
Abstract
Background
For parkinsonian disorders, progressive supranuclear palsy (PSP) continues to be significant for differential diagnosis. PSP presents a range of ocular abnormalities that have been suggested as optional tools for its early detection, apart from the principal characteristic of postural unsteadiness. Nonetheless, such symptoms may be difficult to identify, particularly during the early onset stage of the disorder. It may also be problematic to recognize these symptoms for general practitioners who lack the required experience or physicians who are not specifically educated and proficient in ophthalmology or neurology.
Main body
Thus, here, a methodical evaluation was carried out to identify seven oculomotor abnormalities occurring in PSP, comprising square wave jerks, the speed and range of saccades (slow saccades and vertical supranuclear gaze palsy), ‘round the houses’ sign, decreased blink rate, blepharospasm, and apraxia of eyelid opening. Inspections were conducted using direct visual observation. An approach to distinguish these signs during a bedside examination was also established. When presenting in a patient with parkinsonism or dementia, the existence of such ocular abnormalities could increase the risk of PSP. For the distinction between PSP and other parkinsonian disorders, these signs hold significant value for physicians.
Conclusion
The authors urge all concerned physicians to check for such abnormalities with the naked eye in patients with parkinsonism. This method has advantages, including ease of application, reduced time-consumption, and requirement of minimal resources. It will also help physicians to conduct efficient diagnoses since many patients with PSP could intially present with ocular symptoms in busy outpatient clinics.
Similar content being viewed by others


Background
Progressive supranuclear palsy (PSP, Steele-Richardson-Olszewski syndrome) is a neurodegenerative parkinsonian disorder, classically described as a syndrome of postural instability, supranuclear vertical gaze palsy, dysarthria, dystonic rigidity of the neck and trunk, mild dementia, and pseudobulbar palsy [1]. It represents an important differential diagnosis of Parkinson’s disease (PD), accounting for approximately 6% of all cases of degenerative parkinsonism referred to specialist clinics [2]. The National Institute of Neurological Disorders and Stroke (NINDS) developed a set of clinical research criteria of the syndrome that emphasize the presence of vertical supranuclear gaze palsy or slow vertical saccades as well as early falls due to postural instability as the most useful physical signs that distinguish PSP from PD [3, 4]. Nevertheless, the rate of misdiagnosis of PSP remains high, with up to 50% of cases in routine clinical practice not recognized, partly due to the absence of falls and gaze palsy in many patients [4, 5]. To improve sensitivity toward the different variants of PSP, the new MDS clinical diagnostic criteria of PSP, in combination with specific combinations of certain features, may help optimize early clinical diagnosis of PSP [6].
Supranuclear vertical gaze palsy has been recognized as one of the cardinal features of PSP. However, a clinicopathological study that involved a comprehensive review of case notes of family doctors and medical specialists as well as in-patient notes and consultations indicated that in more than half of the cases early in the disease process, no detailed examination of the eye signs was recorded, beyond a statement of ‘cranial nerve examination normal’ [5]. This may be due to a number of reasons, including a lack of awareness of useful eye findings as part of the diagnostic work-up among treating physicians as well as a lack of eye complaints from patients, resulting in incomplete physical examination. In addition, many general physicians may feel that eye examinations are irrelevant to them and not part of their routine examination, as they consider them to fall under the expertise of eye specialists who try to identify subtle findings.
The early identification of patients with PSP has several benefits to both patients and caregivers, including early referral to specialists, focus treatments being tailored to specific symptoms that impair the patients’ quality of life, and appropriate counselling. Moreover, careful examinations are likely to prevent unnecessary investigations of patients. Eye examinations, particularly eye movement tests, should thus be performed as part of the general examination in patients who primarily present with akinetic-rigid syndromes [7]. Therefore, in this article, we present a systematic review of the existing evidence in the literature on common eye signs in PSP, and propose practical bedside methods on how to elicit them. In many instances, screening eye examinations in patients with parkinsonism are not difficult to perform by general physicians if they know what to look for, and do not take much of the consulting time. Appropriate referrals to specialists, including movement disorder neurologists and ophthalmologists, for specific examinations and management can then be considered in suspected cases of PSP.
Main text
Progressive supranuclear palsy for general physicians
As the name entails, PSP is an adult-onset progressive neurodegenerative disorder associated with supranuclear gaze palsy. It is diagnosed in approximately 6% of all parkinsonian patients being referred to a specialized clinic [2]. In the classification of parkinsonism, PSP falls under the category of multiple system degenerations or ‘parkinsonism-plus syndromes’, with the ‘plus’ feature being oculomotor dysfunction, postural disability, akinesia, and cognitive dysfunction [6, 8]. PSP occurs more often in men, with a mean onset age of 63 years, 10 years later than the typical onset of PD [9]. It has distinct neuropathological features associated with tau pathology at different levels of the central nervous system (CNS), leading to an akinetic-rigid syndrome with oculomotor dysfunction, postural instability, and frontal lobe and bulbar dysfunction. Despite the name, supranuclear gaze palsy is not always present, and clinical manifestations, particularly in the early stage, can be variable and subtle, thus making the diagnosis difficult if certain physical signs (i.e. eye signs) are not specifically looked for. Mobility problems, described as recurrent falls, are a common early feature, and visual symptoms, including diplopia, photophobia, and eyelid apraxia are often functionally disabling (see Additional file 1) [10]. In simple terms, the diagnosis of PSP should be considered in all patients presenting with parkinsonism not responding to levodopa, postural instability with falls, executive dysfunction, dysarthria, and, importantly, eye movement abnormalities.
In order to diagnose PSP clinically, the National Institute of Neurological Disorders and Stroke and Society for Progressive Supranuclear Palsy (NINDS-SPSP) has proposed probable and possible clinical diagnostic criteria that have been shown to provide high sensitivity and high positive predictive values suitable for routine clinical care [3, 11]. The NINDS-SPSP criteria rely on the identification of a progressive disorder with onset age after 40 years with a combination of two cardinal features, namely postural instability with falls during the first year of the disease and slow vertical saccades or supranuclear gaze palsy [3]. However, the validation of these criteria in an independent set of patients has demonstrated low sensitivity in patients in the early stage of the disease and in patients presenting with variant PSP syndromes that differ from the classical form of PSP [3, 12, 13].
The classic form of PSP is also called Richardson PSP phenotype (PSP-RS) (also known as Steele-Richardson-Olszewski syndrome); patients report early difficulties with vertical gaze and pseudobulbar palsy, nuchal dystonia, and dementia [14]. Patients with PSP with non-classical presentations have been reported, which account for up to 76% of autopsy-confirmed PSP populations [15]. For non-classical variants (PSP-ocular motor dysfunction, PSP-postural instability, PSP-parkinsonism, PSP-frontal, PSP-progressive gait freezing, PSP-corticobasal syndrome, PSP-primary lateral sclerosis, PSP-cerebellar, and PSP-speech/language disorders), clinical manifestations are much more diverse, with supranuclear gaze paresis developing late in the majority of cases (Table 1) [6, 16]. PSP-parkinsonism represents the most common form among these variants (32%), characterized by an asymmetric onset, tremor, and a moderate initial therapeutic response to levodopa, and is frequently confused with PD [5]. Patients with these variants may not be diagnosable using the current clinical diagnostic criteria [16]. The progression of PSP is more rapid and severe than that of PD, given the variability described among the different clinical subtypes. PSP has a median survival rate of 7.4 years, with the mode of death commonly related to respiratory compromise or falls [9]. Predictors of progression include Richardson syndrome, male gender, and disease onset in older age [17].
One of the main reasons why the diagnosis of PSP is delayed by several years after the disease onset is that the oculomotor cardinal features (slow vertical saccades and supranuclear gaze palsy) may develop late or never during the course of the disease [6, 15, 18]. A list of differential diagnosis is presented in Table 2. Consequently, the new MDS clinical diagnostic criteria of PSP have been proposed and stratified by three degrees of diagnostic certainty (probable PSP, possible PSP, and suggestive PSP). These criteria are related to the four functional domains where characteristic clinical manifestations of PSP appear, including ocular motor dysfunction, postural instability, akinesia, and cognitive dysfunction [6]. Based on the current available evidence, these criteria provide an optimized early, sensitive, and specific clinical diagnosis of PSP [6].
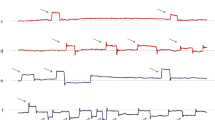
Based on distinctive neuropathological features, PSP can be divided into several clinical subtypes, providing some guidance on the prognosis and natural history [5, 6, 16]. Patients with the classic form of PSP (PSP-RS) usually report early difficulties with balance, personality changes, and visual disturbances in association with initial square-wave jerks, slow vertical saccades evolving into hypometric saccades, supranuclear vertical gaze palsy, and eventually eyelid abnormalities (Fig. 1) [3, 4, 19]. From this systematic review, we are able to identify the occurrence of ocular abnormalities in chronological order as shown in Fig. 1. While their presence may evolve in the given fashion, this is not the rule and variations may indeed occur.

Evolution of ocular abnormalities in PSP presented in chronological order. The clinical stages are divided into three stages related to clinical appearance, namely (1) early stage: clinical features apparent in less than 4 years of disease duration; (2) middle stage: clinical feature apparent for between 4 and 8 years of disease duration; and (3) late stage: clinical feature apparent for more than 8 years of disease duration. Solid white rectangles indicate timeline occurrences of each ocular abnormality, presented in years after onset of disease. Solid arrows indicate the presence ophthalmoplegia and square wave jerks, which continue to be present in the late disease stage
Neuropathological correlations of oculomotor abnormalities in PSP
The constellation of parkinsonism and cognitive, speech, and oculomotor features in PSP reflect marked neuronal degeneration at different levels of the CNS, including the basal nucleus of Meynert, the pallidum, subthalamic nucleus, superior colliculi, mesencephalic tegmentum, substantia nigra (both pars compacta and reticulata), locus coeruleus, red nucleus, reticular formation, vestibular nuclei, cerebellum, and spinal cord (Fig. 2) [14, 20, 21].

Sagittal section of human brainstem showing the locations of the degeneration of brainstem nuclei responsible for the presence of ocular abnormalities in PSP. Black circles represent the degeneration of some specific brainstem structures resulting in ocular abnormalities. Abbreviations: riMLF: rostral interstitial nucleus of the medial longitudinal fasciculus, RN: red nucleus, INC: interstitial nucleus of Cajal, PPRF: paramedian pontine reticular formation, MLF: medial longitudinal fasciculus, SC: superior colliculus, IC: inferior colliculus, 3: nucleus of the oculomotor nerve, 4: nucleus of the trochlear nerve, 6: nucleus of the abducens nerve, 7: nucleus of the facial nerve
In PSP, two oculomotor defects are prominent, namely, a restricted range of vertical movement and vertical saccadic slowing [19]. Slow vertical saccades, a cardinal early feature of PSP, reflect abnormalities in the brainstem neural network affecting primarily burst neurons in the rostral interstitial nucleus of the medial longitudinal fasciculus (riMLF) [22, 23]. As the interstitial nucleus of Cajal (INC) contributes to vertical gaze holding, inactivation of this structure causes a restricted range of vertical eye movement without inducing slow saccades [24]. As the disease progresses, slowing of horizontal saccades is also evident, suggesting the involvement of burst neurons in the parapontine reticular formation (PPRF) as well [23, 24]. These structures are located in the mesencephalic reticular formation, which is involved in PSP. Table 3 summarizes the correlations between neural substrates and oculomotor signs in PSP.
Different types of oculomotor abnormalities in progressive supranuclear palsy: a systematic review
To determine different types of oculomotor abnormalities that were reported in patients with PSP, we conducted a systematic review by performing an electronic database search on PubMed, EMBASE, Cochrane Library, and life science journals. The following terms were used for the literature search: Progressive supranuclear palsy AND gaze OR gaze palsy OR saccades OR slow saccades OR fixation OR saccadic intrusion OR square wave jerks OR round the houses sign OR apraxia of eyelid opening OR blepharospasm OR blink rate OR eye blink OR decreased blink rate OR eye OR eyelid OR ocular OR visual OR vision OR signs OR symptoms OR abnormalities OR impairment OR disorders. A target search of bibliographies of relevant articles was also performed to identify additional articles for inclusion. Titles and abstracts were identified from database searches and chosen according to the aforementioned selection criteria. Additional abstracts were identified from manual searches (authors’ libraries and additional papers quoted in the retrieved papers). Only original full-text articles on PSP related to searching items published in English between January 1973 and December 2018 were included in this review. Reviews and editorial articles presenting no original data were excluded. Both assessors (OP, RB) independently screened each paper and were required to agree on each study in order to be included in this review. We screened 2745 titles and abstracts, from which 117 full-length articles were selected for further review. Of these, 45 articles fulfilled the selection criteria (see Additional file 2). Findings on oculomotor abnormalities were categorized into disorders of ocular fixation, eye movements, and eyelids. Seven oculomotor abnormalities, including square wave jerks, speed and range of saccades (slow saccades and vertical supranuclear gaze palsy), ‘round the houses’ sign, decreased blink rate, blepharospasm, and apraxia of eyelid opening, were selected based on their prevalence, distinctive features, and simplicity to elicit during a bedside examination. To take into account clinical relevance, we structured the presentation of each abnormality according to its definition, examination techniques with supplemented video clips (see Additional files 3 and 4), and clinical implications.
Additional file 3: Video of the common seven eye signs of patients with progressive supranuclear palsy ordered as following; square wave jerks, slow saccades, ‘round the houses’ sign, vertical supranuclear gaze palsy, decreased blink rate, blepharospasm, and apraxia of eyelid opening. (MP4 14427 kb)
Square wave jerks
What are square wave jerks?
Square wave jerks (SWJs) represent the most common type of saccadic intrusions, characterized by conjugated couplets of horizontal back-to-back microsaccades (0.5–5°), taking the eye from the fixation point and back onto it in an involuntary manner, with a normal intersaccadic interval of about 200 ms [25]. Saccadic intrusions are involuntary conjugate saccades that interrupt fixation, and can be sub-classified by the presence (SWJs, square wave pulses, and macrosaccadic oscillations) or absence (ocular flutter and opsoclonus) of an intersaccadic interval [25].
How to elicit square wave jerks
SWJs can be identified by asking the patients to maintain their gaze fixation to a static object for the duration of 10 to 30 s. Alternatively, SWJs can be observed with Frenzel’s goggles or an ophthalmoscope, by asking the patients either to hold their gaze steadily at the primary position or to shift their gaze towards eccentric positions. On clinical inspection, SWJs are visualized as saccade pairs that appear purely horizontal: the first saccade moves the eye away from the fixation target, and, after a short interval, the second saccade brings it back to the target [26]. In addition to SWJs, microsaccades may be observed, but they are likely to be too small (< 0.5°) to be evident during clinical examinations in normal subjects [27]. While SWJs appear to be purely horizontal on clinical inspection, microsaccades often display an oblique trajectory [28]. Recent studies suggested that both microsaccades and SWJs are generated by the same neural circuit, and that larger saccades are more likely to show SWJs [26, 29].
Clinical implications of square wave jerks
It is important to understand that occasional SWJs can occur in preschool children and healthy elderly subjects [30, 31]. The typical frequency of SWJs is less than 9/min in healthy subjects, but the frequency seems to increase with age [32]. However, certain observations, such as large (> 5°), frequent (> 16 per minute during fixation or > 20 per minute in the dark), multiplanar, or disconjugate SWJs, should alert physicians of an underlying neurological dysfunction [33]. While SWJs have been reported in patients with PSP, they are not specific to this disorder, and have been observed in a number of disorders affecting the functions of brainstem circuits, particularly the cerebellum and its output through the superior cerebellar peduncle. However, certain characteristics of SWJs such as macro SWJs were included in the core clinical features suggestive of SPS from the new MDS clinical diagnostic criteria of PSP, which may be useful in the differentiation of individuals with PSP from normal subjects or those with other parkinsonian disorders [6]. These disorders include Friedreich’s ataxia, Huntington’s disease, oculomotor apraxia type 2, PD, multiple system atrophy (MSA), corticobasal degeneration (CBD), and recessive spinocerebellar ataxias [34, 35].
Out of 45 articles included in this systematic review, nine articles studied the presence of SWJs in patients with PSP using different eye recording techniques [26, 34, 36,37,38,39,40,41,42].
Among various forms of parkinsonian syndromes, SWJs were reported in most patients with PSP (60–100%), followed by MSA (40–60%), CBD (33%), and, to a lesser extent), PD (0–15% of patients) [34, 36, 38]. The frequency of SWJs reported in the literature may vary depending on the respective recording technique. Moreover, SWJs in patients with PSP were identified to be larger than 1°, which is a useful clinical sign for differentiation of PSP from PD, since large SWJs or macro SWJs are considered uncommon among patients with PD [34]. When compared to healthy normal subjects, SWJs in patients with PSP were more frequent, larger, and more markedly horizontal [36, 41]. Microsaccades also lost their vertical component in patients with PSP [26]. The latter finding has been identified as the most distinctive characteristic sign of PSP [26].
Other studies looked further into fixational characteristics among patients with parkinsonian disorders [36, 39, 42]. A high proportion of abnormal ocular fixations was observed in patients with PSP, MSA, or CBD [34, 38]. Patients with PSP exhibited larger SWJs than controls when fixation targets were visible, and this difference was reversed in the absence of a fixation target [39, 40]. In contrast, patients with MSA demonstrated increased SWJs in conditions with and without a visible target [36]. Whether this observation represents a useful tool to discriminate PSP from other parkinsonian disorders in the early stage is still unclear, and these findings need to be replicated in subsequent studies. Another study described the distinguished patterns of SWJs in PSP, which showed larger sizes, higher frequencies, but less vertical components than SWJs in control subjects. The authors noted that this finding is consistent with the typical feature of PSP, vertical supranuclear palsy, and suggested that SWJs should be considered an early eye sign in PSP, due to the short disease duration and early symptom onset (median duration of 4 years) [26].
SWJs can result from either (1) disruption to areas that influence saccadic control, including the cortex, cerebellum, superior colliculus, and basal ganglia, or (2) direct injury to omnipause neurons located in the brainstem [37]. One study presented similar explanations for SWJ mechanisms, and proposed the hypothesis that the abnormality in supranuclear control might interrupt omnipause cell activity in paramedian pontine reticular formation, resulting in the release of saccadic burst units [43]. Another proposed mechanism for SWJs might be reflected in the degeneration involving the cerebellum and its connections, such as to the superior cerebellar peduncle, and result in abnormal to-and-fro eye movements during attempted steady fixation [19, 44]. This hypothesis is supported by the proposed relevance of SWJs in the impairment of visual scanning during attempt fixation among patients with cerebellar ataxia [45, 46].
Slow saccades and vertical supranuclear gaze palsy
What are slow saccades and what is vertical supranuclear gaze palsy?
Saccades are rapid eye movements that redirect the foveal line of sight toward features of interest so that they can be seen optimally [25]. The speed and range of saccades represent important components of saccadic examination, and their abnormalities reflect the dysfunction of the burst neurons (riMLF for vertical saccades and PPRF for horizontal saccades) that are frequently affected by the disease process in PSP [22, 23]. Since saccades are the fastest eye movements (up to 500°/s) with very brief duration (< 100 ms), the examiner should not expect to be able follow the full trajectory of normal saccades with their own eye [25]. The normal duration of a saccade is 30–100 ms for amplitudes ranging from 0.5 to 40° [47]. The larger the saccade, the higher the peal velocity. Saccades are slower when made in darkness to a remembered target location and when made in the opposite direction to a visual stimulus (the ‘antisaccade test’) [48].
In general terms, gaze palsy refers to deficits in conjugate eye movements that affect both eyes. Therefore, gaze palsy, when present, is usually indicative of supranuclear lesions resulting from interruption of the neural pathways that carry commands for voluntary saccades and pursuit before they reach the eye movement ‘generators’ in the brainstem. Dysfunction of the generators, namely the riMLF, results in vertical supranuclear gaze palsy, while dysfunction of the PPRF can lead to horizontal gaze palsy [22].
How to test saccadic velocity and gaze palsy
Testing for the velocity of saccades can be performed when the subject is asked to glance back and forth between two horizontal and vertical targets. Not only the velocity, but also the accuracy and conjugacy of the saccades should be observed. Normal individuals can reach the target with a single fast movement or with one small corrective saccade. It is important for the examiner to ensure that the subjects pay full attention during the performance of saccades, since patients who are drowsy, inattentive, or heavily medicated may produce normal saccades with slow velocity [49]. Moreover, there is a very strong age effect in the performance of saccades with a tendency for elderly subjects (60 years and over) to produce slower and delayed saccades, compared to younger individuals [50].
In order to determine slow saccades, researchers have utilized different quantitative methods of eye movement recordings, ranging from electrooculography, scleral search coil systems, and video-oculography. However, the identification of slow saccades on clinical examination is more challenging, particularly in patients with mildly slow saccades. If the examiner can follow the full trajectory of the patient’s saccades with their own eye, the saccades of that particular patient are likely to be slow [51]. Another useful clinical pearl is that patients with delayed saccadic initiation and slow saccades often blink or orient their head towards the target before making a saccade [52]. Different methods have been proposed to enhance the recognition of slow saccades, including the use of an optokinetic drum, or the use of oblique targets that will result in an L-shape saccade [52]. However, these methods have not been tested in patients with PSP to demonstrate their sensitivity and specificity. In suspected cases of PSP, particular attention should be paid to the velocity of vertical and horizontal saccades independently, since selective slow vertical saccades are observed in the early stage, followed by slow horizontal saccades when the disease is more advanced [23].
The range of motion of both horizontal and vertical saccades can be tested in a self-paced or verbally guided manner. Before determining that gaze palsy is pathological, it is important for physicians to be aware that a limitation of the vertical range of eye movements, especially upgaze, can occur in elderly subjects; the velocity should however not be affected in such cases [53]. In addition to gaze palsy, features suggesting supranuclear lesions should be specifically looked for. This can be accomplished by assessing improvements in the gaze palsy with a vestibuloocular reflex, thereby bypassing volitional pathways.
Clinical implications of slow saccades and vertical supranuclear gaze palsy
Out of 45 articles included in this systematic review, 10 articles studied slow saccades in patients with PSP [10, 23, 37, 38, 40, 41, 54,55,56,57], and 19 articles studied VSP in patients with PSP [1, 4, 10, 15, 18, 19, 36, 37, 41, 54, 58,59,60,61,62,63,64,65,66], identified using different eye recording techniques.
In the setting of parkinsonian syndromes, the identification of VSP is highly specific for the diagnosis of PSP, with the exception of a few case reports of dementia with Lewy bodies and CBD [67,68,69,70]. While VSP constitutes one of the mandatory inclusion criteria for the clinical diagnosis of probable PSP, proposed by the NIND-SPSP [3], a real clinical problem is that not all patients with PSP exhibit the full range of eye movement abnormalities, particularly in the early stage [37, 42, 55, 56]. VSP should, however, not be used as an early sign in PSP, as it usually presents in the advanced disease course (range of follow-up periods: 1–10 years) [5, 19, 54, 60,61,62, 66, 71]. In addition, some patients with PSP, especially those with variants who meet the clinic-pathologic criteria, might be diagnosed based on the pathological confirmation of the diagnosis without existing VSP or in spite of a lack of other eye movement abnormalities [4, 5, 59, 60, 63, 72,73,74,75].
Therefore, the typically slow saccades with curved trajectories are giving a strong indication when observed in patients with early onset parkinsonism, but vertical saccades are more affected than horizontal saccades in PSP [10, 23, 38, 40, 54]. Additionally, both horizontal and vertical saccades are slow and have irregular trajectories and velocity profiles, but deficits have been found to be particularly severe in the vertical axis [57]. A number of clinicopathological studies have been able to correlate the presence of midbrain atrophy with the degeneration of burst neurons in the riMLF, clinically manifested as selective slow vertical saccades rather than horizontal saccades [22, 23]. Several studies of pathologically proven cases of PSP reported a range of 40–90% of patients that were documented to have vertical gaze palsy, implying that vertical gaze palsy was not evident in all patients [4, 5, 15, 58,59,60, 66, 71, 76,77,78,79,80]. For example, some variants of PSP including PSP-frontal, PSP-PLS, and PSP-SL show no eye movement abnormalities during standard clinical examination at an early stage of the disease [80,81,82]. In an autopsy-proven case of PSP-PLS, vertical gaze palsy was absent throughout the 10-year disease course [81].
These pathologically studied cases provide strong evidence that, although vertical supranuclear palsy is relatively specific to PSP in the setting of parkinsonian syndromes, it is not a universal finding in this disorder. Additional involvement of cerebellar signs has been proposed for a new subtype of PSP called ‘PSP-cerebellum’ (PSP-C) [63, 66]. In this group of patients, eye signs can be under-recognized during the first 2 years [63, 66]. The real question to clinicians is what types of oculomotor abnormalities can help increase the diagnostic acumen for different variants and stages of PSP. The typical VSP in PSP can usually be partially improved with vestibule-ocular reflex (VOR) manoeuvres, which show an abnormal reciprocal inhibition of antagonist vertical muscles during attempt voluntary movements, while the reciprocal innervation is well preserved during reflex movements [64]. The supranuclear degeneration of the pathway from the substantia nigra pars reticulata to the superior colliculus may be an important cause of gaze palsy in PSP [83]. Our systematic review provides more evidence that slow saccades and vertical supranuclear palsy do not occur at once in patients with PSP. Rather, they tend to evolve as the disease progresses. One longitudinal study of oculomotor function in patients with PSP suggested that slow saccades occur throughout the disease course with preserved saccadic latency [84].
‘Round the houses’ sign
What is a ‘round the houses’ sign?
Indicating an incapability to make pure vertical saccades along a straight line in the midline and lateral arc, which creates a curve course of oblique saccades, the ‘round the houses’ sign for PSP was mentioned for the first time by Quinn in 1996 [85]. Using the analogy that each eye makes a round excursion in its own ‘house’ (orbit), the sign employs the plural ‘houses’. In addition to midbrain atrophy, the degeneration of the burst neurons in the riMLF likely explains the mechanism of early selective slow vertical saccades rather than horizontal saccades, and may also explain the occurrence of the ‘round the houses’ sign [23]. Furthermore, the paramedian pontine reticular construction is mainly responsible for creating horizontal saccades [22, 85, 86].
How is a ‘round the houses’ sign provoked?
By prompting a patient to carry out quick eye movements to a moving target between a completely upright and downwards position while keeping the head stationary, a ‘round the houses’ sign can be detected. Eye motion following a curved course (oblique saccades), rather than a straight line, could also be an indication for a ‘round the houses’ sign [85, 86].
Clinical implications of a ‘round the houses’ sign
Two articles (one commentary and one case report) reporting on ‘round the houses’ signs in patients with PSP were identified [85, 86]. The signs were mentioned by Quinn in an old comment through his experience with several patients with PSP [85]. The curved course of oblique saccades might be a result of the viewer conducting vertical upward and downward saccades, resulting in an oval-like trajectory by inconsistent reduction of the vertical saccades compared to the horizontal saccades, evenly full gaze existed [85]. A ‘round the houses’ sign has thus been defined as an early eye sign to look for in patients with PSP [85]. It may also occur together with other signs as a late indicator in patients with advanced PSP, unless it results from vertical supranuclear gaze palsy [85]. The sign was recently described in Niemann-Pick disease type C (NPC) [87, 88], while dystonia, hyperreflexia, and hepatosplenomegaly are the usual indicators of adult-onset NPC [87].
Decreased blink rate
What is a decreased blink rate?
By eliminating irritants from the surface of the cornea and conjunctiva as well as aiding to spread tears covering the globe surface to sustain a normally moist state, blinking and secretion of tears are basic protective mechanisms of the eyes [89]. Consisting of a swift lowering of an upper eyelid mediated by a short contraction of the orbicularis oculi (OO) muscle, eye blinking is linked to the reciprocal inactivation of the levator palpebrae superioris (LPS) muscle of the upper eyelid [89, 90].
How is a decreased blink rate assessed?
Bilateral paroxysmal closing of the eyelids (duration: less than 1 s) is the definition of a blink that occurs without being triggered by an outside stimulus. By counting how many times a person blinks per minute during a 5-min conversation, a decreased eye blink rate can be identified [91]. Otherwise, digitally-recorded video images concentrated on the eyes may be utilized. When at least 50% closure of the eye happens during a quick closing and subsequent reopening of the eye, a blink is classified as a ‘full blink’. As prolonged eye closure is typically an intentional act in response to deep thought, it is usually excluded from assessments of blinks. Spontaneous blinks with under 50% of eyelid closure are not considered for the total blink rate [92].
Clinical implications of a decreased blink rate
Our methodical analysis showed that four studies found a noteworthy reduction in blink rates among patients with PSP in comparison to healthy controls in terms of unprompted as well as trained blink responses [91, 93,94,95]. Additionally, patients with PSP exhibited much longer periods of closed eyes during unprompted blinking compared to the healthy controls [95]. Further, patients with PSP showed a significant reduction in their conditioned eye blink response and more verbal memory impairment, resulting in the proposal of a dissociated pattern of learning abilities. In a serial reaction time task, meanwhile, compared to controls, obvious sequence patterns were comparatively sustained [93]. A neurophysiological device employed to gauge brainstem excitability could be advantageous for separating patients with PSP from others with CBS, as found in a study on the benefits of the R2 blink reflex recovery cycle (R2BRRC) [96]. In this study, patients with PSP showed earlier employment of R2BRRC than patients with CBS [96].
Striatal and mesolimbic dopaminergic performance can be assessed via blinking rates. Alterations in rates have been found in various neuropsychiatric disorders, and are thought to come from irregular central dopaminergic functions [97]. Patients with schizophrenia, Tardive dyskinesia, Tourette’s syndrome, and Meige’s syndrome have shown increased blink rates, while those with Parkinson’s disease and PSP exhibit decreased blink rates [95, 97,98,99].
Blepharospasm and apraxia of eyelid opening
What are blepharospasm and apraxia of eyelid opening?
Spontaneous, erratic, and strong contractions of the orbital, preseptal, and pretarsal segments of the orbicularis oculi are characteristic of blepharospasm (BSP) [94]. They are typical dystonic indicators of PSP, often related to visual disabilities including functional blindness [100, 101]. Idiopathic BSP and secondary BSP represent the two major forms. Considering the cooccurrence of BSP and other uncharacteristic physical findings including parkinsonism and dementia may aid physicians to make a conclusive diagnosis of PSP [102]. Limb and axial dystonia, oromandibular dystonia, torticollis, and dysfluency (dystonia of the articulation muscle) represent additional focal dystonias other than BSP in PSP [101, 103]. Botulinum toxin usually results in good responses in patients [102]. BSP can be differentiated from apraxia of eyelid opening (ALO) on the basis of dissimilar responses after botulinum neurotoxin treatment [104].
Usually resulting from a lack of supranuclear control of the levator palpebrae and orbicularis oculi muscles, ALO is marked by the incapacity to open one’s eyelid [105, 106]. The majority of patients with this condition stemming from a lack of eyelid elevation control tend to contract their frontalis muscles more strongly to open their eyes instead of using their orbicularis oculi muscles, causing a noticeable forehead wrinkle. In PSP, ALO is a fairly common form of eyelid disorder [37, 94]. If botulinum neurotoxin cannot improve BSP notwithstanding the construction of eyelid weakness, ALO is a significant factor [104]. Due to the selective contraction of the pretarsal section of the orbicularis oculi potentially being responsible for eyelid closure, apraxia is often mistaken [104].
How can BSP and ALO be provoked?
A physician, typically a neurologist or ophthalmologist, is responsible for making a BSP diagnosis during a clinical assessment. While physical and neurological check-ups help offer a diagnosis, it is usually compulsory for the physician to verify the information given by the patient. By prompting a patient to move his/her face into both spontaneous and trained positions based on the physician’s directives, BSP can be recognized. By asking patients to shut their eyes forcefully, physicians can assess the bilateral contractions of the orbicularis oculi muscles. After patients open their eyes again, BSP may be observed as a continuing contraction of the orbicularis oculi muscles, related to a sinking of the eyebrows under the superior orbital rim (Charcot’s sign) [107].
By asking patients to close their eyes, ALO can be recognized. Patients are asked to open their eyes again quickly after closing them. Any incapacity to lift the eyelid without major orbicularis oculi muscle contraction may also be classified as ALO [105, 108]. While a patient is starting to open their eyes, excessive frontalis muscle contraction is typically observed as a forehead wrinkle.
Clinical effects of BSP and ALO
More prevalent in atypical parkinsonism than PD, BSP is a significant characteristic eye sign in PSP [37, 102, 109]. In varying levels of acuteness, nine articles have described BSP in patients with PSP as follows [37, 59, 61, 94, 100,101,102, 109, 110].
BSP in PSP could be serious and cause acute visual disability [94, 100]. The intensity and occurrence of BSP may also increase under certain conditions, as found in these studies. These conditions include exposure to bright light, watching television, reading, stress, and driving. Prior to the emergence of typical blepharospasm, more than one-third of patients with PSP suffer from excessive eye blinking [100]. Basic visual tasks could improve BSP, while direct commands commonly have no effect, as found by past research [94]. BSP in PSP leads to deteriorating changes involving the upper brainstem, without a true clinicopathological link to particular sites, based on a clinicopathological study [59]. As the result of dopaminergic medication, however, dystonia and BSP have been recounted in PSP [101, 109]. Levodopa administration was found to be linked to roughly 24% of cases with BSP in patients with PSP [101]. Further, previous research has found that BSP incidences occur late in the disease as well as at the same time with ALO [37, 61, 94, 102, 109]. Thus, the overlap of BSP and ALO could be an indication of PSP [109]. Similarly to BSP stemming from other causes, BSP in PSP typically responds to botulinum toxin injections [37]. Nonetheless, botulinum toxin injections may not achieve a full response in patients with PSP with isolated BSP when considering who had both BSP and ALO conditions [110].
Concerning research on ALO in patients with PSP, 14 articles have been identified as follows [18, 37, 58, 59, 62, 94, 105, 106, 108, 111,112,113,114,115].
ALO initially gained focus in 1965 as a non-paralytic abnormality characterized by difficulty in performing eyelid elevation [105]. An interruption in supranuclear control of the levator palpebrae and orbicularis oculi muscles may be a main contributor of ALO, causing difficulty in opening the eyes [106]. The benchmarks of ALO can be described as follows: (a) temporary incapability to raise the eyelids; (b) lack of progressive orbicularis oculi muscle contractions; (c) vigorous frontalis muscle contraction combined with incapacity to raise the eyelids; and (d) lack of oculomotor or ocular sympathetic nerve dysfunction as well as lack of ocular myopathy [94]. The most frequent eye sign in patients with PSP who fulfil the criteria of NINDS-SPSP for identification of PSP continues to be ALO, as long established in pathologically established cases of PSP [18, 58, 59, 62, 111]. Although ALO is a belated occurrence in PSP, in one out of six PSP cases it may represent the initial clinical presentation of PSP, at an average of 3.5 years after disease onset [62]. Further, ALO resulting from dopaminergic medications has been revealed in patients with parkinsonism [114, 115]. In a group of 32 patients with ALO of assorted aetiologies who reacted well to botulinum toxin injections, seven were patients with PSP [108]. This study found that ALO may be caused by focal eyelid dystonia instead of true apraxia, because of positive electrophysiologic findings with remote spasms of the pretarsal segment absent but complete contribution of the orbicularis oculi [108]. An incapability to constrain the voluntary contraction of the pretarsal portion of the orbicularis oculi muscle without clinically ongoing contraction of the orbicularis oculi muscle was detected in three patients with eyelid opening apraxia, termed ‘motor persistence of orbicularis oculi muscle’, comparable to the findings in a case study involving three patients with PSP. Subsequent to botulinum toxin injections into their eyelids, all three patients recovered [112]. The hypothesized eyelid dystonia in a report of two patients with PSP with ALO using a suggested injection site was restricted to the joint of the preseptal and pretarsal part of the orbicularis oculi muscles, as verified by the effective treatment response [113]. However, the benefits of botulinum toxin injections were identified in patients with a combination of ALO and BSP only, as proposed by a contrary study [37].
How can a bedside ocular examination be performed to identify PSP?
The three necessary actions for examination of the eye (see Additional file 5) are described in detail in this section. It should be noted that the existence of any ocular abnormalities could indicate PSP when found in those with parkinsonism or dementia. For the distinction between PSP and other parkinsonian disorders, such abnormalities can be important in aiding the physician’s diagnosis. Bedside ocular examinations are typically carried out in the following three steps:
-
Step 1: By asking the patient to keep their gaze fixated on a stationary object for a period of 10 to 30 s, ocular fixation is established. Initial saccades move the fovea away from the intended position of fixation.
-
Step 2: The patient is prompted to move his/her eyes quickly to a moving target, which enables the establishment of ocular movement. It can then be assessed whether or not the patient is suitable for a performance of full eye excursion in the vertical as well as horizontal direction, while the head remains in a stationary position. The distance and rate of eye motion in both planes are observed in order to determine which plane is more flawed. To evaluate the VOR and decide whether to elicit the full vertical excursion with the eyes fixed on a target, the rotation of the head is assessed.
-
Step 3: Eyelid abnormalities are evaluated by asking the patient to close his/her eyes. After the eyes are closed, the patients will be asked to reopen their eyes again, to gauge the speed at which opening occurs. This enables the observer to check for orbicularis oculi muscle contractions.
Additional file 5: Video demonstrates the three necessary actions for examination of the eye. (MP4 9197 kb)
Conclusions
When occurring in a patient with parkinsonism or dementia, distinct ocular abnormalities, which may include square wave jerks, slow vertical saccades, vertical supranuclear gaze palsy, ‘round the houses’ sign, decreased blink rates, blepharospasm, and apraxia of eyelid opening, could indicate PSP. For the discrimination of PSP from other parkinsonian disorders, such abnormalities can be important in aiding the physician’s diagnosis. The authors thus urge all concerned physicians to check for any abnormalities with the naked eye in patients with parkinsonism. This method has advantages, including ease of application, reduced time-consumption, and requirement of minimal resources. It will also help physicians to conduct efficient diagnoses since many patients with PSP could initially present with ocular symptoms in busy outpatient clinics.
Availability of data and materials
Not applicable.
Abbreviations
- ALO:
-
Apraxia of eyelid opening
- BSP:
-
Blepharospasm
- CBD:
-
Corticobasal degeneration
- CNS:
-
Central nervous system
- LPS:
-
Levator palpebrae superioris
- MLF:
-
Medial longitudinal fasciculus
- MSA:
-
Multiple system atrophy
- NPC:
-
Niemann-Pick disease type C
- PD:
-
Parkinson’s disease
- PPRF:
-
Paramedian pontine reticular formation
- PSP:
-
Progressive supranuclear palsy
- R2BRRC:
-
R2 blink reflex recovery cycle
- riMLF:
-
Rostral interstitial nucleus of the medial longitudinal fasciculus
- SWJs:
-
Square wave jerks
- VSP:
-
Vertical supranuclear palsy
References
Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol. 1964;10:333–59.
Hughes AJ, Ben-Shlomo Y, Daniel SE, Lees AJ. What features improve the accuracy of clinical diagnosis in Parkinson’s disease: a clinicopathologic study. Neurology. 1992;42(6):1142–6.
Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47(1):1–9.
Litvan I, Mangone CA, McKee A, et al. Natural history of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) and clinical predictors of survival: a clinicopathological study. J Neurol Neurosurg Psychiatry. 1996;60(6):615–20.
Williams DR, de Silva R, Paviour DC, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain. 2005;128(Pt 6):1247–58.
Hoglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord. 2017;32(6):853–64.
Abdo WF, van de Warrenburg BP, Burn DJ, Quinn NP, Bloem BR. The clinical approach to movement disorders. Nat Rev Neurol. 2010;6(1):29–37.
Fahn S, Jankovic J, Hallett M. Atypical parkinsonism, parkinsonism-plus syndromes, and secondary parkinsonian disorders. In: Fahn S, Jankovic J, Hallett M, editors. Principles and practice of movement disorders. 2nd ed. United States: Saunders; 2011. p. 197–240.
Golbe LI, Ohman-Strickland PA. A clinical rating scale for progressive supranuclear palsy. Brain. 2007;130(Pt 6):1552–65.
Nath U, Ben-Shlomo Y, Thomson RG, Lees AJ, Burn DJ. Clinical features and natural history of progressive supranuclear palsy: a clinical cohort study. Neurology. 2003;60(6):910–6.
Respondek G, Roeber S, Kretzschmar H, et al. Accuracy of the National Institute for Neurological Disorders and Stroke/Society for Progressive Supranuclear Palsy and neuroprotection and natural history in Parkinson plus syndromes criteria for the diagnosis of progressive supranuclear palsy. Mov Disord. 2013;28(4):504–9.
Lopez OL, Litvan I, Catt KE, et al. Accuracy of four clinical diagnostic criteria for the diagnosis of neurodegenerative dementias. Neurology. 1999;53(6):1292–9.
Osaki Y, Ben-Shlomo Y, Lees AJ, et al. Accuracy of clinical diagnosis of progressive supranuclear palsy. Mov Disord. 2004;19(2):181–9.
Lopez G, Bayulkem K, Hallett M. Progressive supranuclear palsy (PSP): Richardson syndrome and other PSP variants. Acta Neurol Scand. 2016;134(4):242-9.
Respondek G, Stamelou M, Kurz C, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord. 2014;29(14):1758–66.
Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol. 2009;8(3):270–9.
dell’Aquila C, Zoccolella S, Cardinali V, et al. Predictors of survival in a series of clinically diagnosed progressive supranuclear palsy patients. Parkinsonism Relat Disord. 2013;19(11):980–5.
Collins SJ, Ahlskog JE, Parisi JE, Maraganore DM. Progressive supranuclear palsy: neuropathologically based diagnostic clinical criteria. J Neurol Neurosurg Psychiatry. 1995;58(2):167–73.
Chen AL, Riley DE, King SA, et al. The disturbance of gaze in progressive supranuclear palsy: implications for pathogenesis. Front Neurol. 2010;1:147.
Hauw JJ, Daniel SE, Dickson D, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology. 1994;44(11):2015–9.
Lantos PL. The neuropathology of progressive supranuclear palsy. J Neural Transm Suppl. 1994;42:137–52.
Bhidayasiri R, Plant GT, Leigh RJ. A hypothetical scheme for the brainstem control of vertical gaze. Neurology. 2000;54(10):1985–93.
Bhidayasiri R, Riley DE, Somers JT, Lerner AJ, Buttner-Ennever JA, Leigh RJ. Pathophysiology of slow vertical saccades in progressive supranuclear palsy. Neurology. 2001;57(11):2070–7.
Averbuch-Heller L, Helmchen C, Horn AK, Leigh RJ, Buttner-Ennerver JA. Slow vertical saccades in motor neuron disease: correlation of structure and function. Ann Neurol. 1998;44(4):641–8.
Leigh RJ, Zee DS. The neurology of eye movements. 5th ed. Oxford: Oxford University Press; 2015.
Otero-Millan J, Serra A, Leigh RJ, Troncoso XG, Macknik SL, Martinez-Conde S. Distinctive features of saccadic intrusions and microsaccades in progressive supranuclear palsy. J Neurosci. 2011;31(12):4379–87.
Martinez-Conde S, Macknik SL, Troncoso XG, Dyar TA. Microsaccades counteract visual fading during fixation. Neuron. 2006;49(2):297–305.
Martinez-Conde S, Macknik SL, Troncoso XG, Hubel DH. Microsaccades: a neurophysiological analysis. Trends Neurosci. 2009;32(9):463–75.
Otero-Millan J, Schneider R, Leigh RJ, Macknik SL, Martinez-Conde S. Saccades during attempted fixation in parkinsonian disorders and recessive ataxia: from microsaccades to square-wave jerks. PLoS One. 2013;8(3):e58535.
Abadi RV, Gowen E. Characteristics of saccadic intrusions. Vis Res. 2004;44(23):2675–90.
Kowler E, Martins AJ. Eye movements of preschool children. Science. 1982;215(4535):997–9.
Herishanu YO, Sharpe JA. Normal square wave jerks. Invest Ophthalmol Vis Sci. 1981;20(2):268–72.
Shallo-Hoffmann J, Petersen J, Muhlendyck H. How normal are “normal” square wave jerks? Invest Ophthalmol Vis Sci. 1989;30(5):1009–11.
Rascol O, Sabatini U, Simonetta-Moreau M, Montastruc JL, Rascol A, Clanet M. Square wave jerks in parkinsonian syndromes. J Neurol Neurosurg Psychiatry. 1991;54(7):599–602.
Fahey MC, Cremer PD, Aw ST, et al. Vestibular, saccadic and fixation abnormalities in genetically confirmed Friedreich ataxia. Brain. 2008;131(Pt 4):1035–45.
Vidailhet M, Rivaud S, Gouider-Khouja N, et al. Eye movements in parkinsonian syndromes. Ann Neurol. 1994;35(4):420–6.
Friedman DI, Jankovic J, McCrary JA 3rd. Neuro-ophthalmic findings in progressive supranuclear palsy. J Clin Neuroophthalmol. 1992;12(2):104–9.
Gironell A, Kulisevsky J, Roig C, Pascual-Sedano B, Rodriguez-Fornells A, Otermin P. Diagnostic potential of acoustic startle reflex, acoustic blink reflex, and electro-oculography in progressive supranuclear palsy: a prospective study. Mov Disord. 2003;18(11):1273–9.
Pinnock RA, McGivern RC, Forbes R, Gibson JM. An exploration of ocular fixation in Parkinson’s disease, multiple system atrophy and progressive supranuclear palsy. J Neurol. 2010;257(4):533–9.
Rottach KG, Riley DE, DiScenna AO, Zivotofsky AZ, Leigh RJ. Dynamic properties of horizontal and vertical eye movements in parkinsonian syndromes. Ann Neurol. 1996;39(3):368–77.
Troost BT, Daroff RB. The ocular motor defects in progressive supranuclear palsy. Ann Neurol. 1977;2(5):397–403.
Garbutt S, Riley DE, Kumar AN, Han Y, Harwood MR, Leigh RJ. Abnormalities of optokinetic nystagmus in progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 2004;75(10):1386–94.
Zee DS, Robinson DA. A hypothetical explanation of saccadic oscillations. Ann Neurol. 1979;5(5):405–14.
Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol. 2010;23(4):394–400.
Oppenheimer DR. Brain lesions in Friedreich’s ataxia. Can J Neurol Sci. 1979;6(2):173–6.
Fisk JD, Goodale MA, Burkhart G, Barnett HJ. Progressive supranuclear palsy: the relationship between ocular motor dysfunction and psychological test performance. Neurology. 1982;32(7):698–705.
Wray SH. Eye movement disorders in clinical practice. New York: Oxford University Press; 2014.
Antoniades C, Ettinger U, Gaymard B, et al. An internationally standardised antisaccade protocol. Vis Res. 2013;84:1–5.
Schmidt D, Abel LA, Dell’Osso LF, Daroff RB. Saccadic velocity characteristics: intrinsic variability and fatigue. Aviat Space Environ Med. 1979;50(4):393–5.
Munoz DP, Broughton JR, Goldring JE, Armstrong IT. Age-related performance of human subjects on saccadic eye movement tasks. Exp Brain Res. 1998;121(4):391–400.
Termsarasab P, Thammongkolchai T, Rucker JC, Frucht SJ. The diagnostic value of saccades in movement disorder patients: a practical guide and review. J Clin Mov Disord. 2015;2:14.
Danchaivijitr C, Kennard C. Diplopia and eye movement disorders. J Neurol Neurosurg Psychiatry. 2004;75(Suppl 4):iv24–31.
Oguro H, Okada K, Suyama N, Yamashita K, Yamaguchi S, Kobayashi S. Decline of vertical gaze and convergence with aging. Gerontology. 2004;50(3):177–81.
Terao Y, Fukuda H, Shirota Y, et al. Deterioration of horizontal saccades in progressive supranuclear palsy. Clin Neurophysiol. 2013;124(2):354–63.
Garbutt S, Harwood MR, Kumar AN, Han YH, Leigh RJ. Evaluating small eye movements in patients with saccadic palsies. Ann N Y Acad Sci. 2003;1004:337–46.
Pierrot-Deseilligny C, Rivaud S, Pillon B, Fournier E, Agid Y. Lateral visually-guided saccades in progressive supranuclear palsy. Brain. 1989;112(Pt 2):471–87.
Shaikh AG, Factor SA, Juncos J. Saccades in progressive supranuclear palsy - maladapted, irregular, curved, and slow. Mov Disord Clin Pract. 2017;4(5):671–81.
Verny M, Jellinger KA, Hauw JJ, Bancher C, Litvan I, Agid Y. Progressive supranuclear palsy: a clinicopathological study of 21 cases. Acta Neuropathol. 1996;91(4):427–31.
De Bruin VM, Lees AJ. Subcortical neurofibrillary degeneration presenting as Steele-Richardson-Olszewski and other related syndromes: a review of 90 pathologically verified cases. Mov Disord. 1994;9(4):381–9.
Birdi S, Rajput AH, Fenton M, et al. Progressive supranuclear palsy diagnosis and confounding features: report on 16 autopsied cases. Mov Disord. 2002;17(6):1255–64.
Diroma C, Dell’Aquila C, Fraddosio A, et al. Natural history and clinical features of progressive supranuclear palsy: a clinical study. Neurol Sci. 2003;24(3):176–7.
Marra M, Toni V, Trianni G, Coppola G. Progressive supranuclear palsy: analysis of six cases. Neurol Sci. 2003;24(3):186–7.
Kanazawa M, Tada M, Onodera O, Takahashi H, Nishizawa M, Shimohata T. Early clinical features of patients with progressive supranuclear palsy with predominant cerebellar ataxia. Parkinsonism Relat Disord. 2013;19(12):1149–51.
Pinhas I, Pinhas A, Goldhammer Y, Braham J. Progressive supranuclear palsy: electomyographic examinations of eye muscles. Acta Neurol Scand. 1978;58(5):304–8.
Kim C, Lee HW, Park MY. Exodeviated ophthalmoplegia in a patient with progressive supranuclear palsy. J Korean Med Sci. 2009;24(5):982–4.
Kanazawa M, Shimohata T, Toyoshima Y, et al. Cerebellar involvement in progressive supranuclear palsy: a clinicopathological study. Mov Disord. 2009;24(9):1312–8.
Anderson TJ, MacAskill MR. Eye movements in patients with neurodegenerative disorders. Nat Rev Neurol. 2013;9(2):74–85.
Riley DE, Lang AE, Lewis A, et al. Cortical-basal ganglionic degeneration. Neurology. 1990;40(8):1203–12.
Lewis AJ, Gawel MJ. Diffuse Lewy body disease with dementia and oculomotor dysfunction. Mov Disord. 1990;5(2):143–7.
Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114(Pt 5):2283–301.
Williams DR, Lees AJ. What features improve the accuracy of the clinical diagnosis of progressive supranuclear palsy-parkinsonism (PSP-P)? Mov Disord. 2010;25(3):357–62.
Nuwer MR. Progressive supranuclear palsy despite normal eye movements. Arch Neurol. 1981;38(12):784.
Williams DR, Holton JL, Strand K, Revesz T, Lees AJ. Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Mov Disord. 2007;22(15):2235–41.
Cordato NJ, Halliday GM, McCann H, et al. Corticobasal syndrome with tau pathology. Mov Disord. 2001;16(4):656–67.
Wakabayashi K, Takahashi H. Pathological heterogeneity in progressive supranuclear palsy and corticobasal degeneration. Neuropathology. 2004;24(1):79–86.
Daniel SE, de Bruin VM, Lees AJ. The clinical and pathological spectrum of Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy): a reappraisal. Brain. 1995;118(Pt 3):759–70.
Tsuboi Y, Josephs KA, Boeve BF, et al. Increased tau burden in the cortices of progressive supranuclear palsy presenting with corticobasal syndrome. Mov Disord. 2005;20(8):982–8.
Ling H, O'Sullivan SS, Holton JL, et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain. 2010;133(Pt 7):2045–57.
Josephs KA, Katsuse O, Beccano-Kelly DA, et al. Atypical progressive supranuclear palsy with corticospinal tract degeneration. J Neuropathol Exp Neurol. 2006;65(4):396–405.
Josephs KA, Duffy JR. Apraxia of speech and nonfluent aphasia: a new clinical marker for corticobasal degeneration and progressive supranuclear palsy. Curr Opin Neurol. 2008;21(6):688–92.
Nagao S, Yokota O, Nanba R, et al. Progressive supranuclear palsy presenting as primary lateral sclerosis but lacking parkinsonism, gaze palsy, aphasia, or dementia. J Neurol Sci. 2012;323(1–2):147–53.
Boeve B, Dickson D, Duffy J, Bartleson J, Trenerry M, Petersen R. Progressive nonfluent aphasia and subsequent aphasic dementia associated with atypical progressive supranuclear palsy pathology. Eur Neurol. 2003;49(2):72–8.
Halliday GM, Hardman CD, Cordato NJ, Hely MA, Morris JG. A role for the substantia nigra pars reticulata in the gaze palsy of progressive supranuclear palsy. Brain. 2000;123(Pt 4):724–32.
Rivaud-Pechoux S, Vidailhet M, Gallouedec G, Litvan I, Gaymard B, Pierrot-Deseilligny C. Longitudinal ocular motor study in corticobasal degeneration and progressive supranuclear palsy. Neurology. 2000;54(5):1029–32.
Quinn N. The “round the houses” sign in progressive supranuclear palsy. Ann Neurol. 1996;40(6):951.
Bhattacharjee S, Ng KL. “Round the houses” sign and apraxia of eyelid opening in a 62-year old man. Neurol India. 2017;65(5):1157.
Crespi J, Brathen G, Quist-Paulsen P, Pagonabarraga J, Roig-Arnall C. Facial dystonia with facial grimacing and vertical gaze palsy with “round the houses” sign in a 29-year-old woman. Neuroophthalmology. 2016;40(1):31–4.
Eggink H, Brandsma R, van der Hoeven JH, Lange F, de Koning TJ, Tijssen MA. Teaching video NeuroImages: the “round the houses” sign as a clinical clue for Niemann-Pick disease type C. Neurology. 2016;86(19):e202.
Davson H. The protective mechanism. In: Davson H, editor. Physiology of the eye fourth edition. United States: Academic; 1980. p. 492.
VanderWerf F, Brassinga P, Reits D, Aramideh M, Ongerboer de Visser B. Eyelid movements: behavioral studies of blinking in humans under different stimulus conditions. J Neurophysiol. 2003;89(5):2784–96.
Reddy VC, Patel SV, Hodge DO, Leavitt JA. Corneal sensitivity, blink rate, and corneal nerve density in progressive supranuclear palsy and Parkinson disease. Cornea. 2013;32(5):631–5.
Fitzpatrick E, Hohl N, Silburn P, O’Gorman C, Broadley SA. Case-control study of blink rate in Parkinson’s disease under different conditions. J Neurol. 2012;259(4):739–44.
Sommer M, Grafman J, Litvan I, Hallett M. Impairment of eyeblink classical conditioning in progressive supranuclear palsy. Mov Disord. 2001;16(2):240–51.
Golbe LI, Davis PH, Lepore FE. Eyelid movement abnormalities in progressive supranuclear palsy. Mov Disord. 1989;4(4):297–302.
Bologna M, Agostino R, Gregori B, et al. Voluntary, spontaneous and reflex blinking in patients with clinically probable progressive supranuclear palsy. Brain. 2009;132(Pt 2):502–10.
Sciacca G, Nicoletti A, Mostile G, et al. Blink reflex recovery cycle to differentiate progressive supranuclear palsy from corticobasal syndrome. Eur J Neurol. 2018;25(8):1100–e1185.
Sandyk R. The significance of eye blink rate in parkinsonism: a hypothesis. Int J Neurosci. 1990;51(1–2):99–103.
Karson CN, Burns RS, LeWitt PA, Foster NL, Newman RP. Blink rates and disorders of movement. Neurology. 1984;34(5):677–8.
Tamer C, Melek IM, Duman T, Oksuz H. Tear film tests in Parkinson’s disease patients. Ophthalmology. 2005;112(10):1795.
Grandas F, Elston J, Quinn N, Marsden CD. Blepharospasm: a review of 264 patients. J Neurol Neurosurg Psychiatry. 1988;51(6):767–72.
Barclay CL, Lang AE. Dystonia in progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 1997;62(4):352–6.
Rana AQ, Kabir A, Dogu O, Patel A, Khondker S. Prevalence of blepharospasm and apraxia of eyelid opening in patients with parkinsonism, cervical dystonia and essential tremor. Eur Neurol. 2012;68(5):318–21.
Janati A. Progressive supranuclear palsy: report of a case with torticollis, blepharospasm, and dysfluency. Am J Med Sci. 1986;292(6):391–2.
Hallett M, Evinger C, Jankovic J, Stacy M, Workshop BI. Update on blepharospasm: report from the BEBRF international workshop. Neurology. 2008;71(16):1275–82.
Goldstein JE, Cogan DG. Apraxia of lid opening. Arch Ophthalmol. 1965;73:155–9.
Lepore FE, Duvoisin RC. “Apraxia” of eyelid opening: an involuntary levator inhibition. Neurology. 1985;35(3):423–7.
Elston JS. A new variant of blepharospasm. J Neurol Neurosurg Psychiatry. 1992;55(5):369–71.
Krack P, Marion MH. “Apraxia of lid opening”, a focal eyelid dystonia: clinical study of 32 patients. Mov Disord. 1994;9(6):610–5.
Yoon WT, Chung EJ, Lee SH, Kim BJ, Lee WY. Clinical analysis of blepharospasm and apraxia of eyelid opening in patients with parkinsonism. J Clin Neurol. 2005;1(2):159–65.
Rana AQ, Shah R. Combination of blepharospasm and apraxia of eyelid opening: a condition resistant to treatment. Acta Neurol Belg. 2012;112(1):95–6.
Esteban A, Gimenez-Roldan S. Involuntary closure of eyelids in parkinsonism. Electrophysiological evidence for prolonged inhibition of the levator palpebrae muscles. J Neurol Sci. 1988;85(3):333–45.
Aramideh M, Ongerboer de Visser BW, Koelman JH, Speelman JD. Motor persistence of orbicularis oculi muscle in eyelid-opening disorders. Neurology. 1995;45(5):897–902.
Piccione F, Mancini E, Tonin P, Bizzarini M. Botulinum toxin treatment of apraxia of eyelid opening in progressive supranuclear palsy: report of two cases. Arch Phys Med Rehabil. 1997;78(5):525–9.
Lamberti P, De Mari M, Zenzola A, Aniello MS, Defazio G. Frequency of apraxia of eyelid opening in the general population and in patients with extrapyramidal disorders. Neurol Sci. 2002;23(Suppl 2):S81–2.
Defazio G, De Mari M, De Salvia R, Lamberti P, Giorelli M, Livrea P. “Apraxia of eyelid opening” induced by levodopa therapy and apomorphine in atypical parkinsonism (possible progressive supranuclear palsy): a case report. Clin Neuropharmacol. 1999;22(5):292–4.
Acknowledgements
We thank Miss Kamolwan Boonpang and Mr. Chanawat Anan for their assitance on the production of ocular examination video clips.
Funding
The study has been funded by the Chulalongkorn Academic Advancement Fund into Its second Century Project of Chulalongkorn University (2300042200), and a Center of Excellence grant from Chulalongkorn University (GCE 6100930004–1).
Author information
Authors and Affiliations
Contributions
OP, study concept and design, systematic review, interpretation of data, and drafting of the manuscript and figures. RB, study concept and design, acquisition of data, study supervision, and critical revision of the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All consents for publication were used with our institutional consent form and were obtained from that person, or their legal guardian.
Competing interests
The authors declare that they have no competing interests.
Additional files
Additional file 1:
The various types of ocular abnormalities in PSP from systematic review. (DOCX 15 kb)
Additional file 2:
Searching flow of systematic review. (PDF 156 kb)
Additional file 4:
Video legend of the common seven eye signs of patients with progressive supranuclear palsy. (DOCX 15 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Phokaewvarangkul, O., Bhidayasiri, R. How to spot ocular abnormalities in progressive supranuclear palsy? A practical review. Transl Neurodegener 8, 20 (2019). https://doi.org/10.1186/s40035-019-0160-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-019-0160-1