
Abstract
Glutamate is the most commonly engaged neurotransmitter in the mammalian central nervous system, acting to mediate excitatory neurotransmission. However, high levels of glutamatergic input elicit excitotoxicity, contributing to neuronal cell death following acute brain injuries such as stroke and trauma. While excitotoxic cell death has also been implicated in some neurodegenerative disease models, the role of acute apoptotic cell death remains controversial in the setting of chronic neurodegeneration. Nevertheless, it is clear that excitatory synaptic dysregulation contributes to neurodegeneration, as evidenced by protective effects of partial N-methyl-D-aspartate receptor antagonists. Here, we review evidence for sublethal excitatory injuries in relation to neurodegeneration associated with Parkinson’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis and Huntington’s disease. In contrast to classic excitotoxicity, emerging evidence implicates dysregulation of mitochondrial calcium handling in excitatory post-synaptic neurodegeneration. We discuss mechanisms that regulate mitochondrial calcium uptake and release, the impact of LRRK2, PINK1, Parkin, beta-amyloid and glucocerebrosidase on mitochondrial calcium transporters, and the role of autophagic mitochondrial loss in axodendritic shrinkage. Finally, we discuss strategies for normalizing the flux of calcium into and out of the mitochondrial matrix, thereby preventing mitochondrial calcium toxicity and excitotoxic dendritic loss. While the mechanisms that underlie increased uptake or decreased release of mitochondrial calcium vary in different model systems, a common set of strategies to normalize mitochondrial calcium flux can prevent excitatory mitochondrial toxicity and may be neuroprotective in multiple disease contexts.
Similar content being viewed by others


Background
Glutamate is the most commonly engaged neurotransmitter in the mammalian central nervous system, acting to mediate excitatory neurotransmission. However, excessive glutamatergic input elicits excitotoxicity, contributing to neuronal cell death following acute hypoxic-ischemic insults [1] or traumatic brain injury [2]. Moreover, sublethal glutamatergic injury contributes to dendritic degeneration [3] accompanied by mitochondrial calcium toxicity [4].
Glutamate binds to calcium-permeable ionotropic receptors called N-methyl-D-aspartate (NMDA) receptors (NMDARs) and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors (AMPARs) due to their ability to be activated by NMDA or AMPA, respectively. NMDAR and AMPAR are localized predominantly to dendritic spines, but also exist in perisynaptic and extrasynaptic regions [5]. Glutamate signaling is terminated to some extent by re-uptake into presynaptic terminals of neurons; however, glial cells play a predominant role in scavenging free glutamate via high-affinity glutamate transporters. Glutamate is converted into glutamine within glial cells, before being released for neuronal uptake as a starting material to replenish both excitatory (glutamate) and inhibitory (GABA) neurotransmitters (reviewed in [6]).
In classic excitotoxicity, ischemic injury results in ATP depletion and impaired glutamate transporter function. The resulting elevation in extracellular glutamate elicits a massive influx of sodium and calcium into neurons via NMDARs [7]. Sodium influx results in swelling of neurons, which is often reversible, whereas elevated calcium contributes ultimately to irreversible excitotoxic injury (reviewed in [8]). In addition to calpains and other degradative enzymes, death associated protein kinase 1 (DAPK1) and neuronal nitric oxide synthase (nNOS), which are associated with the NMDAR cytosolic tail, are activated [9, 10], while CREB signaling is downregulated [11]. Following the initial glutamate-stimulated calcium influx, other mechanisms that elevate cytosolic calcium are triggered, contributing to neuronal cell death [12]. Interestingly, synaptic NMDAR-mediated calcium flux elicits greater neurotoxicity when compared to other calcium sources [7], such as extrasynaptic NMDARs [13] or voltage-gated calcium channels [14]. A similar level of calcium elevation that is toxic after NMDAR activation may not be toxic when calcium enters via voltage-gated calcium channels. These observations suggest that the elevated calcium levels may interact with other factors regulated by the route of entry to determine toxicity.
The role of acute apoptotic [15,16,17,18] or non-apoptotic cell death [18,19,20] is less certain in chronic neurodegenerative diseases than in stroke and trauma. Indeed, loss of synaptic function and synaptic/dendritic atrophy likely occur well before the onset of cell death, along with interruptions in axonal transport [21], protein and organellar homeostasis [22], and chronic mitochondrial dysfunction [23, 24]. While excitotoxic cell death has also been reported in models of Parkinson’s disease (PD) [25], 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) intoxication is relatively acute, mediated by inhibition of mitochondrial complex I activity. Nevertheless, it is clear that excitatory synaptic dysregulation contributes to neurodegeneration, as evidenced by the protective effects of partial NMDAR antagonists [3, 26,27,28]. In general, synaptic activity is regulated by a balance of excitatory and inhibitory inputs. Synaptic hyperactivity may result from changes that favor excitatory inputs and/or increased responsiveness to a given stimulus, or from inhibitory deficits. While physiological bursts in activity trigger dendritic remodeling that underlies plasticity, chronically elevated synaptic activity may trigger pathological effects. In particular, an emerging body of literature implicates a role for mitochondrial calcium dysregulation downstream of synaptic hyperactivity.
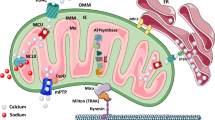
In the following sections, we will discuss the regulation of cytosolic and mitochondrial calcium in neurons. We will review the evidence for sublethal excitatory injuries in models of familial neurodegeneration associated with PD, Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS) and Huntington’s disease (HD), which highlight the key role of mitochondrial calcium dysregulation in excitatory post-synaptic injury that leads to dendritic degeneration. While the mechanisms underlying the increased mitochondrial calcium uptake or decreased release vary among the different model systems (Fig. 1), a common set of strategies to normalize mitochondrial calcium flux appears to be neuroprotective in several disease contexts.

A combination of factors conspire to promote excitatory mitochondrial toxicity (EMT). (1) Pathophysiological processes linked to neuronal hyperexcitability have been identified in several neurodegenerative disorders. Chronic elevations in cytosolic calcium flux due to neuronal hyperexcitability elicit increased demand for mitochondrial calcium buffering. When hyperexcitability is combined with altered mitochondrial calcium handling and/or other factors that enhance susceptibility to mitochondrial injury, EMT may occur. (2) A set of intrinsic changes in mitochondrial calcium handling are triggered by familial neurodegeneration gene mutations that elicit increased mitochondrial calcium uptake and/or decreased mitochondrial calcium release. Mutations in LRRK2 and SOD1 increase MCU expression, while PINK1 loss-of-function results in decreased phosphorylation and activities of NCLX and LETM1. NCLX is also decreasd in sporadic AD brains and tau inhibits NCLX activity. In some contexts, MCU expression is decreased rather than increased. This has been proposed as a compensatory response, but may still contribute to EMT by exacerbating cytosolic calcium elevations. Alternatively, MCU may be regulated differently in neurons than in glia. (3) Post-synaptic mitochondria are most vulnerable to EMT, and injured mitochondria are removed from dendrites by mitophagy. Elevated mitophagy that is not balanced by mitochondrial replacement results in mitochondrial depletion from dendrites. LOF mutations in the genes encoding PINK1 and Parkin impair mitochondrial biogenesis, while PINK1 mutations and hyperphosphorylation of tau contribute to impaired mitochondrial transport. The resulting loss of mitochondrial support leads to dendritic atrophy, an early sign of neurodegeneration. (4) Many neurodegenerative disease-associated genes also elicit altered calcium handling in other organelles. These alterations, particularly those at the ER-mitochondrial contact sites, may also contribute to dysregulation of cytosolic and/or mitochondrial calcium, although the exact relationships have not been directly addressed. Neurodegenerative disease-linked proteins (genes) include amyloid precursor protein (APP), glucocerebrosidase (GBA), huntingtin (HTT), leucine-rich repeat kinase 2 (LRRK2), Tau (MAPT), PTEN-induced kinase 1 (PINK), presenilin 1 and 2 (PSEN1/2), alpha-synuclein (SNCA) and superoxide dismutase 1 (SOD1). Mut, mutation; LOF, loss-of-function
Calcium regulation and mitochondria in neuronal sub-compartments
Calcium plays an important role in multiple signaling cascades within neurons. It is essential for both pre-synaptic and post-synaptic processes, as well as vesicular transport, cytoplasmic motility and cell death, which require exquisitely precise, spatially separated waves of calcium increase and decrease. Dendritic spines serve to isolate and concentrate calcium signals arising from synaptic activity [29]. Mitochondria contribute to rapid, post-stimulatory calcium recovery by taking up massive amounts of calcium [30] and then releasing calcium more gradually back into the cytosol. Other factors that are important for calcium signal recovery include NMDAR/AMPAR channel inactivation, calcium sequestration into the endoplasmic reticulum, and extrusion of calcium from neurons via plasma membrane sodium-calcium exchangers. Mitochondrial function is also important for powering ATP-dependent calcium pumps [31].
Healthy mitochondria in the perisynaptic region, particularly in excitatory neurons, act to transiently take up calcium and slowly release it back to the cytoplasm [32]. Cytosolic calcium concentration is usually 50–100 nM at baseline. When high-calcium microdomains are generated near mitochondria, as may be observed when the overall level of calcium exceeds 500 nM, mitochondrial calcium uptake is triggered [33]. Free calcium within the mitochondrial matrix is buffered by binding to proteins and chemicals to form insoluble calcium phosphate and calcium carbonate precipitates. This allows mitochondria to hold higher levels of calcium for longer periods while continuing to respond to transient elevations of cytosolic calcium. Once the free mitochondrial calcium rises to a threshold, it will be released back to the cytosol through efflux mechanisms. Thus, mitochondria have an immense capability to fine-tune cytosolic and mitochondrial calcium fluxes in a localized fashion within neurons.
The major protein complex involved in mitochondrial calcium uptake is the mitochondrial calcium uniporter (MCU) [34, 35]. This is a high-capacity, low-affinity uptake system, ensuring that the rapid, high-capacity calcium uptake into mitochondria is not triggered until the mitochondria experience a spike of high calcium levels in the vicinity, such as during synaptic transmission [36]. The thresholds and levels of MCU function are regulated by accessory proteins, including mitochondrial calcium uptake 1 (MICU1) [37], MICU2 [38], essential MCU regulator (EMRE) [39], mitochondrial calcium uniporter regulator 1 (MCUR1) [40] and MCUb [41]. Interestingly, the overall affinity of the MCU complex to calcium is higher in neurons compared to other cell types, due to the expression of a brain specific MICU3 isoform [42]. This allows axonal mitochondria to take up calcium in response to smaller changes in cytoplasmic calcium, thus facilitating calcium-dependent acceleration of local ATP synthesis.
Mitochondrial calcium uptake is balanced by the activity of proteins such as mitochondrial sodium calcium exchanger (NCLX) [43] and leucine zipper EF-hand containing transmembrane protein 1 (LETM1) [44], which release calcium back into the cytosol. NCLX is a sodium/calcium antiporter at the inner mitochondrial membrane that is important for mitochondrial calcium release in excitable cells such as neurons and muscle cells [45,46,47]. While LETM1 mediates calcium uptake in response to moderate increases in cytosolic calcium, it has also been proposed to show reversible activity allowing for calcium extrusion from the matrix [36]. The endoplasmic reticulum-mitochondrial contact sites also act to regulate mitochondrial calcium homeostasis [48].
Role of mitochondrial calcium homeostasis in synaptic transmission
Calcium impacts various mitochondrial processes including the activation of several metabolic enzymes to enhance ATP generation, increasing buffering capacity to prevent neuronal toxicity, and modulating mitochondrial transport (anterograde/retrograde) within the cell [49,50,51,52]. During synaptic transmission, perisynaptic mitochondria can buffer changes in calcium levels while maintaining the ATP balance [53]. Physiological levels of calcium uptake into the mitochondrial matrix enhance respiratory function, promoting ATP generation near sites of synaptic activity [54,55,56]. Increased cytosolic calcium generated during synaptic activity also acts to halt mitochondrial trafficking, resulting in mitochondrial accumulation near active synapses [57,58,59]. Recruitment of mitochondria to presynaptic terminals results in homeostatically decreased neurotransmitter release in response to prolonged activity [60]. Dysregulation of mitochondrial buffering capacity in conditional Liver kinase B1 (LKB1)-knockout neurons, which have impaired MCU activity [61, 62], leads to neuron hyperexcitability. LKB1 conditional knockout neurons show reduced axonal branching and decreased immobilization of mitochondria at nascent presynaptic sites [63]. In neurons, dendritic spines undergo waves of calcium transients. While free intracellular cytosolic calcium levels remain relatively low, approximately 50–100 nM, the localized calcium levels within spines may reach 10 μM during activation [64]. Taken together, these studies indicate that the mitochondrial calcium buffering capacity acts to prevent neuronal hyperexcitability [61].
In contrast to transient physiological calcium fluxes, sustained high levels of cytosolic calcium may lead to mitochondrial injury from calcium overload [65]. During classic excitotoxicity, excess mitochondrial calcium uptake can elicit reactive oxygen species (ROS) production [66], permeability transition pore opening [67], and cell death [68]. Further promoting mitochondrial calcium uptake via MCU overexpression exacerbates the NMDAR-mediated mitochondrial depolarization and excitotoxic cell death [69]. NADPH oxidase has been reported as a source of ROS following NMDAR activation [70]. Indeed, cell death caused by NMDA/AMPA/kainate receptor activation is attributed to ROS generation [71]. In the absence of oxygen, the agonist-induced cell death is attenuated, although cells are still capable of increasing intracellular calcium levels. This suggests that ROS and reactive nitrogen species (RNS) may be important down-stream effectors of toxicity elicited by increased intracellular calcium. Interestingly, nNOS-expressing neurons are resistant to the increase in NO mediated by NMDAR activation, possibly because of the increased expression of the antioxidant Mn superoxide dismutase in these cells [72]. Finally, protein–protein interactions involving NMDAR may also modulate the excitotoxic responses. The interaction of postsynaptic density-95 (PSD95) with NMDAR enhances the synthesis of NO, and suppression of PSD95 expression reverses the effects on NO without affecting cytosolic calcium uptake [73]. Similary, inhibiting the interaction of PSD95 with NMDAR using peptides prevents the ischemic brain damage induced by excitotoxicity [74]. Therefore, while elevated intracellular calcium is necessary for excitotoxic injury, it is not the only factor that must be considered.
Mitochondrial calcium dysregulation in neurodegenerative diseases
While the role of acute excitotoxic cell death remains unclear under chronic neurodegenerative conditions, recent studies have indicated a pathogenic role for excitatory mitochondrial calcium dysregulation in mediating sublethal dendritic atrophy observed in chronic neurodegenerative diseases. Simplification and atrophy of dendritic structures are observed in post-mortem studies of AD, PD, and ALS [75,76,77,78] and their animal models [79,80,81,82]. Inhibiting calcium uptake is frequently reported to be neuroprotective [3, 83,84,85], and MCU inhibitors are neuroprotective in multiple genetic models of chronic neurodegenerative diseases [4, 86,87,88,89].
These studies implicate mitochondrial calcium dysregulation, rather than inappropriate activation of cytosolic calcium-dependent enzymes, as the major mechanism by which increased excitatory neurotransmission triggers mitochondrial depletion from and retraction of dendritic structures. Below, we review the evidence that excitatory mitochondrial toxicity (EMT) mediates the phenotype of excitotoxic dendritic degeneration observed in several familial neurodegenerative disease models.
LRRK2 and excitatory mitochondrial toxicity
LRRK2, which encodes leucine-rich repeat kinase 2, is mutated in autosomal dominant PD. Dendritic atrophy is a prominent feature of neurons expressing disease-linked mutations in LRRK2 [3, 4, 80, 84, 90,91,92], in which alterations of microtubule dynamics, endolysosomal dynamics and/or autophagy have been implicated (reviewed in [93]).
Electrophysiological studies indicate that mutant LRRK2 elicits hyperexcitability in cortical and hippocampal neurons through postsynaptic mechanisms [3, 94]. Presynaptic mechanisms have also been proposed given that LRRK2 is identified in synaptic vesicle fractions [95], and overexpression of LRRK2 G2019S decreases endocytosis and increases exocytosis [96]. A major group of LRRK2 targets involves a family of proteins called Rabs (reviewed by [97]), which are involved in membrane trafficking and recycling of synaptic vesicles [98, 99]. Proteomic analysis of Drosophila neurons expressing LRRK2-R1441C has identified differentially expressed or phosphorylated proteins involved in synaptic vesicle transmission [100]. LRRK2 has also been shown to regulate the voltage-gated calcium channels (CaV2.1), which are responsible for the influx of calcium ions in response to membrane depolarization [101].
The process of EMT as a mediator of excitatory dendritic injury was first described in mutant LRRK2 models. Primary cortical neurons transfected with either LRRK2-G2019S or LRRK2-R1441C exhibit increased activity-dependent calcium influx through glutamate receptors and L-type calcium channels [3, 84]. Decreased density of mitochondria is observed specifically in post-synaptic structures, which precedes dendritic retraction [84]. The dendritic shrinkage can be blocked by inhibiting autophagy [84] or expressing an inactivating phosphomimic of the autophagy protein LC3 (microtubule-associated protein 1 light chain 3) at S/T12 [102], a site near the binding motif for cardiolipin-mediated mitophagy [103]. The use of genetically encoded calcium sensors established that both LRRK2-G2019S and LRRK2-R1441C elicited increased calcium uptake into mitochondria following neuronal depolarization [4]. The increased dendritic mitochondrial calcium uptake was mediated by transcriptional upregulation of MCU and MICU1 in mutant LRRK2 patient fibroblasts, and persisted even in permeabilized neurons exposed to identical “cytosolic” calcium concentrations. There were no changes in the expression of MICU2, which elevates the threshold for MCU activation, or NCLX, which extrudes calcium from mitochondria [4]. Neurons treated with MCU inhibitors, MCU RNAi or constitutively active forms of NCLX were protected from dendritic mitophagy and dendritic atrophy [4]. These data implicate calcium-dependent injury to dendritic mitochondria and their elimination by autophagy, mechanisms that link neuronal hyperexcitability to dendritic simplification.
PINK1, Parkin and excitatory mitochondrial toxicity
PTEN (Phosphatase and tensin homolog)-induced kinase 1 (PINK1) and Parkin are products of two recessive PD genes that have been heavily implicated in regulating one of the pathways of selective mitophagy (reviewed in [104]). Both enzymes also have other functions, some of which impact calcium handling in neurons.
Like primary neurons expressing mutant LRRK2, Pink1 knockout mice exhibit reduced dendritic arbors in midbrain, cortical and hippocampal neurons [81, 105, 106]. Older, but not young (6-month vs 2-month) Pink1 knockout mice exhibit increased excitatory neurotransmission, accompanied by increased neurotransmitter release on KCl stimulation without changes in protein expression of synaptic vesicle proteins syntaxin1a, Munc-18 or SNAP25 [107]. Moreover, Pink1 is able to modulate sensory dendrite pathology in a Drosophila model of chemotherapy-induced neuropathy [108]. Neurochemical analysis in Pink1 knockout rats showed higher glutamine levels as an indirect measure of glutamate neurotransmission [109]. Moreover, knocking down Pink1 expression increases the density of thin spines and decreases the expression of postsynaptic cluster proteins, PSD95 and Shank. These changes are proposed to increase the susceptibility of Pink1-deficient neurons to excitotoxicity [106]. The reciprocal effect on PSD95 has also been observed, whereby overexpression of PINK1 increases the expression of PSD95 [81]. Pink1 knockout rat models also display increased glutamate release in striatal spiny neurons, accompanied by enhanced frequency and amplitude of spontaneous excitatory postsynaptic currents and increased numbers of synapses as measured by electron microscopy [110].
One of the earliest shRNA studies of PINK1 in SH-SY5Y neuroblastoma cells revealed mitochondrial calcium overload due to deficient function of the Na+/Ca2+ calcium exchanger, lowering the threshold for opening of the mitochondrial permeability transition pore [111]. Additional observations supporting a role for mitochondrial calcium overload derives from observations that inhibiting mitochondrial calcium uptake using ruthenium red restores ΔΨm in cells co-expressing α-syn A53T and Pink1 W437X and rescues neurite outgrowth [112]. Moreover, mitochondria exhibit altered ultrastructure in PINK1-deficient cells [113]. Subsequently, PINK1 was found to facilitate NCLX activation through phosphorylation at a novel site by protein kinase A (PKA) [45]. PINK1 is able to directly phosphorylate the catalytic subunit of PKA [105], and either PKA or the NCLX phosphomimic is able to rescue the effects of PINK1 deficiency in PINK1 knockout primary neurons [45]. Subsequently, it was found that LETM1, another mitochondrial calcium transporter involved in the bidirectional movement of calcium [44], is a direct phosphorylation target of PINK1 [114].
Like neurons expressing mutant LRRK2 or deficient PINK1, loss of Parkin causes increased excitatory neurotransmission, but via different mechanisms. Parkin is an E3 ubiquitin ligase, which has been shown to be important for the pruning and degradation of excitatory synapses [115]. Loss of endogenous Parkin or expression of the PD-linked Parkin mutants increases the vulnerability to synaptic excitotoxicity. Parkin is also necessary for the maintenance of postsynaptic endocytosis of AMPARs [116], and acts to regulate the endoplasmic reticulum-mitochondrial crosstalk, with indirect effects on mitochondrial calcium levels [117]. However, whether or not these mechanisms contribute to EMT in Parkin-deficient models has yet to be studied.
Beta-amyloid (Aβ), tau and excitatory mitochondrial toxicity
Calcium mishandling is also believed to contribute to the pathogenesis of AD [118, 119]. Among the pathological hallmarks of AD is the accumulation of extracellular amyloid plaques and intracellular tangles of tau.
Oligomeric Aβ elicits elevated intracellular calcium levels accompanied by spine loss in vivo [120]. Accumulation of Aβ on synaptic mitochondria leads to mitochondrial calcium overload and loss of mitochondrial membrane potential [121,121,123]. In addition, overexpression of Aβ results in decreased pre-synaptic mitochondria, decreased synaptic vesicles and elevated synaptic fatigue in Drosophila neurons [124]. In the APP/PS1 transgenic mouse model of AD, secreted Aβ elevates the calcium concentrations in mitochondria in neurons, which can be ameliorated by treatment with an MCU inhibitor [125]. Interestingly, post-mortem AD brains exhibit downregulated expression of calcium influx genes, such as MCU, MICU1 and MICU2, but upregulated expression of NCLX. The authors of this study noted that such gene expression changes could be due to a compensatory response to chronic mitochondrial calcium overload. Only mRNA was examined in this study and it was unknown what stage(s) of disease or brain region(s) were analyzed. In contrast, another study showed decreased NCLX protein in both the 3xTg-AD mouse model and the frontal cortex of sporadic AD patients [126].
AD mouse models also show evidence of excitatory hyperactivity accompanied by decreases in mitochondrial buffering capacity [127]. In mice co-expressing mutated APPswe and mutated PS1G384A, spontaneous increases in hyperactive neurons were observed early in the hippocampus, prior to the formation of plaques [128]. Mutations in presenilins promote calcium leakage into the cytosol through endoplasmic reticulum calcium overload, accompanied by post-translational modification and enhanced recruitment of neuronal ryanodine receptors [118, 129, 130]. These mutations also exaggerate the inositol triphosphate-evoked calcium release, without affecting the cytosolic calcium buffering [131]. It has also been suggested that mutations in presenilins affect intracellular calcium levels [132], and that elevations in intracellular calcium play key roles in promoting tau pathology [133] through activation of microtubule affinity-regulating kinase, cyclin-dependent kinase 5 or AMP-activated kinase [134]. Interestingly, tau inhibits the activity of NCLX, leading to a decrease in mitochondrial calcium efflux and activation of apoptotic cell death [135]. Loss of NCLX function has been shown to accelerate pathology and memory decline in 3xTg-AD mice [126]. In contrast, in AD models expressing a mutant form of PS2, decreased rather than enhanced mitochondrial calcium uptake was linked to glutamate-induced excitotocity. This may be attributed to energy-deprivation with reduced capacity to handle stress (in this case a lower dose of glutamate) [136].
Mutant SOD1 and excitatory mitochondrial toxicity
Dysfunctional mitochondria contribute to the pathogenesis of both sporadic and familial ALS (reviewed in [137, 138]). Multiple genetic mutations have been identified in patients with familial ALS, the most common of which are SOD1 and C9orf-72. Mutations in the SOD1 gene encoding the Cu2+, Zn2+-dependent superoxide dismutase are found in 20% of patients with familial ALS [139]. Mice overexpressing mutant SOD1-G93A exhib i t motor neuron degeneration with sympoms relevant to human disease progression. Interestingly, this model also exhibits early deficits in mitochondrial calcium homeostasis. SOD1 G93A tran sgenic mice exhibit a significant decrease in mitochondrial calcium loading capacity in motor neurons, which precedes the disease onset [140]. Furthermore, in a study comparing disease progression between transgenic and non-transgenic animals, increased MCU expression was observed in embryonic SOD1 mutant motor neurons, while MCU downregulation was observed in adult presymptomatic and symptomatic SOD1 mutant animals. The authors suspect that this decrease of MCU is likely a compensatory response to early calcium dyshomeostasis [141]. However, as the number of mitochondria and the mitophagy were not directly studied, it is unknown whether the decrease in MCU expression could be explained by decreased mitochondrial content.
Mutant Huntingtin (mHTT) and excitatory mitochondrial toxicity
Mitochondrial calcium dyshomeostasis is also thought to be one of the causes for striatal neurodegeneration in HD. mHTT contains an abnormal expansion of polyglutamine at the N-terminus of HTT, which is unable to fold and is cleaved to form short peptides with a high propensity to aggregate. Mitochondria from HD patients exhibit decreased buffering capacity when challenged with a bolus of calcium compared to healthy controls, with mitochondria from juvenile-onset cases markedly worse than those from adult-onset cases [142]. Rodent mHtt transgenic models also exhibit mitochondrial calcium buffering dysfunction [143], which, interestingly, precedes the onset of motor dysfunction [142].
However, experiments using isolated mitochondria are limited by the removal of cellular contexts, and other factors such as ROS/RNS may modulate cellular susceptibility to excitotoxic injury. Within intact cells, mitochondria exhibit high respiratory control, and in this context, moderate expression of mHtt does not significantly impair mitochondrial respiration in resting young neurons. Yet, when challenged with calcium by transiently activating NMDARs, mitochondria from striatal neurons expressing mHtt fail to reestablish calcium homeostasis compared to wildtype [144]. Interestingly, a subset of neurons are spared in HD. Studies by Canzoniero et al. demonstrated that these neurons express high levels of nNOS. While they show similar calcium uptake and NMDAR-evoked responses as other neurons, these neurons have decreased ROS generation in response to NMDAR activation compared to cultured neurons expressing lower levels of nNOS [145].
Similarities and differences between excitotoxicity and EMT
In classic excitotoxicity, the cytosolic calcium overload results in enzyme activation and acute cell death [8]. This is not the situation observed in most genetic models of neurodegenerative diseases, in which cell death is often not a prominent feature. This may be related to the difference in the level of calcium disruption. Much greater fluxes are triggered throughout the neuron during classic excitotoxic injury due to ischemia-mediated failure of ATP-dependent pumps, compared to the effects of chronically elevated excitatory postsynaptic potential frequency observed in several genetic models of neurodegeneration.
As discussed above, excitatory injury may be either exacerbated by or ameliorated by inhibition of mitochondrial calcium uptake via MCU. This may be related to the presumed site of calcium toxicity. In classic excitotoxicity, high cytosolic levels of calcium result in enzyme activation, leading to cell death. For example, in Gaucher’s disease (GBA−/−), increased sensitivity to excitotoxic injury has been attributed to a decrease of mitochondrial calcium uptake due to the decreased MCU expression [146]. Notably, Gaucher’s disease involves infantile presentation of disease, whereas heterozygous GBA mutations have been implicated in PD. Insufficient mitochondrial calcium buffering would be predicted to contribute to increased sensitivity to classic cytosolic calcium toxicity mechanisms as opposed to excitatory mitochondrial toxicity. In this context, enhanced uptake of calcium by mitochondria, endoplasmic reticulum and other organelles acts to reduce injury by reducing the cytosolic calcium level.
Yet elevated calcium uptake via MCU can also lead to cell death during acute injury. In stroke, there is a biphasic response to CsA treatment, suggesting an initial phase during which glutamate toxicity can be reversed, which is not dependent on the energy reserve of the cell. However, after prolonged exposure, there is a collapse in the ATP content of the cell. Under these circumstances, mitochondrial calcium uptake activates PARP (poly-adenosine ribosyl polymerase) to trigger NADH depletion and mitochondrial membrane potential collapse [147].
In EMT, it is the mitochondria themselves that are injured, triggering mitophagy, mitochondrial depletion from dendrites and dendritic atrophy (Fig. 1). The injured mitochondria undergo mitophagy, leading to insufficient mitochondrial support of dendritic structures. Inhibiting mitochondrial calcium uptake by MCU, or facilitating recovery by regulated efflux through NCLX, can protect dendritic mitochondria and dendritic structures.
While several genetic mutations discussed above result in hyperexcitability of synaptic function, which will cause localized increases in intracellular calcium, calcium levels alone do not determine toxicity. Instead, emerging evidence implicates multifactorial pathogenesis. As discussed above, the site of calcium entry and the presence or absence of antioxidant defenses against ROS/RNS may also regulate susceptibility to excitatory injury.
Likewise, while hyperexcitability is a driving factor for EMT, it is not the only factor. For instance, elevated synaptic activity may be combined with intrinsic dysregulation of mitochondrial calcium transporters (Fig. 1). Permeabilization experiments have shown that the increased mitochondrial calcium uptake in the dendrites of mutant LRRK2- expressing neurons persists even after the elevations of cytosolic calcium caused by synaptic hyperexcitability have been controlled [4]. Moreover, a recent study demonstrated that MCU overexpression is sufficient to cause pathology in the absence of synaptic hyperactivity [148]. This may reflect changes in mitochondrial calcium transport protein expression, post-translational modification and/or function that are induced by genetic mutations.
Finally, the potential contributions of altered endoplasmic reticulum, Golgi, lysosomal and plasma membrane calcium pump function remain to be studied in the context of EMT. Disruptions in calcium handling by lysosomes have been repo rted in models of PD and ALS [149, 150]. Certainly, any change that promotes even mild, chronic elevations in cytosolic calcium has the potential to enhance calcium uptake by mitochondria and/or to im pair their rec overy from an episode of calcium buffering.
Neuroprotective strategies for EMT
Neuroprotection against EMT and excitatory dendritic degeneration could conceivably be achieved by targeting multiple processes. These include strategies to normalize the changes observed in the different model systems at the level of (1) presynaptic vesicle exocytosis, (2) post-synaptic calcium uptake, (3) enhanced mitochondrial quality, (4) preventing overactivation of mitochondrial calcium uptake, and (5) promoting the regulated release of calcium out of mitochondria.
As mechanisms underlying increased excitatory neurotransmission are fairly different from model to model, the first strategy of reducing excess glutamate release may be difficult to achieve. In contrast, numerous studies have shown the benefit of blocking calcium uptake through either glutamate receptors or L-type calcium channels [3, 83, 84, 151,151,153]. Yet, the impact of inhibiting these pathways in the more complex human in vivo setting is difficult to predict, particularly if the mechanism of injury involves intrinsic changes to mitochondrial calcium handling or mitochondrial dynamics (Fig. 1) that are not corrected by blocking extracellular calcium uptake. Clinical trials on most NMDAR antagonists have failed to demonstrate benefit for stroke and traumatic brain injury, with side effects triggering early termination [154]. Given the differences in mechanism that may be triggered during acute brain injury relative to chronic neurodegeneration, it is interesting to note that the low-affinity NMDAR antagonist memantine did show small, but significant benefits for moderate to severe AD [27] and has been approved for clinical use.
Mitochondrial quality control is mediated by localized degradation and repair, including the mitochondrial unfolded protein response, or by whole organelle turnover through autophagy [155]. Basal mitophagy is undoubtedly important for maintaining mitochondrial resilience. Impaired calcium recovery [156] and elevated basal mitophagy have been observed in neuronal cells [113, 155] and in pancreatic beta cells in vivo [157], evidently through one of the several PINK1- and Parkin-independent mechanisms [103, 158, 159]. Basal mitophagy in vivo also appears to be mediated by PINK1/Parkin-independent pathways [157, 160,160,162]. Induced mitophagy can be regulated by at least three distinct mechanisms: PINK1/Parkin-induced mitochondrial ubiquitination, externalization of the inner mitochondrial lipid cardiolipin, and transcriptional upregulation of mitophagy receptors such as FUNDC1 [104]. In settings where PINK1 and Parkin are not mutated, small molecules that serve to enhance endogenous PINK1 activity [163] or expression [164] may show beneficial effects.
However, the impact of augmenting injury-induced mitophagy is not straightforward to predict. While mitophagy generally promotes cell survival, a potential side effect of excessive mitophagy in neurons is the loss of synaptic structures due to mitochondrial depletion, particularly under disease conditions that impair mitochondrial biogenesis [165,165,167]. Dendritic degeneration in culture models of LRRK2- and PINK1-related neurodegeneration is preceded by the loss of dendritic mitochondria [81, 84]. Enhanced autophagy has been shown to contribute to excitotoxic lesions in neonatal rats [168]. Parkin-mediated mitophagy has also been implicated in glutamate excitotoxicity in culture systems [169]. These factors may explain the perhaps unexpected observation that inhibiting autophagy or mitophagy acts to preserve dendritic arbors [170, 171]. Furthermore, the potential effects of modulating autophagy or mitophagy on other cell types in the brain, such as glial and vascular cells, remain to be delineated.
Perhaps the most promising avenue for protecting against neuronal atrophy and synaptic loss due to EMT involves the normalization of mitochondrial calcium flux. Knockdown of MCU and application of MCU inhibitors are effective in multiple neurodegenerative disease models [4, 88, 89, 172]. This is the case even when the primary mechanism of EMT involves impaired NCLX activity [45, 126]. Conversely, overexpressing NCLX or its constitutively active phosphomimic NCLX-S258D, confers protection against neuronal atrophy and dendritic loss even when the primary mechanism involves hyperactivation of MCU [4, 173]. In the appropriate familial disease setting, it may also be possible to use LRRK2 kinase inhibitors [174] or small molecules that augment PINK1 expression [164] to reverse downstream changes in mitochondrial calcium flux.
The potential contributions of other brain cell types must also be considered. While global MCU knockout does not show protection against hypoxia ischemic injury [175], neuronal specific conditional knockdown or siRNA-mediated knockdown of MCU has been shown to be neuroprotective in this injury model [176]. Furthermore, there may be tissue-specific minor pathways through with calcium ions can enter the mitochondria. In MCU-KO mice, isolated mitochondria from nonsynaptic and synaptic regions show slowed calcium uptake, which is inhibited by Ru360. However, mitochondria isolated from liver, heart and skeletal muscle tissue show completely inhibited calcium uptake [177].
Although numerous studies have shown that inhibiting MCU may be neuroprotective, it is important to keep in mind that these strategies may exacerbate classic excitotoxic mechanisms. Several acute and some chronic disease contexts involve decreased MCU activity [136, 146], and impaired mitochondrial uptake to begin with. Thus, it would be important to continue to define which steps of calcium homeostasis are affected in a given familial or sporadic neurodegeneration context (Fig. 1) to delineate those settings involving EMT-driven pathogenic mechanisms.
Conclusion
Neurons respond differently to acute vs chronic excitotoxic stimuli. During acute excitotoxicity stimuli, due to energy crisis, neurons undergo cell death. This is preceded by a rapid influx of calcium into the cells, which further exacerbates the situation. In contrast, chronic excitotoxicity is elicited by a sublethal increases in excitatory neurotransmission. There is a gradual loss of neuronal function and dendritic atrophy, eventually leading to neuron loss.
Recent studies have conclusively shown that in addition to cytosolic calcium, mitochondrial calcium also plays an important role in regulating synaptic activity. Apart from being an ATP generator, mitochondria are important for calcium buffering, particularly in excitatory neurons.
Shortening of dendrites and loss of dendritic arborization can occur independently of or preceding neuronal cell death. Dendrites are important for segregating calcium signals and dendritic mitochondria play a vital role in buffering calcium during synaptic activity. Localized increases in calcium levels at synapses halt mitochondrial transport, accumulating them near synapses. Synapses lacking mitochondria in their vicinity show increased hyperexcitability, with loss of homeostatic downregulating mechanisms. Hyperactive neurons are often accompanied by sustained higher cytosolic calcium levels leading to mitochondrial calcium overload, which in turn increases mitochondrial ROS production and mitochondrial damage, leading to enhanced mitochondrial removal by autophagy.
Neurodegenerative diseases that show loss of mitochondria are particularly susceptible to excitotoxicity. For example, neurons expressing LRRK2 mutants (G2019S or R1441C) show decreased dendritic mitochondrial content as well as increased mitochondrial calcium overload which can be prevented by inhibiting MCU activity [4]. Similarly, loss of PINK1 function induces hyperexcitability as evidenced by an increase in glutamine [109]. Recent studies have implicated PINK1 in the regulation of mitochondrial calcium homeostasis by regulating mitochondrial NCLX function through phosphorylation [45]. Like PINK1, loss-of-function mutation in Parkin also increases the vulnerability of neurons to synaptic excitotoxicity [115].
The role of calcium in the pathogenesis of AD, ALS and HD is also beginning to emerge. Exposure to oligomeric Aβ causes spine loss which might be associated with mitochondrial calcium overload, and loss of functional mitochondria. Deletion of NCLX accelerates memory decline and AD-type pathology in mouse models [126]. Improper calcium handling has also been shown to play a role in tau-mediated pathology [135]. Considering all the evidence, dysregulation of mitochondrial calcium homeostasis makes neuronal cells susceptible to excitotoxic injury. From a therapeutic point of view, preservation of mitochondrial function by targeting the mitochondrial calcium homeostasis machinery provides an attractive target to develop the next generation of drugs that can prevent or slow these devastating neurodegenerative disorders.
Availability of data and materials
Not applicable.
Abbreviations
- Aβ:
-
Amyloid beta
- EMT:
-
Excitatory mitochondrial toxicity
- LRRK2:
-
Leucine-rich repeat kinase 2
- MCU:
-
Mitochondrial calcium uniporter
- NCLX:
-
Mitochondrial sodium calcium exchanger
- NMDA:
-
N-Methyl-d-aspartic acid
- NMDAR:
-
N-Methyl-d-aspartic acid receptor
- PINK1:
-
PTEN induced kinase 1
- AMPA:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionate
- AMPAR:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor
- GABA:
-
γ-Aminobutyric acid
- LETM1:
-
Leucine zipper and EF-hand containing transmembrane protein 1
- LKB1:
-
Liver kinase B1
References
Gwag BJ, Lobner D, Koh JY, Wie MB, Choi DW. Blockade of glutamate receptors unmasks neuronal apoptosis after oxygen-glucose deprivation in vitro. Neuroscience. 1995;68(3):615–9.
Guerriero RM, Giza CC, Rotenberg A. Glutamate and GABA imbalance following traumatic brain injury. Curr Neurol Neurosci Rep. 2015;15(5):27.
Plowey ED, Johnson JW, Steer E, Zhu W, Eisenberg DA, Valentino NM, et al. Mutant LRRK2 enhances glutamatergic synapse activity and evokes excitotoxic dendrite degeneration. Biochim Biophys Acta. 2014;1842(9):1596–603.
Verma M, Callio J, Otero PA, Sekler I, Wills ZP, Chu CT. Mitochondrial calcium dysregulation contributes to dendrite degeneration mediated by PD/LBD-associated LRRK2 mutants. J Neurosci. 2017;37(46):11151–65.
Okubo Y, Sekiya H, Namiki S, Sakamoto H, Iinuma S, Yamasaki M, et al. Imaging extrasynaptic glutamate dynamics in the brain. Proc Natl Acad Sci U S A. 2010;107(14):6526–31.
Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65(1):1–105.
Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993;13(5):2085–104.
Choi DW. Excitotoxicity: still hammering the Ischemic brain in 2020. Front Neurosci. 2020;14:579953.
Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, et al. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010;140(2):222–34.
Martel MA, Ryan TJ, Bell KF, Fowler JH, McMahon A, Al-Mubarak B, et al. The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron. 2012;74(3):543–56.
Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5(5):405–14.
Randall RD, Thayer SA. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci. 1992;12(5):1882–95.
Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters CA, Andrews SB. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc Natl Acad Sci U S A. 2009;106(24):9854–9.
Sattler R, Charlton MP, Hafner M, Tymianski M. Distinct influx pathways, not calcium load, determine neuronal vulnerability to calcium neurotoxicity. J Neurochem. 1998;71(6):2349–64.
Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Brück W, Jellinger K, et al. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease. evidence for apoptotic cell death. Am J Pathol. 1999;155(5):1459–66.
Rohn TT, Rissman RA, Davis MC, Kim YE, Cotman CW, Head E. Caspase-9 activation and caspase cleavage of tau in the Alzheimer’s disease brain. Neurobiol Dis. 2002;11(2):341–54.
Hartmann A, Michel PP, Troadec JD, Mouatt-Prigent A, Faucheux BA, Ruberg M, et al. Is Bax a mitochondrial mediator in apoptotic death of dopaminergic neurons in Parkinson’s disease? J Neurochem. 2001;76(6):1785–93.
Iannielli A, Bido S, Folladori L, Segnali A, Cancellieri C, Maresca A, et al. Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in Parkinson’s disease models. Cell Rep. 2018;22(8):2066–79.
Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, et al. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5(1):101–6.
Lassmann H, Bancher C, Breitschopf H, Wegiel J, Bobinski M, Jellinger K, et al. Cell death in Alzheimer’s disease evaluated by DNA fragmentation in situ. Acta Neuropathol. 1995;89(1):35–41.
Guo W, Stoklund Dittlau K, Van Den Bosch L. Axonal transport defects and neurodegeneration: molecular mechanisms and therapeutic implications. Semin Cell Dev Biol. 2020;99:133–50.
Ciechanover A, Kwon YT. Protein quality control by molecular chaperones in neurodegeneration. Front Neurosci. 2017;11:185.
Grünewald A, Rygiel KA, Hepplewhite PD, Morris CM, Picard M, Turnbull DM. Mitochondrial DNA depletion in respiratory chain-deficient Parkinson disease neurons. Ann Neurol. 2016;79(3):366–78.
Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–95.
Caudle WM, Zhang J. Glutamate, excitotoxicity, and programmed cell death in Parkinson disease. Exp Neurol. 2009;220(2):230–3.
Vanle B, Olcott W, Jimenez J, Bashmi L, Danovitch I, IsHak WW. NMDA antagonists for treating the non-motor symptoms in Parkinson’s disease. Transl Psychiatry. 2018;8(1):117.
Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med. 2003;348(14):1333–41.
Trudler D, Sanz-Blasco S, Eisele YS, Ghatak S, Bodhinathan K, Akhtar MW, et al. α-Synuclein oligomers induce glutamate release from astrocytes and excessive extrasynaptic NMDAR activity in neurons, thus contributing to synapse loss. J Neurosci. 2021;41(10):2264–73.
Koch C, Zador A, Brown TH. Dendritic spines: convergence of theory and experiment. Science. 1992;256(5059):973–4.
White RJ, Reynolds IJ. Mitochondria accumulate Ca2+ following intense glutamate stimulation of cultured rat forebrain neurones. J Physiol. 1997;498(Pt 1):31–47.
Budd SL, Nicholls DG. A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. J Neurochem. 1996;66(1):403–11.
Tang Y, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18(3):483–91.
Rizzuto R, Simpson AW, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature. 1992;358(6384):325–7.
Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476(7360):341–5.
De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476(7360):336–40.
De Marchi U, Santo-Domingo J, Castelbou C, Sekler I, Wiederkehr A, Demaurex N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J Biol Chem. 2014;289(29):20377–85.
Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, et al. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010;467(7313):291–6.
Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One. 2013;8(2):e55785.
Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013;342(6164):1379–82.
Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. 2012;14(12):1336–43.
Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, et al. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013;32(17):2362–76.
Ashrafi G, de Juan-Sanz J, Farrell RJ, Ryan TA. Molecular tuning of the axonal mitochondrial Ca(2+) uniporter ensures metabolic flexibility of neurotransmission. Neuron. 2020;105(4):678-687.e675.
Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A. 2010;107(1):436–41.
Doonan PJ, Chandramoorthy HC, Hoffman NE, Zhang X, Cardenas C, Shanmughapriya S, et al. LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. Faseb J. 2014;28(11):4936–49.
Kostic M, Ludtmann MH, Bading H, Hershfinkel M, Steer E, Chu CT, et al. PKA phosphorylation of NCLX reverses mitochondrial calcium overload and depolarization, promoting survival of PINK1-deficient dopaminergic neurons. Cell Rep. 2015;13(2):376–86.
Britti E, Delaspre F, Tamarit J, Ros J. Calpain-inhibitors protect frataxin-deficient dorsal root ganglia neurons from loss of mitochondrial Na(+)/Ca(2+) exchanger, NCLX, and apoptosis. Neurochem Res. 2021;46(1):108–19.
De La Fuente S, Lambert JP, Nichtova Z, Fernandez Sanz C, Elrod JW, Sheu SS, et al. Spatial separation of mitochondrial calcium uptake and extrusion for energy-efficient mitochondrial calcium signaling in the heart. Cell Rep. 2018;24(12):3099-3107.e3094.
Raffaello A, Mammucari C, Gherardi G, Rizzuto R. Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem Sci. 2016;41:1035–49.
Schulz JB. Mechanisms of neurodegeneration in idiopathic Parkinson’s disease. Parkinsonism Relat Disord. 2007;13(Suppl 3):S306-308.
Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6(3):337–50.
Celsi F, Pizzo P, Brini M, Leo S, Fotino C, Pinton P, et al. Mitochondria, calcium and cell death: a deadly triad in neurodegeneration. Biochim Biophys Acta. 2009;1787(5):335–44.
Ureshino RP, Erustes AG, Bassani TB, Wachilewski P, Guarache GC, Nascimento AC, et al. The interplay between Ca(2+) signaling pathways and neurodegeneration. Int J Mol Sci. 2019;20(23):6004.
Pathak D, Shields LY, Mendelsohn BA, Haddad D, Lin W, Gerencser AA, et al. The role of mitochondrially derived ATP in synaptic vesicle recycling. J Biol Chem. 2015;290(37):22325–36.
Bianchi K, Rimessi A, Prandini A, Szabadkai G, Rizzuto R. Calcium and mitochondria: mechanisms and functions of a troubled relationship. Biochim Biophys Acta. 2004;1742(1–3):119–31.
Vos M, Lauwers E, Verstreken P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci. 2010;2:139.
Rangaraju V, Calloway N, Ryan TA. Activity-driven local ATP synthesis is required for synaptic function. Cell. 2014;156(4):825–35.
Cai Q, Davis ML, Sheng ZH. Regulation of axonal mitochondrial transport and its impact on synaptic transmission. Neurosci Res. 2011;70(1):9–15.
Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, et al. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61(4):541–55.
Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, et al. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132(1):137–48.
Vaccaro V, Devine MJ, Higgs NF, Kittler JT. Miro1-dependent mitochondrial positioning drives the rescaling of presynaptic Ca2+ signals during homeostatic plasticity. EMBO Rep. 2017;18(2):231–40.
Kwon SK, Sando R 3rd, Lewis TL, Hirabayashi Y, Maximov A, Polleux F. LKB1 regulates mitochondria-dependent presynaptic calcium clearance and neurotransmitter release properties at excitatory synapses along cortical axons. PLoS Biol. 2016;14(7):e1002516.
Marland JR, Hasel P, Bonnycastle K, Cousin MA. Mitochondrial calcium uptake modulates synaptic vesicle endocytosis in central nerve terminals. J Biol Chem. 2016;291(5):2080–6.
Courchet J, Lewis TL Jr, Lee S, Courchet V, Liou DY, Aizawa S, et al. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell. 2013;153(7):1510–25.
Higley MJ, Sabatini BL. Calcium signaling in dendritic spines. Cold Spring Harb Perspect Biol. 2012;4(4):a005686.
Pivovarova NB, Nguyen HV, Winters CA, Brantner CA, Smith CL, Andrews SB. Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J Neurosci. 2004;24(24):5611–22.
Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15(5 Pt 1):3318–27.
Li V, Brustovetsky T, Brustovetsky N. Role of cyclophilin D-dependent mitochondrial permeability transition in glutamate-induced calcium deregulation and excitotoxic neuronal death. Exp Neurol. 2009;218(2):171–82.
Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1(5):366–73.
Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, et al. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun. 2013;4:2034.
Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, et al. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12(7):857–63.
Dubinsky JM, Kristal BS, Elizondo-Fournier M. An obligate role for oxygen in the early stages of glutamate-induced, delayed neuronal death. J Neurosci. 1995;15(11):7071–8.
Gonzalez-Zulueta M, Ensz LM, Mukhina G, Lebovitz RM, Zwacka RM, Engelhardt JF, et al. Manganese superoxide dismutase protects nNOS neurons from NMDA and nitric oxide-mediated neurotoxicity. J Neurosci. 1998;18(6):2040–55.
Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284(5421):1845–8.
Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, et al. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science. 2002;298(5594):846–50.
Baloyannis SJ, Costa V, Michmizos D. Mitochondrial alterations in Alzheimer’s disease. Am J Alzheimers Dis Demen. 2004;19(2):89–93.
Patt S, Gertz HJ, Gerhard L, Cervos-Navarro J. Pathological changes in dendrites of substantia nigra neurons in Parkinson’s disease: a Golgi study. Histol Histopathol. 1991;6(3):373–80.
Stephens B, Mueller AJ, Shering AF, Hood SH, Taggart P, Arbuthnott GW, et al. Evidence of a breakdown of corticostriatal connections in Parkinson’s disease. Neuroscience. 2005;132(3):741–54.
Hammer RP Jr, Tomiyasu U, Scheibel AB. Degeneration of the human Betz cell due to amyotrophic lateral sclerosis. Exp Neurol. 1979;63(2):336–46.
Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, et al. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30(7):2636–49.
MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52(4):587–93.
Dagda RK, Pien I, Wang R, Zhu J, Wang KZ, Callio J, et al. Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J Neurochem. 2014;128(6):864–77.
Fogarty MJ, Klenowski PM, Lee JD, Drieberg-Thompson JR, Bartlett SE, Ngo ST, et al. Cortical synaptic and dendritic spine abnormalities in a presymptomatic TDP-43 model of amyotrophic lateral sclerosis. Sci Rep. 2016;6:37968.
Ilijic E, Guzman JN, Surmeier DJ. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiol Dis. 2011;43(2):364–71.
Cherra SJ III, Steer E, Gusdon AM, Kiselyov K, Chu CT. Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol. 2013;182(2):474–84.
Esposito Z, Belli L, Toniolo S, Sancesario G, Bianconi C, Martorana A. Amyloid beta, glutamate, excitotoxicity in Alzheimer’s disease: are we on the right track? CNS Neurosci Ther. 2013;19(8):549–55.
Soman S, Keatinge M, Moein M, Da Costa M, Mortiboys H, Skupin A, et al. Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons in pink1(-/-) zebrafish. Eur J Neurosci. 2017;45(4):528–35.
Sharma Y, Garabadu D. Ruthenium red, mitochondrial calcium uniporter inhibitor, attenuates cognitive deficits in STZ-ICV challenged experimental animals. Brain Res Bull. 2020;164:121–35.
Soman SK, Bazała M, Keatinge M, Bandmann O, Kuznicki J. Restriction of mitochondrial calcium overload by mcu inactivation renders a neuroprotective effect in zebrafish models of Parkinson's disease. Biol Open. 2019;8(10).
Xie N, Wu C, Wang C, Cheng X, Zhang L, Zhang H, et al. Inhibition of the mitochondrial calcium uniporter inhibits Aβ-induced apoptosis by reducing reactive oxygen species-mediated endoplasmic reticulum stress in cultured microglia. Brain Res. 2017;1676:100–6.
Ramonet D, Daher JP, Lin BM, Stafa K, Kim J, Banerjee R, et al. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One. 2011;6(4):e18568.
Reinhardt P, Schmid B, Burbulla LF, Schondorf DC, Wagner L, Glatza M, et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell. 2013;12(3):354–67.
Winner B, Melrose HL, Zhao C, Hinkle KM, Yue M, Kent C, et al. Adult neurogenesis and neurite outgrowth are impaired in LRRK2 G2019S mice. Neurobiol Dis. 2011;41(3):706–16.
Verma M, Steer EK, Chu CT. ERKed by LRRK2: a cell biological perspective on hereditary and sporadic Parkinson’s disease. Biochim Biophys Acta. 2014;1842(8):1273–81.
Sweet ES, Saunier-Rebori B, Yue Z, Blitzer RD. The Parkinson’s disease-associated mutation LRRK2-G2019S impairs synaptic plasticity in mouse hippocampus. J Neurosci. 2015;35(32):11190–5.
Piccoli G, Onofri F, Cirnaru MD, Kaiser CJ, Jagtap P, Kastenmüller A, et al. Leucine-rich repeat kinase 2 binds to neuronal vesicles through protein interactions mediated by its C-terminal WD40 domain. Mol Cell Biol. 2014;34(12):2147–61.
Pan PY, Li X, Wang J, Powell J, Wang Q, Zhang Y, et al. Parkinson’s disease-associated LRRK2 hyperactive kinase mutant disrupts synaptic vesicle trafficking in ventral midbrain neurons. J Neurosci. 2017;37(47):11366–76.
Kuwahara T, Iwatsubo T. The emerging functions of LRRK2 and Rab GTPases in the endolysosomal system. Front Neurosci. 2020;14:227.
Yun HJ, Kim H, Ga I, Oh H, Ho DH, Kim J, et al. An early endosome regulator, Rab5b, is an LRRK2 kinase substrate. J Biochem. 2015;157(6):485–95.
Shin N, Jeong H, Kwon J, Heo HY, Kwon JJ, Yun HJ, et al. LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res. 2008;314(10):2055–65.
Islam MS, Nolte H, Jacob W, Ziegler AB, Pütz S, Grosjean Y, et al. Human R1441C LRRK2 regulates the synaptic vesicle proteome and phosphoproteome in a Drosophila model of Parkinson’s disease. Hum Mol Genet. 2016;25(24):5365–82.
Bedford C, Sears C, Perez-Carrion M, Piccoli G, Condliffe SB. LRRK2 regulates voltage-gated calcium channel function. Front Mol Neurosci. 2016;9:35.
Cherra SJ, Kulich SM, Uechi G, Balasubramani M, Mountzouris J, Day BW, Chu CT. Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol. 2010;190(4):533–9. https://doi.org/10.1083/jcb.201002108.
Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15(10):1197–205.
Chu CT. Mechanisms of selective autophagy and mitophagy: implications for neurodegenerative diseases. Neurobiol Dis. 2019;122:23–34.
Wang KZQ, Steer E, Otero PA, Bateman NW, Cheng MH, Scott AL, et al. PINK1 interacts with VCP/p97 and activates PKA to promote NSFL1C/p47 phosphorylation and dendritic arborization in neurons. eNeuro. 2018;5(6).
Hernández CJ, Báez-Becerra C, Contreras-Zárate MJ, Arboleda H, Arboleda G. PINK1 silencing modifies dendritic spine dynamics of mouse hippocampal neurons. J Mol Neurosci. 2019;69(4):570–9.
Feligioni M, Mango D, Piccinin S, Imbriani P, Iannuzzi F, Caruso A, et al. Subtle alterations of excitatory transmission are linked to presynaptic changes in the hippocampus of PINK1-deficient mice. Synapse. 2016;70(6):223–30.
Kim YY, Yoon JH, Um JH, Jeong DJ, Shin DJ, Hong YB, et al. PINK1 alleviates thermal hypersensitivity in a paclitaxel-induced Drosophila model of peripheral neuropathy. PLoS One. 2020;15(9):e0239126.
Ren X, Hinchie A, Swomley A, Powell DK, Butterfield DA. Profiles of brain oxidative damage, ventricular alterations, and neurochemical metabolites in the striatum of PINK1 knockout rats as functions of age and gender: relevance to Parkinson disease. Free Radic Biol Med. 2019;143:146–52.
Creed RB, Roberts RC, Farmer CB, McMahon LL, Goldberg MS. Increased glutamate transmission onto dorsal striatum spiny projection neurons in Pink1 knockout rats. Neurobiol Dis. 2021;150:105246.
Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009;33(5):627–38.
Marongiu R, Spencer B, Crews L, Adame A, Patrick C, Trejo M, et al. Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J Neurochem. 2009;108:1561–74.
Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284(20):13843–55.
Huang E, Qu D, Huang T, Rizzi N, Boonying W, Krolak D, et al. PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat Commun. 2017;8(1):1399.
Helton TD, Otsuka T, Lee MC, Mu Y, Ehlers MD. Pruning and loss of excitatory synapses by the parkin ubiquitin ligase. Proc Natl Acad Sci U S A. 2008;105(49):19492–7.
Cortese GP, Zhu M, Williams D, Heath S, Waites CL. Parkin deficiency reduces hippocampal glutamatergic neurotransmission by impairing AMPA receptor endocytosis. J Neurosci. 2016;36(48):12243–58.
Cali T, Ottolini D, Negro A, Brini M. Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim Biophys Acta. 2013;1832(4):495–508.
Popugaeva E, Pchitskaya E, Bezprozvanny I. Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease—a therapeutic opportunity? Biochem Biophys Res Commun. 2017;483(4):998–1004.
Lopez JR, Lyckman A, Oddo S, Laferla FM, Querfurth HW, Shtifman A. Increased intraneuronal resting [Ca2+] in adult Alzheimer’s disease mice. J Neurochem. 2008;105(1):262–71.
Arbel-Ornath M, Hudry E, Boivin JR, Hashimoto T, Takeda S, Kuchibhotla KV, et al. Soluble oligomeric amyloid-beta induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol Neurodegener. 2017;12(1):27.
Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010;107(43):18670–5.
Keller JN, Lauderback CM, Butterfield DA, Kindy MS, Yu J, Markesbery WR. Amyloid beta-peptide effects on synaptosomes from apolipoprotein E-deficient mice. J Neurochem. 2000;74(4):1579–86.
Sanz-Blasco S, Valero RA, Rodríguez-Crespo I, Villalobos C, Núñez L. Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS One. 2008;3(7):e2718.
Zhao XL, Wang WA, Tan JX, Huang JK, Zhang X, Zhang BZ, et al. Expression of beta-amyloid induced age-dependent presynaptic and axonal changes in Drosophila. J Neurosci. 2010;30(4):1512–22.
Calvo-Rodriguez M, Hou SS, Snyder AC, Kharitonova EK, Russ AN, Das S, et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat Commun. 2020;11(1):2146.
Jadiya P, Kolmetzky DW, Tomar D, Di Meco A, Lombardi AA, Lambert JP, et al. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat Commun. 2019;10(1):3885.
Du H, Guo L, Zhang W, Rydzewska M, Yan S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol Aging. 2011;32(3):398–406.
Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, et al. Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2012;109(22):8740–5.
Lacampagne A, Liu X, Reiken S, Bussiere R, Meli AC, Lauritzen I, et al. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017;134(5):749–67.
Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci. 2006;26(19):5180–9.
Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004;24(2):508–13.
Pizzo P, Basso E, Filadi R, Greotti E, Leparulo A, Pendin D, et al. Presenilin-2 and calcium handling: molecules, organelles, cells and brain networks. Cells. 2020;9(10):2166.
Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30(36):11938–50.
Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Abeta oligomers through Tau phosphorylation. Neuron. 2013;78(1):94–108.
Britti E, Ros J, Esteras N, Abramov AY. Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell Calcium. 2020;86:102150.
Rossi A, Rigotto G, Valente G, Giorgio V, Basso E, Filadi R, et al. Defective mitochondrial pyruvate flux affects cell bioenergetics in alzheimer’s disease-related models. Cell Rep. 2020;30(7):2332-2348.e2310.
Smith EF, Shaw PJ, De Vos KJ. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett. 2019;710:132933.
Swerdlow RH, Parks JK, Pattee G, Parker WD Jr. Role of mitochondria in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(3):185–90.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62.
Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, et al. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem. 2006;96(5):1349–61.
Tadić V, Adam A, Goldhammer N, Lautenschlaeger J, Oberstadt M, Malci A, et al. Investigation of mitochondrial calcium uniporter role in embryonic and adult motor neurons from G93A(hSOD1) mice. Neurobiol Aging. 2019;75:209–22.
Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, et al. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5(8):731–6.
Gellerich FN, Gizatullina Z, Nguyen HP, Trumbeckaite S, Vielhaber S, Seppet E, et al. Impaired regulation of brain mitochondria by extramitochondrial Ca2+ in transgenic Huntington disease rats. J Biol Chem. 2008;283(45):30715–24.
Oliveira JM, Jekabsons MB, Chen S, Lin A, Rego AC, Gonçalves J, et al. Mitochondrial dysfunction in Huntington’s disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. J Neurochem. 2007;101(1):241–9.
Canzoniero LM, Granzotto A, Turetsky DM, Choi DW, Dugan LL, Sensi SL. nNOS(+) striatal neurons, a subpopulation spared in Huntington’s disease, possess functional NMDA receptors but fail to generate mitochondrial ROS in response to an excitotoxic challenge. Front Physiol. 2013;4:112.
Plotegher N, Perocheau D, Ferrazza R, Massaro G, Bhosale G, Zambon F, et al. Impaired cellular bioenergetics caused by GBA1 depletion sensitizes neurons to calcium overload. Cell Death Differ. 2020;27(5):1588–603.
Abramov AY, Duchen MR. Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity. Biochim Biophys Acta. 2008;1777(7–8):953–64.
Granatiero V, Pacifici M, Raffaello A, De Stefani D, Rizzuto R. Overexpression of mitochondrial calcium uniporter causes neuronal death. Oxid Med Cell Longev. 2019;2019:1681254.
Hockey LN, Kilpatrick BS, Eden ER, Lin-Moshier Y, Brailoiu GC, Brailoiu E, et al. Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J Cell Sci. 2015;128(2):232–8.
Tedeschi V, Petrozziello T, Secondo A. Calcium dyshomeostasis and lysosomal Ca(2+) dysfunction in amyotrophic lateral sclerosis. Cells. 2019;8(10):1216.
Stevens TR, Krueger SR, Fitzsimonds RM, Picciotto MR. Neuroprotection by nicotine in mouse primary cortical cultures involves activation of calcineurin and L-type calcium channel inactivation. J Neurosci. 2003;23(31):10093–9.
Ortner NJ. Voltage-gated Ca(2+) channels in dopaminergic substantia nigra neurons: therapeutic targets for neuroprotection in parkinson’s disease? Front Synaptic Neurosci. 2021;13:636103.
Nimmrich V, Eckert A. Calcium channel blockers and dementia. Br J Pharmacol. 2013;169(6):1203–10.
Muir KW. Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr Opin Pharmacol. 2006;6(1):53–60.
Chu CT. A pivotal role for PINK1 and autophagy in mitochondrial quality control: implications for Parkinson disease. Hum Mol Genet. 2010;19(R1):R28-37.
Heeman B, Van den Haute C, Aelvoet SA, Valsecchi F, Rodenburg RJ, Reumers V, et al. Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J Cell Sci. 2011;124(Pt 7):1115–25.
McWilliams TG, Prescott AR, Montava-Garriga L, Ball G, Singh F, Barini E, et al. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 2018;27(2):439-449.e435.
Bhujabal Z, Birgisdottir AB, Sjottem E, Brenne HB, Overvatn A, Habisov S, et al. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017;18(6):947–61.
Strappazzon F, Nazio F, Corrado M, Cianfanelli V, Romagnoli A, Fimia GM, et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015;22(3):419–32.
Allen GF, Toth R, James J, Ganley IG. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013;14(12):1127–35.
Pogson JH, Ivatt RM, Sanchez-Martinez A, Tufi R, Wilson E, Mortiboys H, et al. The complex I subunit NDUFA10 selectively rescues Drosophila pink1 mutants through a mechanism independent of mitophagy. PLoS Genet. 2014;10(11):e1004815.
Lee JJ, Sanchez-Martinez A, Martinez Zarate A, Benincá C, Mayor U, Clague MJ, et al. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol. 2018;217(5):1613–22.
Hertz NT, Berthet A, Sos ML, Thorn KS, Burlingame AL, Nakamura K, et al. A neo-substrate that amplifies catalytic activity of parkinson’s-disease-related kinase PINK1. Cell. 2013;154(4):737–47.
Liu Y, Lear TB, Verma M, Wang KZ, Otero PA, McKelvey AC, et al. Chemical inhibition of FBXO7 reduces inflammation and confers neuroprotection by stabilizing the mitochondrial kinase PINK1. JCI Insight. 2020;5(11):e131834.
Zhu JH, Gusdon AM, Cimen H, Van Houten B, Koc E, Chu CT. Impaired mitochondrial biogenesis contributes to depletion of functional mitochondria in chronic MPP+ toxicity: dual roles for ERK1/2. Cell Death Dis. 2012;3(5):e312.
Verma M, Zhu J, Wang KZQ, Chu CT. Chronic treatment with the complex I inhibitor MPP(+) depletes endogenous PTEN-induced kinase 1 (PINK1) via up-regulation of Bcl-2-associated athanogene 6 (BAG6). J Biol Chem. 2020;295(23):7865–76.
Wang KZ, Zhu J, Dagda RK, Uechi G, Cherra SJ 3rd, Gusdon AM, et al. ERK-mediated phosphorylation of TFAM downregulates mitochondrial transcription: implications for Parkinson’s disease. Mitochondrion. 2014;17:132–40.
Descloux C, Ginet V, Rummel C, Truttmann AC, Puyal J. Enhanced autophagy contributes to excitotoxic lesions in a rat model of preterm brain injury. Cell Death Dis. 2018;9(9):853.
Van Laar VS, Roy N, Liu A, Rajprohat S, Arnold B, Dukes AA, et al. Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and in the presence of N-acetyl cysteine, mitophagy. Neurobiol Dis. 2015;74:180–93.
Su YC, Qi X. Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum Mol Genet. 2013;22(22):4545–61.
Cherra SJ 3rd, Chu CT. Autophagy in neuroprotection and neurodegeneration: a question of balance. Future Neurol. 2008;3(3):309–23.
Yu S, Zheng S, Leng J, Wang S, Zhao T, Liu J. Inhibition of mitochondrial calcium uniporter protects neurocytes from ischemia/reperfusion injury via the inhibition of excessive mitophagy. Neurosci Lett. 2016;628:24–9.
Ludtmann MHR, Kostic M, Horne A, Gandhi S, Sekler I, Abramov AY. LRRK2 deficiency induced mitochondrial Ca(2+) efflux inhibition can be rescued by Na(+)/Ca(2+)/Li(+) exchanger upregulation. Cell Death Dis. 2019;10(4):265.
Korecka JA, Talbot S, Osborn TM, de Leeuw SM, Levy SA, Ferrari EJ, et al. Neurite collapse and altered ER Ca(2+) control in human parkinson disease patient iPSC-derived neurons with LRRK2 G2019S mutation. Stem Cell Rep. 2019;12(1):29–41.
Nichols M, Elustondo PA, Warford J, Thirumaran A, Pavlov EV, Robertson GS. Global ablation of the mitochondrial calcium uniporter increases glycolysis in cortical neurons subjected to energetic stressors. J Cereb Blood Flow Metab. 2017;37(8):3027–41.
Nichols M, Pavlov EV, Robertson GS. Tamoxifen-induced knockdown of the mitochondrial calcium uniporter in Thy1-expressing neurons protects mice from hypoxic/ischemic brain injury. Cell Death Dis. 2018;9(6):606.
Hamilton J, Brustovetsky T, Rysted JE, Lin Z, Usachev YM, Brustovetsky N. Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria. J Biol Chem. 2018;293(40):15652–63.
Acknowledgements
Not applicable.
Funding
The Chu Laboratory is supported in part by the National Institutes of Health (AG026389, NS065789, NS101628) and the Helen Mendel Fund. Dr. Chu holds the A. Julio Martinez Chair in Neuropathology at the University of Pittsburgh. The sources of funding played no part in the analysis of the scientific literature or the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
MV and CTC wrote the initial manuscript, contributed to revisions, and approved the final manuscript. BNL contributed to the revised manuscript and figure, and approved the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Studies involving animals
Not applicable.
Consent for publication
Not applicable.
Competing interests
CTC is named on a patent application targeting FBX07 (PCT/US2018/039327) submitted jointly by the University of Pittsburgh and the US Department of Veterans Affairs. The other authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Verma, M., Lizama, B.N. & Chu, C.T. Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration. Transl Neurodegener 11, 3 (2022). https://doi.org/10.1186/s40035-021-00278-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-021-00278-7