromal stage [1]. Sleep disorders are common non-motor symptoms, including excessive daytime sleepiness (EDS), insomnia, restless legs syndrome (RLS), circadian rhythm disorders, sleep attacks, obstructive sleep apnea and rapid-eye-movement behavior disorders (RBD). In addition, some sleep disorders such as RBD are considered as a prodrome of α-synucleinopathies [2], suggesting that sleep disorders are closely related to the pathophysiology of PD.
Currently, multiple variants of genes, such as alpha-synuclein gene (SNCA), leucine-rich repeat kinase 2 gene (LRRK2), glucocerebrosidase gene (GBA) and parkin gene (PRKN), have been reported to be causes of PD. Different variants in such causative genes also play a role in the discrepancies in clinical manifestations and prognosis of PD. As one of the most common non-motor symptoms of PD, sleep disorders are strongly associated with some genes, since a growing number of genome-wide association studies have identified genetic risks for sleep disorders [3]. Therefore, sleep disorders in patients carrying PD-related gene mutations have received much attention for research. However, studies have not reached a consensus regarding the genetic risk factors for sleep disorders in PD or prodromal PD. Some studies have found that PD patients carrying GBA variants seem to develop RBD more frequently than patients without GBA variants [4]. However, another cohort study did not find any difference in the risk of RBD between patients with and without GBA variants [5]. Inconsistent results have been found on the prevalence and the severity of other sleep disorders, such as EDS and RLS, from studies on LRRK2 [6] or PRKN variants [7, 8]. Moreover, controversial conclusions regarding the role of genetic variants on sleep disorders exist when comparing asymptomatic carriers of causative gene variants with non-carrier healthy controls (HCs) [9, 10]. Differences in the sites of variants of causative genes, disease durations of participants in cohorts, and the study design could contribute to such discrepancies. Therefore, the association of genetic heterogeneity with the risk of sleep disorders may differ, especially at different stages of PD, such as prodromal and clinical stages.
In this context, we systematically reviewed the genetic variants associated with the risk of sleep disorders in PD patients and asymptomatic PD genetic carriers to elucidate this inconsistency.
Methods
This meta-analysis was conducted in accordance with the Meta-analysis of Observational Studies in Epidemiology (MOOSE) guidelines and Preferred Reporting Items for Systematic Reviews and Meta-analyses guidelines (PRISMA) [11].
Search strategy and literature selection
Literature search was performed in the MEDLINE/PubMed, EMBASE and PsychINFO databases by the date of July 1, 2021, using search terms “(((Parkinsonian Disorders) OR (Parkinson disease)) AND (((((genetic variation) OR (gene)) OR (genetic)) OR (inherited)) OR (familial))) AND (((((((((((sleep) OR (sleep wake disorders)) OR (sleep apnea syndromes)) OR (Sleep Initiation and Maintenance Disorders)) OR (Disorders of Excessive Somnolence)) OR (excessive daytime sleepiness)) OR (EDS)) OR (REM Sleep Behavior Disorder)) OR (RBD)) OR (Restless Legs Syndrome)) OR (RLS))”. Studies were selected based on the following inclusion criteria: (1) observational studies, namely cohort, case–control or cross-sectional studies; (2) studies providing specific genotyping methods; and (3) trials incorporating outcomes of sleep disorders by validated tests and comparing the outcomes of PD causative gene variant carriers with those of idiopathic PD (iPD) or HC. The following exclusion criteria were applied: (1) in vitro studies of gene variants; (2) case reports, abstracts for conferences, reviews or meta-analyses; and (3) samples incorporated in multiple cohorts by the same institutions.
To ensure complete retrieval, at preliminary screening we focused on studies related to patients with PD or asymptomatic carriers of gene variants that are considered causative or not. We then narrowed down the selected studies to focus on the relationships between PD pathogenic genes and sleep disorders in PD patients and asymptomatic gene carriers. Quantitative analysis was performed when there were three or more studies on the same gene variant; otherwise, narrative analysis was performed for the same gene variant.
Risk-of-bias assessments
Two researchers (JH and YC) independently assessed the methodological quality of the included studies using the Newcastle–Ottawa Quality Assessment Scale. Studies with scores > 6 were considered to have a low risk of bias [12].
Data extraction
Two researchers (JH and YC) independently extracted the following information from each study: the first author, publication year, country, age at assessment, sex, disease duration, disease severity and cognitive status. The sample size, the mean (standard deviation, SD) value of the sleep disturbance test, and the number of sleep disorder events were obtained as primary outcomes. Discrepancies in data extraction were resolved by discussion with a third investigator (CL).
Statistical analysis
To investigate the relationship between PD causative genes and sleep disorders at the symptomatic and prodromal stages of PD, we divided the studies into two groups: studies on gene variants in patients with PD and studies on gene variants in asymptomatic carriers. In the meta-analysis, we calculated the odds ratio (OR) for binary variables and the standard mean difference (SMD) for continuous variables with 95% confidence intervals (CI) using the Mantel–Haenszel statistical method. Statistical heterogeneity between studies was evaluated used the I2 test. We adopted the fixed-effect model for pooled analysis when I2 < 50% and P > 0.1, while the random effect model was chosen when I2 > 50% or P < 0.1, considering the relatively high heterogeneity between studies. Subgroup and meta-regression analyses were conducted to explore potential sources of heterogeneity. For publication bias, we adopted Egger’s test and funnel plot asymmetry to evaluate the effect of publication bias when the number of recruited trials was greater than or equal to 10, according to the Cochrane Handbook recommendations [13]. We performed a sensitivity analysis by omitting one study at a time, to ensure the stability of results. All data were analyzed using the STATA (version 16.0) software, and statistical significance was set at P < 0.05.
Results
Characteristics of the selected studies
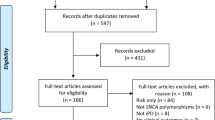
A total of 845 records from MEDLINE (through PubMed), 839 records from EMBASE, and 615 records from PsychINFO were identified at initial search. After removal of duplicates and exclusion by titles and abstracts, 155 articles were retrieved through a full-text review. Moreover, 12 studies regarding non-pathogenic genes of PD in sleep disorders were excluded after further extraction (Additional file 1: Table S1). Finally, 40 studies were selected for quantitative analysis, including studies on GBA, LRRK2 and PRKN genes, and three studies on SNCA gene were used for qualitative analysis (Fig. 1). Twelve studies were included to compare patients with and without heterozygous GBA carriers [5, 14,15,16,17,18,19,20,21,22,23,24], 17 studies to compare patients with and without heterozygous LRRK2 carriers [15, 16, 20,21,22, 25,26,27,28,29,30,31,32,33,34,35,36] and seven studies to compare patients with and without homozygous or compound heterozygous PRKN carriers [7, 8, 15, 37,38,39,40]. Six studies comparing asymptomatic heterozygous GBA carriers with non-carrier HCs were selected [9, 10, 20, 41,42,43], and 11 studies compared asymptomatic heterozygous LRRK2 carriers with non-carrier HCs [9, 20, 29,30,31, 42, 44,45,46,47,48]. The study qualities were evaluated, and one study [18] was considered to have a moderate risk of bias (Additional file 1: Table S2). The general characteristics and outcomes of each study are summarized in Additional file 1: Tables S3 and S4.

Flowchart of the literature search according to Preferred Reporting Items for Systematic Review and Meta-Analyses (PRISMA). PD Parkinson’s disease, GBA glucocerebrosidase gene, LRRK2 Leucine-rich repeat kinase 2 gene, PRKN parkin gene
GBA variants and the risk of sleep disorders in patients with PD
A total of 12 studies compared sleep disturbance between patients with and without heterozygous GBA variants (7 studies on the risk of RBD [5, 14, 16, 18, 19, 21, 22], 7 on the RBD Screening Questionnaire (RBDSQ) score [14, 15, 17, 20, 21, 23, 24] and 2 on the risk of EDS [15, 21]). A random-effect meta-analysis indicated that PD patients carrying heterozygous GBA variants had a significantly higher risk of RBD (OR, 1.82; 95% CI, 1.21–2.74, I2 = 55.8%) (Fig. 2a) and a higher RBDSQ score (SMD, 0.33; 95% CI, 0.21–0.45; I2 = 0.0%) (Fig. 2b). In the subgroup analysis, the PD patients with N370S [19, 21] and L444P variants [19, 22] of GBA were at a higher risk of developing RBD than patients without these variants (N370S: OR, 1.64; 95% CI, 1.03–2.61; I2 = 0.0; L444P: OR, 2.02; 95% CI, 1.08–3.75; I2 = 37.9%) (Fig. 3a). Additionally, PD patients carrying the GBA N370S variant had higher RBDSQ scores than those without the variant (SMD, 0.24; 95% CI, 0.09–0.39; I2 = 0.0%) (Fig. 3b) [17, 20, 21, 23]. However, we found that the E326K and T369M variants of GBA [5, 19] did not affect the risk of RBD in PD patients (OR, 1.43; 95% CI, 0.98–2.07; I2 = 83.4%) (Fig. 3a). Sensitivity analysis performed by removing each study demonstrated robustness of the results that GBA variants increased the risk and severity of RBD in PD. Nevertheless, no significant difference was found in the severity of EDS between patients with and without GBA variants (Additional file 1: Fig. S1). Moderate heterogeneity (I2 = 55.8%, P = 0.035) was found for the risk of RBD in PD patients carrying GBA variants. The heterogeneity was diminished when the GBA variants were divided into subgroups except for GBA E326K and T369M subgroups (GBA E326K and T369M, I2 = 83.4%, P = 0.014; GBA N370S, I2 = 0.0%, P = 0.396; GBA L444P, I2 = 0.0%, P = 0.204). Meta-regression analyses did not identify confounding factors for the outcome of the quantitative analyses (Additional file 1: Table S5). All comparative outcomes of the meta-analysis are displayed in Table 1.

Forest plot of the risk and severity of RBD in PD patients with GBA variants. Forest plots display meta-analysis results on the risk (a) and severity (b) of RBD in PD patients with GBA variants compared with iPD. PD Parkinson’s disease, iPD idiopathic Parkinson’s disease, GBA glucocerebrosidase gene, OR odds ratio, SMD standard mean difference

Forest plots of meta-analysis results on the risk (a) and severity (b) of RBD in PD patient subgroups carrying GBA variants compared with iPD. PD Parkinson’s disease, iPD idiopathic Parkinson’s disease, GBA glucocerebrosidase gene, OR odds ratio, SMD standard mean difference
LRRK2 variants and the risk of sleep disorders in patients with PD
Pooled analysis of 13 studies [16, 21, 22, 25,26,27,28,29, 31,32,33,34, 36] showed that there was no significant difference in the risk (OR, 0.69; 95% CI, 0.48–1.00; I2 = 71.7%) of RBD between patients with and without LRRK2 variants. There was also no significant difference in the severity of RBD (SMD, −0.06; 95% CI, − 0.32 to 0.19; I2 = 78.6%) from seven studies [15, 20, 21, 28, 30, 31, 35] between patients with and without LRRK2 variants (Fig. 4a, b). High heterogeneity was found in the studies on the prevalence and severity of RBD. Subgroup analysis of LRRK2 G2019S [21, 22, 27, 29, 31,32,33,34] and LRRK2 G2385R variants [25, 26, 28, 36] indicated that PD patients carrying LRRK2 G2019S had 51% lower odds of the risk of RBD compared to those without LRRK2 G2019S (OR, 0.49; 95% CI, 0.39–0.61; I2 = 0.0%) (Fig. 5a), whereas LRRK2 G2385R had no effect on the risk of RBD (OR, 1.53; 95% CI, 0.75–3.13; I2 = 75.5%) (Fig. 5b). PD patients carrying the LRRK2 G2019S variant [20, 21, 30, 31] had lower RBDSQ scores than those without LRRK2 G2019S (SMD, − 0.33; 95% CI, − 0.47 to − 0.20; I2 = 0.0%) (Fig. 5c), while there was no difference in RBDSQ score between patients with and without LRRK2 G2385R [28, 35] (Fig. 5d). There was no risk of EDS in patients with LRRK2 variants (Additional file 1: Fig. S2a, S2b), or in the subgroup of patients with the LRRK2 G2019S variant (Additional file 1: Fig. S2c). There were no differences in the Parkinson Disease Sleep Scale score or the risk of RLS between patients with and without LRRK2 variants (Additional file 1: Fig. S3a, S3b). Sensitivity analysis demonstrated that the pooled ORs and SMDs of RBD in patients with and without LRRK2 variants were stable. Egger’s test showed no publication bias (P = 0.672). Funnel plot of studies on RBD risk in PD patients carrying LRRK2 G2019S is shown in Additional file 1: Fig. S4. High heterogeneity was discovered in the risk and severity of RBD in patients with PD carrying LRRK2 variants (risk of RBD, I2 = 71.7%, P = 0.000; severity of RBD, I2 = 78.6%, P = 0.000). Subgroup analysis revealed that there was no heterogeneity in LRRK2 G2019S variants (risk of RBD, I2 = 0.0%, P = 0.639; severity of RBD, I2 = 0.0%, P = 0.596) while high heterogeneity still existed in LRRK2 G2385R variant among studies (risk of RBD, I2 = 75.5%, P = 0.007; severity of RBD, I2 = 71.1%, P = 0.063). Age, sex, disease duration, disease stage and cognitive status were not confounding factors for the results of meta-regression of LRRK2 G2385R groups (Additional file 1: Table S5).

Forest plots displaying meta-analysis results on the risk (a) and severity (b) of RBD in PD patients with LRRK2 variants compared with iPD. PD Parkinson’s disease, iPD idiopathic Parkinson’s disease, LRRK2 Leucine-rich repeat kinase 2 gene, OR odds ratio, SMD standard mean difference

Forest plots displaying meta-analysis results on the risk of RBD in PD patients with LRRK2 G2019S variant (a) and LRRK2 G2385R variant (b) compared with iPD, as well as severity of RBD in PD patients with LRRK2 G2019S variant (c) and LRRK2 G2385R variant (d) compared with iPD. PD Parkinson’s disease, iPD idiopathic Parkinson’s disease, LRRK2 Leucine-rich repeat kinase 2 gene, OR odds ratio, SMD standard mean difference
PRKN variants and risk of sleep disorders in patients with PD
We found no significant difference in the risk of RBD (OR, 0.74; 95% CI, 0.29–1.86; I2 = 60.9%), EDS (OR, 0.21; 95% CI, 0.04–1.23; I2 = 0.0%), or RLS (OR, 1.01; 95% CI, 0.08–13.22; I2 = 74.6%) between PD patients carrying homozygous or compound heterozygous PRKN variants and iPD (Additional file 1: Figs. S5a, S6a, S7). PRKN variants did not worsen or alleviate the severity of RBD or EDS (Additional file 1: Figs. S5b, S6b).
Genetic heterogeneity and risk of sleep disorders in asymptomatic carriers
Pooled analysis of cross-sectional studies demonstrated that there were no significant differences in the risk of RBD (OR, 1.04; 95% CI, 0.68–1.59; I2 = 0.0%) [9, 10, 41, 42] (Additional file 1: Fig. S8a) and RBDSQ score (SMD, 0.22; 95% CI, −0.08 to 0.52; I2 = 0.0%) [20, 41] between asymptomatic heterozygous GBA carriers and non-carrier HCs (Additional file 1: Fig. S8b). However, quantitative analysis of longitudinal cohort studies demonstrated an increased RBDSQ score over follow-up (SMD, 0.63; 95% CI, 0.18–1.08; I2 = 0.0%) in asymptomatic heterozygous GBA carriers than in the non-carrier HCs (Additional file 1: Fig. S8d), despite no difference at baseline between the two groups [10, 43] (SMD, −0.26; 95% CI, −0.69 to 0.18; I2 = 0.0%) (Additional file 1: Fig. S8c). LRRK2 variant did not affect the risk and severity of RBD or EDS in asymptomatic LRRK2 carriers (Additional file 1: Fig. S9 and S10).
Abnormal sleep architecture in PD patients and asymptomatic carriers of SNCA variant
Three small-sized studies investigated the association between p.A53T (p.Ala53Thr, c.209G > A) variant of SNCA and sleep disturbance in PD patients or asymptomatic carriers. Simitsi et al. [49] recruited 15 PD patients carrying the SNCA p.A53T variant and found a higher prevalence of RBD in these patients than in iPD. However, Koros et al. [50] did not find a difference in sleep disturbances including insomnia, RBD and EDS between patients with and without SNCA p.A53T. Regarding the prodromal stages of PD, two studies found a low prevalence of RBD in SNCA p.A53T asymptomatic carriers [49, 51].
Discussion
In this systematic review and meta-analysis, we examined the associations between variants of causative genes of PD and sleep disturbance in both PD patients and asymptomatic carriers at the prodromal phase of PD. Our findings suggest that variants of the causative genes play a role in sleep disturbances in PD. We found that GBA variants increased the risk and severity of RBD in patients with PD, while the LRRK2 G2019S variant reduced the risk and severity of RBD. In addition, GBA variants worsened the RBD symptoms at the prodromal stage of PD, while variants of GBA and LRRK2 did not influence the risk of EDS in patients with PD. PRKN variants did not influence the risk of RBD, EDS, or RLS in PD.
Our study revealed relationships between GBA variants and RBD in patients with PD. GBA encodes beta-glucocerebrosidase, and biallelic pathogenic variants of GBA1 cause Gaucher disease (GD), a lysosomal disorder. Both homozygous and heterozygous GBA variants increase the risk of PD [52]. GBA variants are classified into severe variants (such as L444P, W291X, H225Q, and IVS2 + 1G > A) and mild variants (such as N370S). In addition, several non-pathogenic variants of GBA in GD, such as E326K and T369M, have been found to increase the risk of PD [53]. PD patients with severe variants of GBA have a younger age of onset, faster progression and more severe cognitive impairment than those with mild variants [54]. In this meta-analysis, we divided GBA variants into groups based on the severity of GBA variants (L444P regarded as the severe variant, N370S as the mild variant, and E326K & T369M as the non-pathogenic variants), and showed that the pathogenic variants of GBA increase the risk of RBD. Notably, the OR of GBA L444P variant (2.02) [19, 22] for RBD in PD patients was higher than that of the GBA N370S variant (OR 1.68) [19, 21]. However, there were no differences in the risk of RBD between patients with and without GBA E326K or T369M variant [5, 19]. Our current findings indicate that the role of GBA variants in increasing the risk of RBD in PD differs depending on the severity of GBA variants. RBD is considered as a prodrome of α-synucleinopathy, and GBA gene is closely correlated with α-synuclein. Impaired proteolysis caused by GCase deficiency preferentially increases α-synuclein deposition and spread of α-synuclein pathology, while elevated α-synuclein decreases the GCase activity [55]. Lower levels of α-synuclein in CSF have been detected in PD patients carrying GBA variants compared to iPD patients, with a downward trend depending on the order of severe, mild and risk variants [19]. Differences in α-synuclein expression in different GBA variants may elucidate the discrepancy in phenotypes.
For the prodromal stage of PD, our meta-analysis found that GBA variant carriers had worse RBD symptoms during the follow-up period [10, 43]. Interestingly, no difference was found in the severity of RBD between GBA variant carriers and HCs at baseline [10, 43]. Statistical analysis of cross-sectional studies failed to find any difference in risk [9, 10, 41, 42] or severity [20, 41] between asymptomatic carriers of GBA variants and HCs. These findings suggest that GBA variant is a risk factor for the development of RBD at the prodromal stage with disease duration.
LRRK2 is one of the causative genes for autosomal dominant PD. Previous cross-sectional studies have indicated that patients with PD carrying the LRRK2 G2019S variant, the most common variant, exhibit slower disease progression and milder motor symptoms than iPD patients [56]. Our meta-analysis found that LRRK2 G2019S decreased the risk and severity of RBD [21, 22, 27, 29,30,31,32,33,34], but LRRK2 G2385R did not affect the risk of RBD in patients with PD [25, 26, 28, 36]. In prodromal stages, our pooled analysis suggested that LRRK2 G2019S did not increase or decrease the risk and severity of RBD in asymptomatic carriers, consistent with a review by Tolosa et al. [57]. In addition, our meta-analysis found no associations between other sleep disturbances, including EDS and RLS, and LRRK2 G2019S and G2385R variants. Further studies are needed to elucidate the associations between EDS/RLS and LRRK2 variants.
The pathophysiological mechanisms of LRRK2 variants in PD are related to a variety of pathways such as kinase activity, autophagy and oxidative stress. The interplay between α-synuclein and LRRK2 mutations accelerates the progression of α-synuclein-mediated neurodegeneration, with Rab proteins and chaperones serving as mediators [58]. However, only half of PD patients carrying LRRK2 variants were found to contain brainstem synucleinopathy in postmortem studies, suggesting that some LRRK2 variants may not be involved in central synucleinopathy [59]. Most PD patients with LRRK2 variants display loss of dopaminergic neurons in the substantia nigra. Interestingly, patients carrying the LRRK2 G2019S variant show more Lewy body pathology than patients with other LRRK2 variants [60]. Skin biopsy revealed no difference in deposition of phosphorylated α-synuclein between LRRK2 G2385R carriers and non-carriers [25], whereas high levels of α-synuclein deposition in sympathetic noradrenergic nerves in skin biopsies were found in patients with LRRK2 G2019S and R1441G variants [59]. Reasons for the low risk of RBD in PD patients carrying the LRRK2 G2019S variant require more pathological research for interpretation.
PRKN is one of the most frequently mutated genes in patients with early-onset PD [15] and is involved in the mitochondrial function of neural cells. Hatice Kumru found that six out of 10 PD patients carrying PRKN variants (7 carrying homozygous PRKN variants) developed mild RBD as assessed by video polysomnography [61]. However, our pooled analysis of cross-sectional studies showed no correlation between PRKN variant and the risk and severity of sleep disturbances including RBD [7, 8, 15, 37,38,39,40], EDS [7, 8, 15, 37, 38, 40] and RLS [7, 8, 40] in patients with PD. At the prodromal stage of PD, only one study showed no difference in the risk of RBD between asymptomatic carriers with and without PRKN variants [62]. Large-scale studies are urgently required to dispel the mist of inconsistent results on sleep disorders in patients with PRKN variants.
SNCA encodes α-synuclein proteins, which abnormally accumulate in PD as a major component of Lewy bodies. Observational studies in cases and families have reported RBD in PD patients carrying various variants of SNCA, such as A53T variant, duplications [63] and triplications [64]. However, two studies on the risk of RBD in PD patients carrying SNCA A53T [49, 50] demonstrated contradictory results. As for the prodromal stage of PD, carriers of SNCA A53T reported RBD before the appearance of motor symptoms in a longitudinal study [51]. Moreover, SNCA methylation may play a role in the process of SNCA expression, which is associated with RBD. Previous studies have found hypomethylation in the promoter and intron 1 regions of SNCA in idiopathic RBD patients and PD patients compared to HCs [65].
Patients carrying GBA, LRRK2, PRKN, or SNCA variants have different manifestations and pathogeneses in essence. Patients with SNCA, LRRK2, or GBA variants have peripheral synucleinopathy, while patients with PRKN variants do not [59]. Moreover, PD patients with PRKN variants mostly lack α-synuclein deposition in the brain and part of PD patients with LRRK2 variants have α-synuclein deposition in the central nervous system. GBA, LRRK2 and SNCA variants are associated with lysosomal dysfunction, while the PRKN variant is directly involved in mitochondrial functions [66]. It has been proposed that patients with GBA or SNCA variants typically have initial pathological α-synuclein originating in the enteric and autonomic nervous systems, whereas patients with LRRK2 or PRKN variants have initial α-synuclein pathology originating in the brain [67].
Limitations
This systematic review and meta-analysis had some limitations. First, the studies included were associated with various disease durations and ages of patients at enrollment. Due to the limited number of studies, we did not perform a pooled analysis stratified by disease duration or age, even though the meta-regression showed that age, sex, disease duration, disease stage and cognitive status did not contribute to the heterogeneity of enrolled studies. Second, the included studies were conducted in different countries, indicating different genetic characteristics due to different regions. It is worth mentioning that studies on LRRK2 G2385R were from Asian regions since this risk variant of PD is more common in individuals of Asian descent. In contrast, the LRRK2 G2019S variant is rare in Asian populations. Third, the limited case–control studies of SNCA variants restricted us from performing quantitative analysis, even though SNCA is a causative gene for PD and directly correlates to pathological hallmark of PD. Moreover, other causative genes of PD, such as PINK1 and DJ-1, were not discussed in this meta-analysis due to the lack of original studies. Fourth, most diagnoses of sleep disturbances in the included studies were made through clinical evaluation scales but not polysomnography; therefore, more accurate techniques such as polysomnography should be used. Moreover, the studies included in the meta-analysis were mostly cross-sectional. A lack of prospective longitudinal study with a larger number of patients may result in a failure to discover the difference in sleep disturbance between cases and controls, especially between genetic asymptomatic carriers and non-carrier controls.
Conclusions
In conclusion, this meta-analysis showed that some PD causative genes are associated with sleep disorders in PD patients at clinical stages, as well as at the asymptomatic stage. GBA variants can increase the risk and severity of RBD in the clinical stages of PD and also increase the severity of RBD in the prodromal stage of PD, while LRRK2 G2019S is negatively associated with RBD in patients with PD. These findings provide evidence that genetic heterogeneities play a role in the development of sleep disturbance, especially RBD, in PD patients and at the prodromal stage of PD. These genetic heterogeneities indicate involvement of several common pathways and pathogenesis between PD and sleep disorders, such as α-synuclein aggregation in both PD and RBD. Longitudinal studies and investigations on more PD causative or risk genes are needed to explore the relations between PD and sleep disorders, leading to precise recognition and target therapy for PD patients and gene carriers prone to sleep disorders, such as RBD in patients and carriers of GBA variants.
Availability of data and materials
All data generated or analyzed during this study are included in Additional file 1.
Abbreviations
- PD:
-
Parkinson’s disease
- EDS:
-
Excessive daytime sleepiness
- RLS:
-
Restless legs syndrome
- RBD:
-
Rapid-eye-movement behavior disorders
- SNCA :
-
Alpha-synuclein gene
- LRRK2 :
-
Leucine-rich repeat kinase 2 gene
- GBA :
-
Glucocerebrosidase gene
- PRKN :
-
Parkin gene
- HC:
-
Healthy controls
- OR:
-
Odds ratios
- SMD:
-
Standard mean difference
- CI:
-
Confidence intervals
- RBDSQ:
-
RBD Screening Questionnaire
References
Berg D, Borghammer P, Fereshtehnejad SM, Heinzel S, Horsager J, Schaeffer E, et al. Prodromal Parkinson disease subtypes—key to understanding heterogeneity. Nat Rev Neurol. 2021;17:349–61.
Miglis MG, Adler CH, Antelmi E, Arnaldi D, Baldelli L, Boeve BF, et al. Biomarkers of conversion to α-synucleinopathy in isolated rapid-eye-movement sleep behaviour disorder. Lancet Neurol. 2021;20(8):671–84.
Byrne EM, Gehrman PR, Medland SE, Nyholt DR, Heath AC, Madden PA, et al. A genome-wide association study of sleep habits and insomnia. Am J Med Genet B Neuropsychiatr Genet. 2013;162b(5):439–51.
Gan-Or Z, Liong C, Alcalay RN. GBA-associated Parkinson’s disease and other synucleinopathies. Curr Neurol Neurosci Rep. 2018;18(8):1–10.
Malek N, Weil RS, Bresner C, Lawton MA, Grosset KA, Tan M, et al. Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J Neurol Neurosurg Psychiatry. 2018;89(7):702–9.
Belarbi S, Hecham N, Lesage S, Kediha MI, Smail N, Benhassine T, et al. LRRK2 G2019S mutation in Parkinson’s disease: a neuropsychological and neuropsychiatric study in a large Algerian cohort. Parkinsonism Relat Disord. 2010;16(10):676–9.
Kim HJ, Kim HJ, Lee JY, Yun JY, Kim SY, Park SS, et al. Phenotype analysis in patients with early onset Parkinson’s disease with and without parkin mutations. J Neurol. 2011;258(12):2260–7.
Kagi G, Klein C, Wood NW, Schneider SA, Pramstaller PP, Tadic V, et al. Nonmotor symptoms in Parkin gene-related parkinsonism. Mov Disord. 2010;25(9):1279–84.
Simuni T, Uribe L, Cho HR, Caspell-Garcia C, Coffey CS, Siderowf A, et al. Clinical and dopamine transporter imaging characteristics of non-manifest LRRK2 and GBA mutation carriers in the Parkinson’s Progression Markers Initiative (PPMI): a cross-sectional study. Lancet Neurol. 2020;19(1):71–80.
Avenali M, Toffoli M, Mullin S, McNeil A, Hughes DA, Mehta A, et al. Evolution of prodromal parkinsonian features in a cohort of GBA mutation-positive individuals: a 6-year longitudinal study. J Neurol Neurosurg Psychiatry. 2019;90(10):1091–7.
Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med. 2009;151(4):264–9.
Wells GA SB, O’Connell D, Peterson J, Welch V, Losos M, Tugwell P. The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses. http://www.ohri.ca/programs/clinical_epidemiology/oxford.asp.
Cumpston M, Li T, Pge MJ, Chandler J, Welch VA, Higgins JP, et al. Updated guidance for trusted systematic reviews: a new edition of the Cochrane Handbook for Systematic Reviews of Interventions. Cochrane Database Syst Rev. 2019;10:Ed000142.
Gan-Or Z, Mirelman A, Postuma RB, Arnulf I, Bar-Shira A, Dauvilliers Y, et al. GBA mutations are associated with rapid eye movement sleep behavior disorder. Ann Clin Transl Neurol. 2015;2(9):941–5.
Zhao Y, Qin L, Pan H, Liu Z, Jiang L, He Y, et al. The role of genetics in Parkinson’s disease: a large cohort study in Chinese mainland population. Brain. 2020;143(7):2220–34.
Chen Y, Gu X, Ou R, Zhang L, Hou Y, Liu K, et al. Evaluating the role of SNCA, LRRK2, and GBA in Chinese patients with early-onset Parkinson’s disease. Mov Disord. 2020;35(11):2046–55.
Thaler A, Bregman N, Gurevich T, Shiner T, Dror Y, Zmira O, et al. Parkinson’s disease phenotype is influenced by the severity of the mutations in the GBA gene. Parkinsonism Relat Disord. 2018;55:45–9.
Bonner N, Bozzi S, Morgan L, Mason B, Peterschmitt MJ, Fischer TZ, et al. Patients’ experiences of Parkinson’s disease: a qualitative study in glucocerebrosidase and idiopathic Parkinson’s disease. J Patient Rep Outcomes. 2020;4(1):65.
Lerche S, Wurster I, Roeben B, Zimmermann M, Riebenbauer B, Deuschle C, et al. Parkinson’s disease: glucocerebrosidase 1 mutation severity is associated with CSF alpha-synuclein profiles. Mov Disord. 2020;35(3):495–9.
Thaler A, Omer N, Giladi N, Gurevich T, Bar-Shira A, Gana-Weisz M, et al. Mutations in GBA and LRRK2 are not associated with increased inflammatory markers. J Parkinsons Dis. 2021.
Simuni T, Brumm MC, Uribe L, Caspell-Garcia C, Coffey CS, Siderowf A, et al. Clinical and dopamine transporter imaging characteristics of leucine rich repeat kinase 2 (LRRK2) and glucosylceramidase beta (GBA) Parkinson’s disease participants in the Parkinson’s progression markers initiative: a cross-sectional study. Mov Disord. 2020;35(5):833–44.
Yahalom G, Greenbaum L, Israeli-Korn S, Fay-Karmon T, Livneh V, Ruskey JA, et al. Carriers of both GBA and LRRK2 mutations, compared to carriers of either, in Parkinson’s disease: risk estimates and genotype-phenotype correlations. Parkinsonism Relat Disord. 2019;62:179–84.
Canu E, Basaia S, Sarasso E, Leocadi M, Piramide N, Filippi M, et al. Longitudinal clinical, cognitive, and neuroanatomical changes over 5 years in GBA-positive Parkinson’s disease patients. J Neurol. 2021;269(3):1485–500.
Caminiti SP, Carli G, Avenali M, Blandini F, Perani D. Clinical and dopamine transporter imaging trajectories in a cohort of Parkinson’s disease patients with GBA mutations. Mov Disord. 2022;37(1):106–18.
Yang J, Wang H, Yuan Y, Fan S, Li L, Jiang C, et al. Peripheral synucleinopathy in Parkinson disease with LRRK2 G2385R variants. Ann Clin Transl Neurol. 2021;8(3):592–602.
Liang D, Shu L, Pan H, Xu Q, Guo J, Yan X, et al. Clinical characteristics of PD patients with LRRK2 G2385R and R1628P variants. Neurosci Lett. 2018;685:185–9.
Marras C, Alcalay RN, Caspell-Garcia C, Coffey C, Chan P, Duda JE, et al. Motor and nonmotor heterogeneity of LRRK2-related and idiopathic Parkinson’s disease. Mov Disord. 2016;31(8):1192–202.
Sun Q, Wang T, Jiang TF, Huang P, Li DH, Wang Y, et al. Effect of a Leucine-rich Repeat Kinase 2 variant on motor and non-motor symptoms in Chinese Parkinson’s disease patients. Aging Dis. 2016;7(3):230–6.
Ehrminger M, Leu-Semenescu S, Cormier F, Corvol JC, Vidailhet M, Debellemaniere E, et al. Sleep aspects on video-polysomnography in LRRK2 mutation carriers. Mov Disord. 2015;30(13):1839–43.
Pont-Sunyer C, Iranzo A, Gaig C, Fernández-Arcos A, Vilas D, Valldeoriola F, et al. Sleep disorders in Parkinsonian and Nonparkinsonian LRRK2 mutation carriers. PLoS ONE. 2015;10(7):e0132368.
Saunders-Pullman R, Alcalay RN, Mirelman A, Wang C, Luciano MS, Ortega RA, et al. REM sleep behavior disorder, as assessed by questionnaire, in G2019S LRRK2 mutation PD and carriers. Mov Disord. 2015;30(13):1834–9.
Trinh J, Amouri R, Duda JE, Morley JF, Read M, Donald A, et al. Comparative study of Parkinson’s disease and leucine-rich repeat kinase 2 p.G2019S parkinsonism. Neurobiol Aging. 2014;35(5):1125–31.
Gaig C, Vilas D, Infante J, Sierra M, García-Gorostiaga I, Buongiorno M, et al. Nonmotor symptoms in LRRK2 G2019S associated Parkinson’s disease. PLoS ONE. 2014;9(10):e108982.
Alcalay RN, Mirelman A, Saunders-Pullman R, Tang MX, Mejia Santana H, Raymond D, et al. Parkinson disease phenotype in Ashkenazi Jews with and without LRRK2 G2019S mutations. Mov Disord. 2013;28(14):1966–71.
Cui SS, Fu R, Du JJ, Lin YQ, Huang P, Gao C, et al. Sex effects on clinical features in LRRK2 G2385R carriers and non-carriers in Parkinson’s disease. BMC Neurosci. 2021;22(1):22.
Li DW, Gu Z, Wang C, Ma J, Tang BS, Chen SD, et al. Non-motor symptoms in Chinese Parkinson’s disease patients with and without LRRK2 G2385R and R1628P variants. J Neural Transm (Vienna). 2015;122(5):661–7.
Zhou XY, Liu FT, Chen C, Luo SS, Zhao J, Tang YL, et al. Quality of life in newly diagnosed patients with parkin-related Parkinson’s disease. Front Neurol. 2020;11:580910.
Song J, Shen B, Yang YJ, Liu FT, Zhao J, Tang YL, et al. Non-motor symptoms in Parkinson’s disease patients with Parkin mutations: more depression and less executive dysfunction. J Mol Neurosci. 2020;70(2):246–53.
Morgante F, Fasano A, Ginevrino M, Petrucci S, Ricciardi L, Bove F, et al. Impulsive-compulsive behaviors in parkin-associated Parkinson disease. Neurology. 2016;87(14):1436–41.
Limousin N, Konofal E, Karroum E, Lohmann E, Theodorou I, Dürr A, et al. Restless legs syndrome, rapid eye movement sleep behavior disorder, and hypersomnia in patients with two parkin mutations. Mov Disord. 2009;24(13):1970–6.
Moran EE, Bressman SB, Ortega RA, Raymond D, Nichols WC, Palmese CA, et al. Cognitive functioning of Glucocerebrosidase (GBA) non-manifesting carriers. Front Neurol. 2021;12:635958.
Chahine LM, Urbe L, Caspell-Garcia C, Aarsland D, Alcalay R, Barone P, et al. Cognition among individuals along a spectrum of increased risk for Parkinson’s disease. PLoS ONE. 2018;13(8):e0201964.
Beavan M, McNeill A, Proukakis C, Hughes DA, Mehta A, Schapira AH. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol. 2015;72(2):201–8.
Pont-Sunyer C, Tolosa E, Caspell-Garcia C, Coffey C, Alcalay RN, Chan P, et al. The prodromal phase of leucine-rich repeat kinase 2-associated Parkinson disease: clinical and imaging studies. Mov Disord. 2017;32(5):726–38.
Mirelman A, Alcalay RN, Saunders-Pullman R, Yasinovsky K, Thaler A, Gurevich T, et al. Nonmotor symptoms in healthy Ashkenazi Jewish carriers of the G2019S mutation in the LRRK2 gene. Mov Disord. 2015;30(7):981–6.
Johansen KK, White LR, Farrer MJ, Aasly JO. Subclinical signs in LRRK2 mutation carriers. Parkinsonism Relat Disord. 2011;17(7):528–32.
van den Heuvel L, Lim AS, Visanji NP, Huang J, Ghate T, Mestre TA, et al. Actigraphy detects greater intra-individual variability during gait in non-manifesting LRRK2 mutation carriers. J Parkinson’s Dis. 2018;8(1):131–9.
Mestre TA, Pont-Sunyer C, Kausar F, Visanji NP, Ghate T, Connolly BS, et al. Clustering of motor and nonmotor traits in leucine-rich repeat kinase 2 G2019S Parkinson’s disease nonparkinsonian relatives: a multicenter family study. Mov Disord. 2018;33(6):960–5.
Simitsi AM, Koros C, Stamelou M, Papadimitriou D, Leonardos A, Bougea A, et al. REM sleep behavior disorder and other sleep abnormalities in p. A53T SNCA mutation carriers. Sleep. 2021;44(5):48.
Koros C, Stamelou M, Simitsi A, Beratis I, Papadimitriou D, Papagiannakis N, et al. Selective cognitive impairment and hyposmia in p.A53T SNCA PD vs typical PD. Neurology. 2018;90(10):e864–9.
Papadimitriou D, Antonelou R, Miligkos M, Maniati M, Papagiannakis N, Bostantjopoulou S, et al. Motor and nonmotor features of carriers of the p.A53T alpha-synuclein mutation: a longitudinal study. Mov Disord. 2016;31(8):1226–30.
Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012;11(11):986–98.
Menozzi E, Schapira AHV. Exploring the genotype-phenotype correlation in GBA-Parkinson disease: clinical aspects, biomarkers, and potential modifiers. Front Neurol. 2021;12:694764.
Petrucci S, Ginevrino M, Trezzi I, Monfrini E, Ricciardi L, Albanese A, et al. GBA-related Parkinson’s disease: dissection of genotype-phenotype correlates in a large Italian cohort. Mov Disord. 2020;35(11):2106–11.
Gegg ME, Verona G, Schapira AHV. Glucocerebrosidase deficiency promotes release of α-synuclein fibrils from cultured neurons. Hum Mol Genet. 2020;29(10):1716–28.
Kestenbaum M, Alcalay RN. Clinical features of LRRK2 carriers with Parkinson’s disease. Adv Neurobiol. 2017;14:31–48.
Tolosa E, Vila M, Klein C, Rascol O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat Rev Neurol. 2020;16(2):97–107.
O’Hara DM, Pawar G, Kalia SK, Kalia LV. LRRK2 and α-synuclein: distinct or synergistic players in Parkinson’s disease? Front Neurosci. 2020;14:577.
Isonaka R, Goldstein DS, Zhu W, Yoon E, Ehrlich D, Schindler AB, et al. α-Synuclein deposition in sympathetic nerve fibers in genetic forms of Parkinson's disease. Mov Disord. 2021.
Kalia LV, Lang AE, Hazrati LN, Fujioka S, Wszolek ZK, Dickson DW, et al. Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol. 2015;72(1):100–5.
Kumru H, Santamaria J, Tolosa E, Valldeoriola F, Muñoz E, Marti MJ, et al. Rapid eye movement sleep behavior disorder in parkinsonism with parkin mutations. Ann Neurol. 2004;56(4):599–603.
Sixel-Döring F, Lohmann K, Klein C, Trenkwalder C, Mollenhauer B. REM sleep-associated motor behaviors in Parkinson’s disease patients with heterozygous Parkin mutations. Mov Disord. 2015;30(4):597–8.
Kielb S, Kisanuki YY, Dawson E. Neuropsychological profile associated with an alpha-synuclein gene (SNCA) duplication. Clin Neuropsychol. 2021:1–12.
Zafar F, Valappil RA, Kim S, Johansen KK, Chang ALS, Tetrud JW, et al. Genetic fine-mapping of the Iowan SNCA gene triplication in a patient with Parkinson’s disease. NPJ Parkinson’s Dis. 2018;4:18.
Zhao A, Li Y, Niu M, Li G, Luo N, Zhou L, et al. SNCA hypomethylation in rapid eye movement sleep behavior disorder is a potential biomarker for Parkinson’s disease. J Parkinson’s Dis. 2020;10(3):1023–31.
Li JL, Lin TY, Chen PL, Guo TN, Huang SY, Chen CH, et al. Mitochondrial function and Parkinson’s disease: from the perspective of the electron transport chain. Front Mol Neurosci. 2021;14:797833.
Horsager J, Knudsen K, Sommerauer M. Clinical and imaging evidence of brain-first and body-first Parkinson’s disease. Neurobiol Dis. 2022;164:1626.
Acknowledgements
We would like to thank all authors of the original research studies included in this meta-analysis.
Funding
This study was supported by the 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (No. ZYJC18038), the Sichuan Science and Technology Program (Grant No. 2021YJ0415) and the Science Foundation of Chengdu Science and Technology Bureau (Grant No. 2019-YF05-00307-SN).
Author information
Authors and Affiliations
Contributions
JXH: Study selections, data collection, and manuscript drafting designed. YFC: Study selections, data collection. CYL: Data collection, statistical analysis. HFS: Study design, statistical analysis and interpretation, and revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Supplementary Information
Additional file 1: Table S1.
Studies excluded because of non-pathogenic genes of PD. Table S2. Study Evaluation according to Newcastle-Ottawa Quality Assessment Scale. Table S3. General characteristics of included studies. Table S4. Primary outcomes of included studies. Table S5. Results of meta-regression in patients with gene variants. Fig. S1. Severity of EDS in PD patients with and without GBA variants. Fig. S2. Risk (a) and severity (b) of EDS in PD patients with and without LRRK2 variants, severity of EDS in PD patients with and without LRRK2 G2019S variants (c). Fig. S3. PDSS score (a) and risk of RLS (b) in PD patients with and without LRRK2 variants. Fig. S4. Funnel plots for the risk of RBD in PD patients with LRRK2 variants. Fig. S5. Risk (a) and severity (b) of RBD in PD patients with PRKN variants. Fig. S6. Risk (a) and severity (b) of EDS in PD patients with PRKN variants. Fig. S7. Risk of RLS in PD patients with and without PRKN variants. Fig. S8. Risk (a) and severity (b) of RBD in asymptomatic carriers with GBA variant and HCs. Fig. S9. Risk (a) and severity (b) of RBD in asymptomatic carriers with LRRK2 G2019S and HCs. Fig. S10. Risk (a) and severity (b) of EDS in asymptomatic carriers with LRRK2 G2019S and HCs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huang, J., Cheng, Y., Li, C. et al. Genetic heterogeneity on sleep disorders in Parkinson’s disease: a systematic review and meta-analysis. Transl Neurodegener 11, 21 (2022). https://doi.org/10.1186/s40035-022-00294-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-022-00294-1