
Abstract
O-GlcNAcylation is a unique monosaccharide modification that is ubiquitously present in numerous nucleoplasmic and mitochondrial proteins. The hexosamine biosynthesis pathway (HBP), which is a key branch of glycolysis, provides the unique sugar donor UDP-GlcNAc for the O-GlcNAc modification. Thus, HBP/O-GlcNAcylation can act as a nutrient sensor to perceive changes in nutrient levels and trigger O-GlcNAc modifications of functional proteins in cellular (patho-)physiology, thereby regulating diverse metabolic processes. An imbalance in O-GlcNAcylation has been shown to be a pathogenic contributor to dysfunction in metabolic diseases, including type 2 diabetes, cancer, and neurodegeneration. However, under acute stress conditions, protein O-GlcNAc modification exhibits rapid and transient upregulation, which is strongly correlated with stress tolerance and cell survival. In this context, we discuss the metabolic, pharmacological and genetic modulation of HBP/O-GlcNAc modification in the biological system, the beneficial role of O-GlcNAcylation in regulating stress tolerance for cardioprotection, and neuroprotection, which is a novel and rapidly growing field. Current evidence suggests that transient activation of the O-GlcNAc modification represents a potent pro-survival signalling pathway and may provide a promising strategy for stress-related disorder therapy.
Similar content being viewed by others

Introduction
Glycosylation is a posttranslational modification (PTM) characterized by the covalent attachment of glycans to proteins, that occurs in 50%-70% of human proteins [1]. Unlike classic protein glycosylation (N-glycosylation), which occurs mostly via an endoplasmic reticulum–Golgi-dependent secretory pathway in the cell, O-linked N-acetylglucosaminylation (O-GlcNAcylation) is a unique PTM that is widely present in the nucleoplasm and mitochondria. O-GlcNAcylation is a highly dynamic signalling modification involving the attachment/removal of N-acetylglucosamine (GlcNAc) via an O-linkage with specific serine and threonine residues on proteins, and its function is similar to that of quintessential protein phosphorylation. Since it was first identified on mouse lymphocytes in 1984 [2], O-GlcNAcylation has been shown to regulate a multitude of cellular (patho)physiologies, including type 2 diabetes, cancer, and neurodegeneration [3]. For example, when cells are exposed to chronic hyperglycaemia, high O-GlcNAc levels reduce the effectiveness of insulin signalling pathways via metabolic regulation at the transcriptional level, leading to insulin resistance and type 2 diabetes [4, 5]. However, an increase in O-GlcNAc levels is an endogenous defence response to stress and initially acts in a protective manner. The beneficial effects of an acute and transient increase in O-GlcNAcylation in mediating stress tolerance and cell survival have recently been recognized. In this review, we discuss the beneficial role and potential mechanisms by which O-GlcNAcylation promotes self-tolerance and maintains cellular homeostasis under stress conditions, with a focus on the cardiovascular and central nervous systems (CNS).
Metabolic, pharmacological, and genetic modulation of O-GlcNAcylation
Unlike kinases and phosphatases with substrate specificities, the recycling of O-GlcNAc on proteins is controlled by only one pair of antagonistic enzymes, O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). This posttranslational modification requires UDP-GlcNAc as its sugar donor, which is synthesized via the hexosamine biosynthesis pathway (HBP), which is a branch of glucose metabolism [6]. The HBP branches off from the beginning stages of glycolysis and ultimately generates UDP-GlcNAc under multistep enzymatic catalysis with the involvement of amino acids (glutamine), fatty acids (acetyl-CoA), and nucleotides (UTP) (Fig. 1). Because multiple metabolites enter the HBP, the levels of UDP-GlcNAc and O-GlcNAc cycling are sensitive to fluctuations in these nutrient intermediates. For example, an increase in HBP flux driven by acute or chronic hyperglycaemia can lead to an increase in UDP-GlcNAc levels, causing the activation of O-GlcNAcylation in multiple cell types [7, 8]. Glutamine is also a potential activator of the HBP. Numerous studies have demonstrated that glutamine enhances stress tolerance and cell survival via HBP flux and increased protein O-GlcNAc levels in the heart and brain [9, 10]. The addition of glucosamine should be an effective means of driving the HBP/O-GlcNAc, since glucosamine can be directly phosphorylated to form glucosamine-6-phosphate by hexokinase, bypassing glutamine-fructose-6-phosphate amidotransferase (GFAT), a key rate-limiting enzyme for the formation of UDP-GlcNAc [11]. Thus, glucosamine is widely and extensively used in the biological systems as a metabolic intervention for functional studies of O-GlcNAcylation. Notably, unlike glucose, high concentrations of glucosamine can overwhelm the biosynthetic capacity of the HBP, causing massive accumulation of glucosamine-6-phosphate, ultimately leading to cellular ATP depletion via allosteric changes in various enzymes. Therefore, the judicious use of glucosamine rather than excessive concentrations of glucosamine facilitates our understanding of insulin resistance induced by hexosamine [12,13,14]. In addition, GlcNAc also significantly contributes to UDP-GlcNAc biosynthesis and serves as an available means for the increase in O-GlcNAc levels [15, 16]. Therefore, O-GlcNAcylation is highly sensitive to metabolite pools via the HBP and responds quickly to metabolic cues (Fig. 1), representing an important posttranslational mechanism for maintaining cellular homeostasis.

A schematic overview of the hexosamine biosynthesis pathway (HBP) and O-GlcNAcylation. Glucose imported into the cells is rapidly converted to fructose-6-phosphate via the beginning stages of glycolysis. Then, under the catalysis of the rate-limiting enzyme GFAT and other enzymes, HBP integrates multiple metabolic nutrients, ultimately generating UDP-GlcNAc, which is a unique monosaccharide donor for O-GlcNAcylation. The O-GlcNAc cycling is a highly dynamic and reversible modification controlled by a pair of antagonistic enzymes, OGT and OGA. The metabolic and pharmacological interventions for studying the functional role of HBP/O-GlcNAcylation are illustrated with blue and green boxes, respectively
In addition to the aforementioned metabolic interventions, the modulation of O-GlcNAc cycling can be achieved by pharmacological manipulation targeting key regulatory enzymes involved in the HBP/O-GlcNAcylation pathway. GFAT, which is the rate-limiting enzyme in the HBP, is key to controlling HBP flux. Since flux through GFAT is glutamine dependent, HBP flux can be inhibited by glutamine analogues, such as 6-diazo-5-oxo-norleucine (DON) or O-diazoacetyl-l-serine (azaserine). Of note, these substrate analogues have too many off-target effects and potential cytotoxicity [17, 18], therefore, new potent and reversible GFAT inhibitors are being developed [19, 20].
In addition to GFAT, the expression or activity control of OGT and OGA are also targets for intervention. OGT has a high affinity for UDP-GlcNAc, and its affinity for peptides is exquisitely modulated by UDP-GlcNAc levels. In fact, several OGT inhibitors are being widely used to pharmacologically modulate O-GlcNAcylation in functional analyses, including alloxan, a UDP-GlcNAc analogue (Ac4-5SGlcNAc), ST045849, and OSMI-1, − 2, − 3, and − 4. Notably, alloxan is now rarely used as an OGT inhibitor due to its dual inhibitory effects on both OGT and OGA [21]. Moreover, there are several pharmacological inhibitors of OGA, including streptozotocin (STZ), PUGNAc, NButGT, GlcNAcstatin, NAG-thiazoline, and thiamet-G (Fig. 1). Despite the widespread use of these OGT and OGA inhibitors, researchers are still concerned about their potential off-target effects. Thus, the use of genetic approaches to manipulate the level of O-GlcNAc modification, such as RNA interference, adenoviral overexpression or transgenic mouse models, are expected to contribute to the understanding of drug targets and off-target-associated safety. The commonly used and emerging metabolic, pharmacological and genetic interventions for functional studies on HBP/O-GlcNAcylation are listed in Table 1.
O-GlcNAcylation and stress tolerance
It is well known that organisms have evolved specific stress adaptation strategies to response to environmental fluctuations [52]. Although chronic hyperglycaemia is a potential risk factor for the severity of multiple diseased organs, early and rapid hyperglycaemia caused by stress has been considered an evolutionarily preserved adaptive response that provides a protective effect and supports survival during acute illnesses [53]. Under acute stress, the neuroendocrine response is characterized by activation of the sympathetic nervous system and the massive release of catecholamines, leading to increased secretion of glucagon. Glucagon promotes excessive gluconeogenesis and glycogenolysis, causing stress-induced hyperglycaemia and providing energy for high-energy organs, such as the brain and heart [54]. Along with providing a ready source of fuel, the hypermetabolic state can inhibit glycolytic flux via reactive oxygen species (ROS), thereby increasing the availability of glucose for the HBP/O-GlcNAc pathway [55]. Thus, O-GlcNAc modification is a nutrient and stress sensor, indicating a potential mechanism linking stress-induced hyperglycaemia with beneficial outcomes.
In the past two decades, the beneficial effects of acute stimulation of protein O-GlcNAc levels in the context of stress tolerance and cell survival have received widespread attention. In 2004, Zachara et al. first proposed that O-GlcNAcylation was a stress signalling through which cells rapidly detected and responded to a diverse array of stress stimuli to survive [56]. In fact, numerous reports have demonstrated that transient activation of O-GlcNAcylation is an endogenous adaptation against stress, and metabolic, pharmacological and genetic augmentations of O-GlcNAc levels promote cellular survival in multiple tissues and organs. Next, we focus our discussion on the beneficial role of O-GlcNAcylation in mediating stress tolerance in the cardiovascular system, as well as neuroprotection, which is a novel and rapidly growing field.
O-GlcNAcylation and cardioprotection
GIK therapy and O-GlcNAcylation
Glucose–insulin–potassium (GIK) therapy has played a beneficial role in acute myocardial infarction and cardiac surgery over the last 50 years [57, 58]. Although the mechanism by which GIK therapy confers cardioprotection is not known, it is widely accepted that increases in glucose uptake and metabolism are common features of this metabolism-based therapy [59]. Other researchers have suggested that the beneficial effects of GIK therapy can be attributed to an increase in O-GlcNAc signalling. In patients undergoing aortic valve replacement surgery, the improved outcome of low cardiac output after GIK therapy is associated with increased AMPK/Akt phosphorylation and O-GlcNAcylation of selected protein bands [60]. Furthermore, in cultured cardiomyocytes exposed to ischaemic shock, the cytoprotective effect of GIK therapy may involve the inhibition of ROS and upregulation of O-GlcNAcylation and OGT expression [61]. In fact, O-GlcNAcylation can act as a signalling molecule to rapidly respond to nutrient status and play a fundamental role in the endogenous defence of cardiomyocyte survival. For example, a series of studies on isolated perfused rat hearts provided early evidence of the functional relevance of HBP/O-GlcNAc flux and cellular stress tolerance, signifying that acute O-GlcNAc activation was an important PTM that regulated stress survival [9, 34, 62]. Subsequently, many studies have investigated changes in O-GlcNAc modification under stress conditions in various in vitro and in vivo models and explored the functional role of O-GlcNAcylation in mediating myocardial stress tolerance via metabolic, pharmacological and genetic interventions (Table 2). Next, we discuss the functional relevance of O-GlcNAc levels and cellular stress resistance, as well as the specific mechanisms through which O-GlcNAc exerts cardioprotection.
Mechanisms by which O-GlcNAcylation confers myocardial stress tolerance
Calcium and redox homeostasis
The severity of myocardial I/R injury is intimately tied to the sustained increase in intracellular calcium levels (calcium overload). O-GlcNAc signalling has been shown to regulate Ca2+-mediated events in cardiomyocytes. In cultured cardiomyocytes acutely treated with glucosamine, an increase in UDP-GlcNAc and O-GlcNAc levels is coupled to the inhibition of calcium overload induced by angiotensin II. This cardioprotection can be simulated by PUGNAc or eliminated by alloxan, indicating a close link between HBP/O-GlcNAc levels and intracellular calcium homeostasis [80]. Subsequently, research on the calcium paradox model of isolated hearts found that short-term high glucose or glucosamine challenge significantly improved cardiac function recovery, while pharmacological inhibition of GFAT or OGT restored sensitivity to the calcium paradox [34]. These cardioprotective mechanisms can be attributed at least in part to the reduction in calcium/calpain-dependent proteolysis, including alpha-fodrin, Ca2+/calmodulin (CaM)-dependent protein kinase (CaMKII) [62], and calcineurin [67]. It is worth noting that in a comparative study of K+ channel remodelling in hearts exposed to acute and chronic hyperglycaemia, O-GlcNAcylation of CaMKII at Ser-280 enhanced the recovery of K+ channels from inactivation during acute hyperglycaemia. However, chronic hyperglycaemia and sustained activation of CaMKII lead to significant arrhythmogenic electrophysiological remodelling [81]. Furthermore, excessive O-GlcNAc modification of CaMKII has been shown to contribute to the induction of ROS, which may exacerbate the pathological consequences of hyperglycaemia in diabetes [7, 82]. Notably, in a recent study on the diabetic heart, the O-GlcNAc modification of the histone deacetylase 4 subdomain at Ser-642, which is an important epigenetic regulator, exerted cardioprotective effects by counteracting pathological CaMKII signalling [83].
Calcium transport pathways are highly sensitive to oxidative stress. Accumulating evidence indicates that ROS and Ca2+ signalling likely play central roles in the pathogenesis of cardiovascular dysfunction [84]. Recent work suggests that a dynamic mitochondrial O-GlcNAcylation system rapidly modulates oxidative phosphorylation and ROS release in the heart [39]. In mice exposed to hypoxic acclimation, O-GlcNAc modification of glucose-6-phosphate dehydrogenase increases the NADPH/NADP+ and GSH/GSSG ratios, contributing to redox homeostasis in the I/R-exposed heart [77]. O-GlcNAc signalling also attenuates hypoxic/H2O2-induced Ca2+ overload in cultured neonatal rat cardiomyocytes [51]. In addition, a similar protective mechanism of O-GlcNAcylation has been found in the neuronal defence against Aβ neurotoxicity [85]. Interestingly, in human corneal endothelial cells exposed to tBHP (an oxidative stress inducer), the increase in O-GlcNAc signalling induced by PUGNAc reduces intracellular ROS and restores cellular viability, and this beneficial effect is due to the maintenance of mitochondrial calcium homeostasis, indicating that mitochondrial calcium signalling may be a key target for O-GlcNAcylation [86]. Paradoxically, high glucose or thiamet-G treatment promotes the excessive ROS generation in cardiomyocytes via CaMKII O-GlcNAcylation-dependent sarcoplasmic reticulum Ca2+ release [7]. The integration of O-GlcNAc into calcium and redox signalling is under intense investigation.
Mitochondrial homeostasis
In 2008, Ngoh et al. first provided evidence that O-GlcNAcylation plays a fundamental role in mitochondrial homeostasis to influence cardiomyocyte survival/death. They reported that an acute increase in OGT exerts a cardioprotective effect by maintaining the mitochondrial permeability transition pore (mPTP) and mitochondrial membrane potential in the myocardium exposed to hypoxia–reoxygenation insult [46]. Further studies investigating the effects of high glucose or hyperglycaemia in diabetes on myocardial function showed that a chronic increase in O-GlcNAcylation causes mitochondrial dysfunction, including the impairment of mitochondrial respiratory complex activity [87], an imbalance in mitochondrial fusion and fission [88], and mitochondrial DNA (mtDNA) damage [89]. Subsequently, Banerjee et al. reported the presence of mitochondrial-specific OGT, OGA, and UDP-GlcNAc transporters and confirmed that the dysregulation of O-GlcNAc cycling within mitochondria contributed to mitochondrial dysfunction associated with diabetic cardiomyopathy [90]. In fact, mitochondria are targets of O-GlcNAc modification [91]. In a mouse model with impaired branched-chain amino acid catabolism, a reduction in HBP/O-GlcNAc levels selectively disrupted the use of mitochondrial pyruvate by inhibiting pyruvate dehydrogenase complex activity, resulting in a significant decrease in glucose oxidation in the heart [92]. In a study of cardiac I/R injury, the beneficial effect of O-GlcNAcylation induced by hypoxic acclimation was partly attributed to mitochondrial preservation, including effects on mitochondrial ultrastructure, mitochondrial respiration, mtDNA, and mitochondrial redox homeostasis [77]. Redox and calcium handling may be key regulators of mPTP-dependent apoptosis cascade events that occur in mitochondria impaired by stress. The transient opening of the mPTP allows for the release of mitochondrial contents and the activation of intrinsic apoptosis pathways. In cardiomyocytes subjected to hypoxia or oxidative stress, pharmacological and genetic manipulation of OGT and OGA confirmed that O-GlcNAcylation alleviated the formation of mPTP by inhibiting ROS generation and calcium overload [51]. Proteome and O-GlcNAcome analysis of cardiac mitochondria from thiamet-G-treated rats revealed that many mitochondrial proteins, especially those in the oxidative phosphorylation system, are major targets for O-GlcNAcylation. Although certain sites of specific proteins exhibit decreases in O-GlcNAc modification, global protein O-GlcNAc levels are increased, which leads to the enhancement of mitochondrial bioenergetics and the threshold for mPTP opening in the presence of calcium [93].
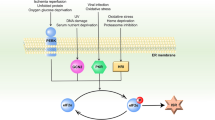
Voltage-dependent anion channel (VDAC), which is the primary channel for Ca2+ influx and efflux through the outer mitochondrial membrane (OMM) [94], is a target for O-GlcNAcylation. Functional biochemical assessments indicate that the enhanced resistance of mitochondria to mPTP formation induced by calcium is intimately associated with the increase in the number of O-GlcNAc-modified VDACs [70], while inhibiting VDAC O-GlcNAc modification makes mitochondria sensitive to calcium-induced mPTP opening [46]. The functional relevance of VDAC O-GlcNAcylation to mPTP inhibition and cellular tolerance has also been confirmed by the protective effect of the volatile anaesthetic isoflurane on myocardial I/R stress [68]. Although VDAC may not be an essential component of the mPTP, the fact that VDAC participates in mitochondrial membrane permeability and apoptosis signalling by modulating mitochondrial Ca2+ flux cannot be ignored [95]. The Bcl-2 family appears to be responsible for the regulation of mitochondrial Ca2+ transport systems, including VDAC and the mPTP [96]. The dynamic interactions between Bcl-2 family proteins induce conformational changes in proteins, leading to oligomerization (homologous or heterologous) and membrane insertion, thereby regulating the permeabilization of the OMM and apoptosis [97]. There is evidence that the beneficial effect of O-GlcNAc modification on the maintenance of mitochondrial membrane potential and cytochrome c in stressed cardiomyocytes can be attributed to an increase in mitochondrial Bcl-2 translocation rather than changes in BAD or Bax [66]. However, in H9c2 cardiomyoblasts exposed to chronic hyperglycaemia, excessive O-GlcNAcylation of the proapoptotic protein BAD has been shown to contribute to the formation of the BAD-Bcl-2 dimer, thus enhancing cellular apoptosis [98]. Mitochondrial dysfunction associated with an imbalance in O-GlcNAcylation in the context of glucose toxicity due to hyperglycaemia cannot be ignored. Overall, these studies highlight a profound impact of O-GlcNAcylation on mitochondrial homeostasis, including mitochondrial structure, mitochondrial bioenergy, redox signalling, calcium handling, and the mitochondrial apoptosis pathway (Fig. 2). Further investigation of the multiple layers of complexity between O-GlcNAcylation and mitochondrial homeostasis is needed.

Schematic representation of key targets for O-GlcNAc cycling on mitochondrial homeostasis, including mitochondrial structure, mitochondrial bioenergy, redox signalling, calcium handling, and the mitochondrial apoptosis pathway
Endoplasmic reticulum stress
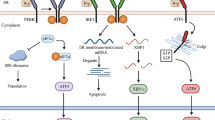
The endoplasmic reticulum (ER) possesses a strict quality control system for protein folding, posttranslational modification, and assembly. The quality control capability of ER is limited. Under pathological conditions, large amounts of unfolded or misfolded proteins accumulate in the ER, resulting in ER stress and the unfolded protein response (UPR). The adaptive UPR plays a beneficial role in restoring protein homeostasis in the ER, while the maladaptive or terminal UPR is involved in the destruction of ER integrity and cellular defects. In metazoans, the UPR includes three signalling pathways: the membrane-anchored transcription factor ATF6, the inositolase IRE1, and the protein kinase PERK [99]. In 2009, Ngoh et al. first proposed that ER stress was a key pathological factor in cardiomyocyte death induced by hypoxia, and increasing O-GlcNAc levels by pharmacological or genetic manipulation mitigated the death of cardiomyocytes exposed to ER stress inducers [71]. In constitutive cardiomyocyte-specific OGT-KO mice, gradual and progressive cardiomyopathy is accompanied by increased expression of ER stress markers, suggesting a close link between ER function and O-GlcNAcylation [100]. Notably, transcriptional activation of the UPR/HBP axis in various stress conditions has been confirmed. The UPR triggers the transcription of key members (GFAT1, GNPNAT1, and PGM3) of the HBP via its most conserved signal transducer spliced X-box binding protein 1 (xbp1s, a transcription factor), leading to the activation of HBP and O-GlcNAcylation, thus providing robust cardioprotection in mice (Fig. 3) [69]. Similar to xbp1s, the ER resident transcription factor spermatogenesis 40 (Tisp40) transcriptionally activates the HBP in conditions of cardiac stress [101]. In turn, O-GlcNAcylation can modulate cellular homeostasis in response to ER stress by modulating eukaryotic translation initiation factor 2α (eIF2α), which is one of the UPR branches [102]. However, this regulatory mechanism has not yet been demonstrated in the cardiovascular system.

A schematic overview of the XBP1s/HBP/O-GlcNAc axis in the heart and brain
Inflammation
Although it has been reported that the beneficial effects of O-GlcNAc stimulation on improving survival and cardiac function in septic shock are independent of inflammation [38], numerous studies have shown that there is an intimate relationship between inflammation and O-GlcNAcylation in stressed hearts. Glucosamine exerts anti-inflammatory effects on various cell types and models, including systemic inflammation [103] and osteoarthritis [104]. In a rat model of trauma-induced haemorrhage, the increase in O-GlcNAc levels induced by glucosamine or PUGNAc has been shown to improve survival, organ perfusion, and cardiac function. One mechanism of these beneficial outcomes is the attenuation of circulating inflammatory cytokines [36, 37, 79, 105]. Furthermore, the protective effect of the increase in protein O-GlcNAc modification on vascular inflammation and vascular dysfunction has also been confirmed [65, 73, 78]. Importantly, the nuclear factor NF-κB, which is a prototypical proinflammatory signalling factor, is a key molecular bridge linking O-GlcNAcylation and inflammation. For example, an increase in O-GlcNAc levels weakens NF-κB nuclear translocation and subsequent TNF-α and IL-6 expression, thus improving cardiac function following trauma-induced haemorrhage [23]. It is also noteworthy that the NF-κB pathway is involved in vascular inflammation, and glucosamine or thiamet-G treatment has been shown to alleviate inflammation-induced vascular damage by antagonizing the NF-κB signalling cascade [72, 106]. The NF-κB subunit p65 is a target for O-GlcNAcylation, and glycosylation of this factor inhibits self-phosphorylation, thereby preventing p65 downstream signalling [107]. Furthermore, acute O-GlcNAcylation can reduce inducible nitric oxide synthase (iNOS) by inhibiting the NF-κB pathway, thus alleviating oxidative stress-induced vascular dysfunction [73]. Paradoxically, O-GlcNAcylation of NF-κB may also contribute to the lipopolysaccharide (LPS)-induced endothelial inflammatory response [108]. Thus, a deeper investigation of the nuanced relationship between O-GlcNAc and inflammation and how this association impacts cardiac and vascular function under stress conditions is needed.
Heat shock response
The heat shock response (HSR) is an ancient defence signalling pathway that maintains proteostasis to cope with a variety of cellular stresses. In the HSR, diverse heat shock factors are recruited to control the heat shock protein (HSP)/chaperone network to help modulate protein folding and repair [109]. O-GlcNAc appears to improve the tolerance of cardiomyocytes to multiple forms of stress by upregulating the rates and extent of HSP induction [56, 63, 110, 111], including HSP70, HSP40, HSP72, and aB-crystallin (HSPB5). Many studies associated with protection strategies against myocardial stress have focused on HSP70, a master regulator of protein degradation. Activation of HSP70 enhances the stress adaptation of the myocardium to I/R injury via multiple mechanisms, including oxidative stress, calcium overload, apoptosis, autophagy, and inflammatory responses [112, 113]. Pharmacological enhancement with glutamine induces HSP70 expression and the activation of key transcription factors in the HSP70 pathway in animal models of inflammatory responses [10, 114, 115]. HSP70 exhibits adjustable lectinic activity that depends on glucose concentrations and O-GlcNAc levels [116, 117]. Protein misfolding triggers the release of Hsp70-GlcNAc-binding activity in response to a wide variety of cellular stresses [118]. Importantly, O-GlcNAc signalling prevents proteasome degradation by modifying the specific interactions of HSP70 family members [119]. In addition to HSP70, other HSPs are targets for O-GlcNAcylation, including Hsp90β [120], HSP28 [121], and HSPA6 [122]. However, further studies are needed to determine the role of O-GlcNAcylation in the functional regulation of these HSPs during cellular stress.
Collectively, these results demonstrate that O-GlcNAcylation is a pro-survival signal that mediates myocardial stress tolerance via multiple mechanisms, including calcium and redox homeostasis, mitochondrial homeostasis, ER stress, inflammation, and the HSR.
O-GlcNAcylation and neuroprotection
Inspired by the beneficial effects of O-GlcNAcylation on improving cardiac function under stress, the discovery and knowledge of the pro-survival response of O-GlcNAcylation in the CNS has exploded recently (Table 3). Extensive work has focused on in vivo cerebral I/R injury experiments in which the activation of O-GlcNAcylation has been shown to be an adaptive response to improve cellular stress tolerance, and increasing this PTM might be a promising strategy for stroke therapy.
Age-related activation of O-GlcNAcylation
Stroke is an acute cerebrovascular accident that primarily impacts elderly individuals, and clinical evidence shows that the recovery of neurological function worsens with age [139]. Therefore, researchers have focused on comparing cellular responses to ischaemic challenges in young and aged animals in experimental stroke studies and have attempted to determine the role of ageing in the cellular response to severe forms of stress associated with I/R. In a transient forebrain ischaemia model, Liu et al. analysed the activation of proteostasis-related pathways in young and aged mice and found that the most prominent change in the ageing brain was the inactivation of the O-GlcNAc modification, suggesting that this pathway might be a promising target for stroke therapy [132]. In addition to the brain, impaired age-related activation of O-GlcNAcylation has also been confirmed in the kidney and spinal cord after cardiac arrest and cardiopulmonary resuscitation (CA/CPR) [45], signifying the importance of O-GlcNAcylation as a potential mechanism underlying the impairment of functional recovery in ageing organs/tissues in response to ischaemic challenge. LC–MS/MS analysis showed that the availability of UDP-GlcNAc in the aged brain was impaired both at baseline and after I/R, while metabolic intervention with glucosamine significantly improved the acute outcomes in young and elderly mice [130]. Furthermore, other studies have reported that pharmacological increases in O-GlcNAc levels with thiamet-G improved outcomes after ischaemic stroke or CA/CPR in both young and elderly animals [44, 131]. Therefore, interventions targeting the HBP/O-GlcNAc axis might be a promising therapeutic strategy for stroke.
Mechanisms by which O-GlcNAcylation confers neuronal stress tolerance
Mitochondrial homeostasis
Mitochondria are crucial for maintaining metabolic homeostasis in the high-energy CNS. In the brain, O-GlcNAc cycling participates in the modulation of mitochondrial network homeostasis, which is diverse and includes mitochondrial trafficking, mitochondrial bioenergetics, mitochondrial fission and fusion, and mitochondrial apoptosis [91]. In fact, under ischaemic stress conditions, O-GlcNAcylation-mediated mitochondrial homeostasis and cellular bioenergetics have emerged as potential pharmacological targets for the development of neuroprotective agents. For example, an active component of Gastrodia elata exerts a potent neuroprotective effect by maintaining mitochondrial energy metabolism during cerebral I/R injury. Targeted metabolic profiling suggests that the increased levels of UDP‑GlcNAc and its regulatory enzyme OGT contribute to the beneficial effects of Gastrodia elata on stroke [134, 135]. In our laboratory, the compound SalA-4 g was shown to have neuroprotective effects [124]. Specific mechanisms may involve the O-GlcNAc modification of mitochondria by SalA-4 g, which was shown to exert neuroprotective effects by improving mitochondrial homeostasis and inhibiting mitochondrial apoptosis pathways in neurons exposed to ischaemia-like conditions [125]. Recently, in a mouse model exposed to sevoflurane, the beneficial effects of hypoxia acclimation on anaesthetic sensitivity were attributed to the increase in O-GlcNAc-dependent modulation of glutamatergic synapses and mitochondria [137].
Other studies have focused on the functional effects of O-GlcNAc on individual proteins in mitochondria. In neurons, dynamin-related protein 1 (Drp1), which is a critical protein involved in mitochondrial fission, is a target for O-GlcNAcylation [140]. In cerebral I/R injury, the expression of ogt is significantly upregulated, and ogt knockout reduces the phosphorylation of Drp1 Ser-637, leading to the translocation of Drp1 from the cytosol to mitochondria, thus accelerating mitochondria-dependent apoptosis [133]. Another O-GlcNAcylation target, adenosine 5'-triphosphate synthase subunit α (ATP5A), is critically involved in mitochondrial bioenergetics. The decrease in O-GlcNAc modification of the Thr-432 residue on ATP5A induced by Aβ inhibited ATPase activity and disrupted ATP synthesis in Alzheimer's disease (AD) pathology [128]. In neuronal excitotoxicity, the nitric oxide synthase adaptor (NOS1AP) acts as a ligand of neuronal nitric oxide synthases (nNOS) to participate in NMDA receptor-nNOS signalling. Mass spectrometry identified multiple sites for O-GlcNAc modification of NOS1AP, and an increase in this modification prevented its interaction with nNOS, thus protecting against neuronal excitotoxicity induced by glutamate [129].
XBP1s/HBP/O-GlcNAc axis
The discovery that the UPR branch is involved in the transcriptional activation of HBP/O-GlcNAcylation in cardiac ischaemia has generated a tremendous amount of interest among neuroscientists. In 2017, Jiang and colleagues first reported that the XBP1s/HBP/O-GlcNAc axis was neuroprotective in the context of ischaemic stroke (Fig. 3). They showed that O-GlcNAcylation was activated in an xbp1-dependent manner in the ischaemic penumbra after stroke, and this activation was impaired in the aged brain. Critically, an increase in this response induced by thiamet-G improved short-term stroke outcomes in young and aged mice [131]. Subsequently, further research evaluated and confirmed that thiamet-G improved stroke outcomes in neuron-specific xbp1-knockout mice, including long-term functional recovery. Given the impaired availability of UDP-GlcNAc in the aged brain, the research group further established the beneficial effects of metabolic intervention with glucosamine on stroke models in young and elderly animals [130]. The functional XBP1s/HBP/O-GlcNAc axis, which is a key pro-survival pathway, has also been confirmed in CA/CPR [136] and subarachnoid haemorrhage (SAH) models [126]. Thus, these studies demonstrate that the XBP1s/HBP/O-GlcNAc axis is a promising target for stroke therapy.
Inflammation
O-GlcNAcylation is involved in controlling inflammatory responses in experimental stroke. Acute increases in O-GlcNAc levels induced by glucosamine [127] or thiamet-G [44] exert neuroprotective effects on the ischaemic brain by inhibiting inflammatory cytokine production and microglial activation. The specific mechanism may involve the inhibition of NF-κB p65 signalling. The similar effects of glucosamine and thiamet-G suggest that suppressing inflammation might contribute to the neuroprotective mechanism of O-GlcNAcylation. Notably, a study on inflammatory modulation in macrophages exposed to LPS suggested that glucosamine could regulate inflammation by sensing different energy states. Under normal and high glucose conditions, glucosamine exerted opposite effects on NO/iNOS production stimulated by LPS depending on energy availability. The bidirectional regulatory effects of glucosamine may contribute to understanding the mechanisms by which O-GlcNAcylation affects nutrient sensing and inflammatory responses [141].
Moreover, dysfunctional O-GlcNAcylation-mediated neuroinflammation has been shown to be involved in the pathology of neurodegeneration. OGT protein levels are significantly low in the cortical neurons of severe AD patients, and specific loss of OGT in the forebrain leads to progressive neurodegeneration, including behavioural and histological phenotypes, as well as extensive gliosis and the upregulation of immune-response genes [142]. In an in vivo zebrafish model of hypoxic brain damage, the downregulation of several glucose metabolites and O-GlcNAc levels may be an important cause of brain inflammation and neurodegeneration, and these changes can be reversed by glucosamine supplementation [138]. In addition to neurons, O-GlcNAcylation is essential for inflammatory responses in astrocytes. The O-GlcNAc modification of NF‑κB p65 has been identified in astrocytes in vitro and in vivo, and increasing O-GlcNAcylation with GlcNAc inhibits inflammation and activation of astrocytes in AD mice by repressing the NF-κB signalling pathway [16]. Collectively, these findings illustrate the beneficial effect of O-GlcNAcylation on stress tolerance by modulating neuroinflammation.
Conclusions and perspective
The early and rapid hyperglycaemic response to severe injury or trauma is an important adaptive pro-survival process, which is accompanied by an increase in HBP flux and the activation of O-GlcNAc signalling. In animal models and clinical trials, the exact contribution of the HBP/O-GlcNAc pathway to various metabolic-based therapies (high glucose, GIK, and glutamine) has been confirmed. In fact, O-GlcNAc modification can serve as an environmental sensor in metabolic and stress regulation by directly and dynamically modulating protein functions. Numerous studies have demonstrated that the adaptive enhancement of O-GlcNAcylation is a pro-survival signal under stress, and a transient increase in global O-GlcNAc levels induced by stress or interventions (metabolic, pharmacological, or genetic) contributes to stress tolerance, especially in two high-energy organs: the heart and brain. The specific mechanism may involve calcium and redox homeostasis, mitochondrial homeostasis, ER stress, inflammation, and the HSR.
Although the benefits of O-GlcNAcylation in mediating stress tolerance have been clearly recognized, most functional studies still face many challenges. (1) The duration of changes in O-GlcNAc signalling under pathologic conditions (i.e., glucose toxicity and type II diabetes) may have contrary and deleterious effects. The molecular mechanisms underlying the transition from adaptive and pro-survival pathways to pathological responses are still unknown. (2) Due to the potential off-target effects of existing inhibitors, the development of small molecule kinase inhibitors with high specificity and inhibitory effects may contribute to the understanding of drug targets and off-target-associated safety. (3) The tools for identifying the individual O-GlcNAcylation of specific proteins and site-specific O-GlcNAc proteomics (O-GlcNAcomics) are limited. From this perspective, technical advances in high-throughput glycoproteomic studies will provide in-depth insights into the role of O-GlcNAcylation.
Availability of data and materials
Not applicable.
Abbreviations
- PTM:
-
Posttranslational modification
- O-GlcNAcylation:
-
O-Linked N-acetylglucosaminylation
- GlcNAc:
-
N-Acetylglucosamine
- CNS:
-
Central nervous system
- OGT:
-
O-GlcNAc transferase
- OGA:
-
O-GlcNAcase
- HBP:
-
Hexosamine biosynthesis pathway
- GFAT:
-
Glutamine-fructose-6-phosphate amidotransferase
- DON:
-
6-Diazo-5-oxo-norleucine
- azaserine:
-
O-Diazoacetyl-l-serine
- STZ:
-
Streptozotocin
- HK:
-
Hexokinase
- GPI:
-
Glucose-6-phosphate isomerase
- GNPNAT:
-
Glucosamine-phosphate N-acetyltransferase
- PGM:
-
Phosphoglucomutase
- UAP:
-
UDP-N-acetylglucosamine pyrophosphorylase
- ROS:
-
Reactive oxygen species
- GIK:
-
Glucose–insulin–potassium
- H/R:
-
Hypoxia–reoxygenation
- I/R:
-
Ischaemia–reperfusion
- BfA:
-
Brefeldin A
- TM:
-
Tunicamycin
- IPC:
-
Ischaemic preconditioning
- rIPC:
-
Remote ischaemic preconditioning
- NRVMs:
-
Neonatal rat ventricular myocytes
- NRCMs:
-
Neonatal rat cardiac myocytes
- NMCMs:
-
Neonatal mouse cardiac myocytes
- CSCs:
-
Cardiac stem cells
- HUVECs:
-
Human umbilical vein endothelial cells
- VSMCs:
-
Vascular smooth muscle cells
- CaMKII:
-
Ca2+/calmodulin (CaM)-dependent protein kinase
- mtDNA:
-
Mitochondrial DNA
- mPTP:
-
Mitochondrial permeability transition pore
- VDAC:
-
Voltage-dependent anion channel
- OMM:
-
Outer mitochondrial membrane
- ER:
-
Endoplasmic reticulum
- UPR:
-
Unfolded protein response
- ATF6:
-
Activating transcription factor 6
- IRE1:
-
Inositol-requiring enzyme 1
- PERK:
-
Protein kinase RNA-line ER kinase
- Xbp1:
-
X-box binding protein 1
- Xbp1s:
-
Spliced X-box binding protein 1
- Tisp40:
-
Spermatogenesis 40
- eIF2α:
-
Eukaryotic translation initiation factor 2α
- GNPNAT1:
-
Glucosamine-phosphate N-acetyltransferase 1
- PGM3:
-
Phosphoglucomutase 3
- iNOS:
-
Inducible nitric oxide synthase
- LPS:
-
Lipopolysaccharide
- HSR:
-
Heat shock response
- HSPs:
-
Heat shock proteins
- OGD/R:
-
Oxygen–glucose deprivation/reoxygenation
- tMCAO:
-
Transient middle cerebral artery occlusion
- pMCAO:
-
Permanent middle cerebral artery occlusion
- CA/CPR:
-
Cardiac arrest/cardiopulmonary resuscitation
- RH:
-
Repetitive hypoxia
- SAH:
-
Subarachnoid haemorrhage
- HT22 cells:
-
Mouse hippocampal neuronal cells
- CHO cells:
-
Chinese hamster ovary cells
- PC12 cells:
-
Rat pheochromocytoma cells
- Drp1:
-
Dynamin-related protein 1
- ATP5A:
-
Adenosine 5'-triphosphate synthase subunit α
- AD:
-
Alzheimer's disease
- NOS1AP:
-
Nitric oxide synthase adaptor
- nNOS:
-
Neuronal nitric oxide synthases
References
Schjoldager KT, Narimatsu Y, Joshi HJ, Clausen H. Global view of human protein glycosylation pathways and functions. Nat Rev Mol Cell Biol. 2020;21:729–49.
Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem. 1984;259:3308–17.
Chatham JC, Zhang J, Wende AR. Role of O-Linked N-acetylglucosamine protein modification in cellular (Patho)physiology. Physiol Rev. 2021;101:427–93.
Wells L, Vosseller K, Hart GW. A role for N-acetylglucosamine as a nutrient sensor and mediator of insulin resistance. Cell Mol Life Sci. 2003;60:222–8.
Hart GW. Nutrient regulation of signaling and transcription. J Biol Chem. 2019;294:2211–31.
Bond MR, Hanover JA. A little sugar goes a long way: the cell biology of O-GlcNAc. J Cell Biol. 2015;208:869–80.
Lu S, Liao Z, Lu X, Katschinski DM, Mercola M, Chen J, Heller Brown J, Molkentin JD, Bossuyt J, Bers DM. Hyperglycemia acutely increases cytosolic reactive oxygen species via O-linked GlcNAcylation and CaMKII activation in mouse ventricular myocytes. Circ Res. 2020;126:e80–96.
Durning SP, Flanagan-Steet H, Prasad N, Wells L. O-Linked beta-N-acetylglucosamine (O-GlcNAc) acts as a glucose sensor to epigenetically regulate the insulin gene in pancreatic beta cells. J Biol Chem. 2016;291:2107–18.
Liu J, Marchase RB, Chatham JC. Glutamine-induced protection of isolated rat heart from ischemia/reperfusion injury is mediated via the hexosamine biosynthesis pathway and increased protein O-GlcNAc levels. J Mol Cell Cardiol. 2007;42:177–85.
Wang J, Lu X, Zheng K, Jing L. Glutamine’s protection against brain damage in septic rats via increased protein oxygen-N-acetylglucosamine modification. NeuroReport. 2021;32:214–22.
Virkamaki A, Yki-Jarvinen H. Allosteric regulation of glycogen synthase and hexokinase by glucosamine-6-phosphate during glucosamine-induced insulin resistance in skeletal muscle and heart. Diabetes. 1999;48:1101–7.
Marshall S, Yamasaki K, Okuyama R. Glucosamine induces rapid desensitization of glucose transport in isolated adipocytes by increasing GlcN-6-P levels. Biochem Biophys Res Commun. 2005;329:1155–61.
Marshall S, Nadeau O, Yamasaki K. Dynamic actions of glucose and glucosamine on hexosamine biosynthesis in isolated adipocytes: differential effects on glucosamine 6-phosphate, UDP-N-acetylglucosamine, and ATP levels. J Biol Chem. 2004;279:35313–9.
Marshall S, Nadeau O, Yamasaki K. Glucosamine-induced activation of glycogen biosynthesis in isolated adipocytes. Evidence for a rapid allosteric control mechanism within the hexosamine biosynthesis pathway. J Biol Chem. 2005;280:11018–24.
Johswich A, Longuet C, Pawling J, Abdel Rahman A, Ryczko M, Drucker DJ, Dennis JW. N-glycan remodeling on glucagon receptor is an effector of nutrient sensing by the hexosamine biosynthesis pathway. J Biol Chem. 2014;289:15927–41.
Dong X, Shu L, Zhang J, Yang X, Cheng X, Zhao X, Qu W, Zhu Q, Shou Y, Peng G, Sun B, Yi W, Shu Q, Li X. Ogt-mediated O-GlcNAcylation inhibits astrocytes activation through modulating NF-kappaB signaling pathway. J Neuroinflamm. 2023;20:146.
Low SY, Taylor PM, Ahmed A, Pogson CI, Rennie MJ. Substrate-specificity of glutamine transporters in membrane vesicles from rat liver and skeletal muscle investigated using amino acid analogues. Biochem J. 1991;278(Pt 1):105–11.
Lyons SD, Sant ME, Christopherson RI. Cytotoxic mechanisms of glutamine antagonists in mouse L1210 leukemia. J Biol Chem. 1990;265:11377–81.
Qian Y, Ahmad M, Chen S, Gillespie P, Le N, Mennona F, Mischke S, So SS, Wang H, Burghardt C, Tannu S, Conde-Knape K, Kochan J, Bolin D. Discovery of 1-arylcarbonyl-6,7-dimethoxyisoquinoline derivatives as glutamine fructose-6-phosphate amidotransferase (GFAT) inhibitors. Bioorg Med Chem Lett. 2011;21:6264–9.
Vyas B, Silakari O, Bahia MS, Singh B. Glutamine: fructose-6-phosphate amidotransferase (GFAT): homology modelling and designing of new inhibitors using pharmacophore and docking based hierarchical virtual screening protocol. SAR QSAR Environ Res. 2013;24:733–52.
Lee TN, Alborn WE, Knierman MD, Konrad RJ. Alloxan is an inhibitor of O-GlcNAc-selective N-acetyl-beta-D-glucosaminidase. Biochem Biophys Res Commun. 2006;350:1038–43.
Cardozo CF, Vera A, Quintana-Pena V, Arango-Davila CA, Rengifo J. Regulation of Tau protein phosphorylation by glucosamine-induced O-GlcNAcylation as a neuroprotective mechanism in a brain ischemia-reperfusion model. Int J Neurosci. 2023;133:194–200.
Zou L, Yang S, Champattanachai V, Hu S, Chaudry IH, Marchase RB, Chatham JC. Glucosamine improves cardiac function following trauma-hemorrhage by increased protein O-GlcNAcylation and attenuation of NF-kappaB signaling. Am J Physiol Heart Circ Physiol. 2009;296:H515–23.
Kim SM, Zhang S, Park J, Sung HJ, Tran TT, Chung C, Han IO. REM sleep deprivation impairs learning and memory by decreasing brain O-GlcNAc cycling in mouse. Neurotherapeutics. 2021;18:2504–17.
Cui YL, Xue RQ, Xi H, Ming Z, Yu XJ, Liu LZ, Wu Q, Si Y, Li DL, Zang WJ. Cholinergic drugs ameliorate endothelial dysfunction by decreasing O-GlcNAcylation via M3 AChR-AMPK-ER stress signaling. Life Sci. 2019;222:1–12.
Gelinas R, Mailleux F, Dontaine J, Bultot L, Demeulder B, Ginion A, Daskalopoulos EP, Esfahani H, Dubois-Deruy E, Lauzier B, Gauthier C, Olson AK, Bouchard B, Des Rosiers C, Viollet B, Sakamoto K, Balligand JL, Vanoverschelde JL, Beauloye C, Horman S, Bertrand L. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat Commun. 2018;9:374.
Konrad RJ, Zhang F, Hale JE, Knierman MD, Becker GW, Kudlow JE. Alloxan is an inhibitor of the enzyme O-linked N-acetylglucosamine transferase. Biochem Biophys Res Commun. 2002;293:207–12.
Andres LM, Blong IW, Evans AC, Rumachik NG, Yamaguchi T, Pham ND, Thompson P, Kohler JJ, Bertozzi CR. Chemical modulation of protein O-GlcNAcylation via OGT inhibition promotes human neural cell differentiation. ACS Chem Biol. 2017;12:2030–9.
Gloster TM, Zandberg WF, Heinonen JE, Shen DL, Deng L, Vocadlo DJ. Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat Chem Biol. 2011;7:174–81.
Lima VV, Giachini FR, Carneiro FS, Carvalho MH, Fortes ZB, Webb RC, Tostes RC. O-GlcNAcylation contributes to the vascular effects of ET-1 via activation of the RhoA/Rho-kinase pathway. Cardiovasc Res. 2011;89:614–22.
Nomura A, Yokoe S, Tomoda K, Nakagawa T, Martin-Romero FJ, Asahi M. Fluctuation in O-GlcNAcylation inactivates STIM1 to reduce store-operated calcium ion entry via down-regulation of Ser(621) phosphorylation. J Biol Chem. 2020;295:17071–82.
Ortiz-Meoz RF, Jiang J, Lazarus MB, Orman M, Janetzko J, Fan C, Duveau DY, Tan ZW, Thomas CJ, Walker S. A small molecule that inhibits OGT activity in cells. ACS Chem Biol. 2015;10:1392–7.
Martin SES, Tan ZW, Itkonen HM, Duveau DY, Paulo JA, Janetzko J, Boutz PL, Tork L, Moss FA, Thomas CJ, Gygi SP, Lazarus MB, Walker S. Structure-based evolution of low nanomolar O-GlcNAc transferase inhibitors. J Am Chem Soc. 2018;140:13542–5.
Liu J, Pang Y, Chang T, Bounelis P, Chatham JC, Marchase RB. Increased hexosamine biosynthesis and protein O-GlcNAc levels associated with myocardial protection against calcium paradox and ischemia. J Mol Cell Cardiol. 2006;40:303–12.
Toleman C, Paterson AJ, Shin R, Kudlow JE. Streptozotocin inhibits O-GlcNAcase via the production of a transition state analog. Biochem Biophys Res Commun. 2006;340:526–34.
Zou L, Yang S, Hu S, Chaudry IH, Marchase RB, Chatham JC. The protective effects of PUGNAc on cardiac function after trauma-hemorrhage are mediated via increased protein O-GlcNAc levels. Shock. 2007;27:402–8.
Not LG, Brocks CA, Vamhidy L, Marchase RB, Chatham JC. Increased O-linked beta-N-acetylglucosamine levels on proteins improves survival, reduces inflammation and organ damage 24 hours after trauma-hemorrhage in rats. Crit Care Med. 2010;38:562–71.
Ferron M, Cadiet J, Persello A, Prat V, Denis M, Erraud A, Aillerie V, Mevel M, Bigot E, Chatham JC, Gauthier C, Rozec B, Lauzier B. O-GlcNAc stimulation: a new metabolic approach to treat septic shock. Sci Rep. 2019;9:18751.
Dontaine J, Bouali A, Daussin F, Bultot L, Vertommen D, Martin M, Rathagirishnan R, Cuillerier A, Horman S, Beauloye C, Gatto L, Lauzier B, Bertrand L, Burelle Y. The intra-mitochondrial O-GlcNAcylation system rapidly modulates OXPHOS function and ROS release in the heart. Commun Biol. 2022;5:349.
Dorfmueller HC, Borodkin VS, Schimpl M, Shepherd SM, Shpiro NA, van Aalten DM. GlcNAcstatin: a picomolar, selective O-GlcNAcase inhibitor that modulates intracellular O-glcNAcylation levels. J Am Chem Soc. 2006;128:16484–5.
Dorfmueller HC, Borodkin VS, Schimpl M, van Aalten DM. GlcNAcstatins are nanomolar inhibitors of human O-GlcNAcase inducing cellular hyper-O-GlcNAcylation. Biochem J. 2009;420:221–7.
Laczy B, Marsh SA, Brocks CA, Wittmann I, Chatham JC. Inhibition of O-GlcNAcase in perfused rat hearts by NAG-thiazolines at the time of reperfusion is cardioprotective in an O-GlcNAc-dependent manner. Am J Physiol Heart Circ Physiol. 2010;299:H1715–27.
Macauley MS, Vocadlo DJ. Increasing O-GlcNAc levels: an overview of small-molecule inhibitors of O-GlcNAcase. Biochim Biophys Acta. 2010;1800:107–21.
He Y, Ma X, Li D, Hao J. Thiamet G mediates neuroprotection in experimental stroke by modulating microglia/macrophage polarization and inhibiting NF-kappaB p65 signaling. J Cereb Blood Flow Metab. 2017;37:2938–51.
Shen Y, Yan B, Zhao Q, Wang Z, Wu J, Ren J, Wang W, Yu S, Sheng H, Crowley SD, Ding F, Paschen W, Yang W. Aging is associated with impaired activation of protein homeostasis-related pathways after cardiac arrest in mice. J Am Heart Assoc. 2018;7: e009634.
Ngoh GA, Watson LJ, Facundo HT, Dillmann W, Jones SP. Non-canonical glycosyltransferase modulates post-hypoxic cardiac myocyte death and mitochondrial permeability transition. J Mol Cell Cardiol. 2008;45:313–25.
Rahman MA, Hwang H, Cho Y, Rhim H. Modulation of O-GlcNAcylation regulates autophagy in cortical astrocytes. Oxid Med Cell Longev. 2019;2019:6279313.
Umapathi P, Mesubi OO, Banerjee PS, Abrol N, Wang Q, Luczak ED, Wu Y, Granger JM, Wei AC, Reyes Gaido OE, Florea L, Talbot CC Jr, Hart GW, Zachara NE, Anderson ME. Excessive O-GlcNAcylation causes heart failure and sudden death. Circulation. 2021;143:1687–703.
Tavassoly O, Yue J, Vocadlo DJ. Pharmacological inhibition and knockdown of O-GlcNAcase reduces cellular internalization of alpha-synuclein preformed fibrils. FEBS J. 2021;288:452–70.
Ngoh GA, Facundo HT, Hamid T, Dillmann W, Zachara NE, Jones SP. Unique hexosaminidase reduces metabolic survival signal and sensitizes cardiac myocytes to hypoxia/reoxygenation injury. Circ Res. 2009;104:41–9.
Ngoh GA, Watson LJ, Facundo HT, Jones SP. Augmented O-GlcNAc signaling attenuates oxidative stress and calcium overload in cardiomyocytes. Amino Acids. 2011;40:895–911.
Kultz D. Molecular and evolutionary basis of the cellular stress response. Annu Rev Physiol. 2005;67:225–57.
Marik PE, Bellomo R. Stress hyperglycemia: an essential survival response! Crit Care. 2013;17:305.
Mizock BA. Alterations in fuel metabolism in critical illness: hyperglycaemia. Best Pract Res Clin Endocrinol Metab. 2001;15:533–51.
Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, Wu J, Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A. 2000;97:12222–6.
Zachara NE, O’Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem. 2004;279:30133–42.
Udelson JE, Selker HP, Braunwald E. Glucose-insulin-potassium therapy for acute myocardial infarction: 50 years on and time for a relook. Circulation. 2022;146:503–5.
Li Q, Yang J, Zhang J, Yang C, Fan Z, Yang Y, Zheng T, Yang J. Effect of perioperative glucose-insulin-potassium therapy in patients undergoing on-pump cardiac surgery: a meta-analysis. Heart Surg Forum. 2020;23:E063–9.
van der Horst IC, Zijlstra F. Potential beneficial mechanisms of insulin (glucose-potassium) in acute myocardial infarction. Neth Heart J. 2005;13:233–8.
Howell NJ, Ashrafian H, Drury NE, Ranasinghe AM, Contractor H, Isackson H, Calvert M, Williams LK, Freemantle N, Quinn DW, Green D, Frenneaux M, Bonser RS, Mascaro JG, Graham TR, Rooney SJ, Wilson IC, Pagano D. Glucose-insulin-potassium reduces the incidence of low cardiac output episodes after aortic valve replacement for aortic stenosis in patients with left ventricular hypertrophy: results from the Hypertrophy, Insulin, Glucose, and Electrolytes (HINGE) trial. Circulation. 2011;123:170–7.
Chun WJ, Nah DY, Bae JH, Chung JW, Lee H, Moon IS. Glucose-insulin-potassium solution protects ventricular myocytes of neonatal rat in an in vitro coverslip ischemia/reperfusion model. Korean Circ J. 2015;45:234–41.
Liu J, Marchase RB, Chatham JC. Increased O-GlcNAc levels during reperfusion lead to improved functional recovery and reduced calpain proteolysis. Am J Physiol Heart Circ Physiol. 2007;293:H1391–9.
Krishnamoorthy V, Donofrio AJ, Martin JL. O-GlcNAcylation of alphaB-crystallin regulates its stress-induced translocation and cytoprotection. Mol Cell Biochem. 2013;379:59–68.
Zafir A, Readnower R, Long BW, McCracken J, Aird A, Alvarez A, Cummins TD, Li Q, Hill BG, Bhatnagar A, Prabhu SD, Bolli R, Jones SP. Protein O-GlcNAcylation is a novel cytoprotective signal in cardiac stem cells. Stem Cells. 2013;31:765–75.
Liu H, Wang Z, Yu S, Xu J. Proteasomal degradation of O-GlcNAc transferase elevates hypoxia-induced vascular endothelial inflammatory response. Cardiovasc Res. 2014;103:131–9.
Champattanachai V, Marchase RB, Chatham JC. Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein O-GlcNAc and increased mitochondrial Bcl-2. Am J Physiol Cell Physiol. 2008;294:C1509–20.
Champattanachai V, Marchase RB, Chatham JC. Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein-associated O-GlcNAc. Am J Physiol Cell Physiol. 2007;292:C178–87.
Hirose K, Tsutsumi YM, Tsutsumi R, Shono M, Katayama E, Kinoshita M, Tanaka K, Oshita S. Role of the O-linked beta-N-acetylglucosamine in the cardioprotection induced by isoflurane. Anesthesiology. 2011;115:955–62.
Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, Criollo A, Luo X, Tan W, Jiang N, Lehrman MA, Rothermel BA, Lee AH, Lavandero S, Mammen PPA, Ferdous A, Gillette TG, Scherer PE, Hill JA. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell. 2014;156:1179–92.
Jones SP, Zachara NE, Ngoh GA, Hill BG, Teshima Y, Bhatnagar A, Hart GW, Marban E. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation. 2008;117:1172–82.
Ngoh GA, Hamid T, Prabhu SD, Jones SP. O-GlcNAc signaling attenuates ER stress-induced cardiomyocyte death. Am J Physiol Heart Circ Physiol. 2009;297:H1711–9.
Yao D, Xu L, Xu O, Li R, Chen M, Shen H, Zhu H, Zhang F, Yao D, Chen YF, Oparil S, Zhang Z, Gong K. O-Linked beta-N-Acetylglucosamine modification of A20 enhances the inhibition of NF-kappaB (Nuclear Factor-kappaB) activation and elicits vascular protection after acute endoluminal arterial injury. Arterioscler Thromb Vasc Biol. 2018;38:1309–20.
Hilgers RH, Xing D, Gong K, Chen YF, Chatham JC, Oparil S. Acute O-GlcNAcylation prevents inflammation-induced vascular dysfunction. Am J Physiol Heart Circ Physiol. 2012;303:H513–22.
Fulop N, Zhang Z, Marchase RB, Chatham JC. Glucosamine cardioprotection in perfused rat hearts associated with increased O-linked N-acetylglucosamine protein modification and altered p38 activation. Am J Physiol Heart Circ Physiol. 2007;292:H2227–36.
Jensen RV, Zachara NE, Nielsen PH, Kimose HH, Kristiansen SB, Botker HE. Impact of O-GlcNAc on cardioprotection by remote ischaemic preconditioning in non-diabetic and diabetic patients. Cardiovasc Res. 2013;97:369–78.
Vibjerg Jensen R, Johnsen J, Buus Kristiansen S, Zachara NE, Botker HE. Ischemic preconditioning increases myocardial O-GlcNAc glycosylation. Scand Cardiovasc J. 2013;47:168–74.
Ou W, Liang Y, Qin Y, Wu W, Xie M, Zhang Y, Zhang Y, Ji L, Yu H, Li T. Hypoxic acclimation improves cardiac redox homeostasis and protects heart against ischemia-reperfusion injury through upregulation of O-GlcNAcylation. Redox Biol. 2021;43: 101994.
Xing D, Feng W, Not LG, Miller AP, Zhang Y, Chen YF, Majid-Hassan E, Chatham JC, Oparil S. Increased protein O-GlcNAc modification inhibits inflammatory and neointimal responses to acute endoluminal arterial injury. Am J Physiol Heart Circ Physiol. 2008;295:H335–42.
Yang S, Zou LY, Bounelis P, Chaudry I, Chatham JC, Marchase RB. Glucosamine administration during resuscitation improves organ function after trauma hemorrhage. Shock. 2006;25:600–7.
Nagy T, Champattanachai V, Marchase RB, Chatham JC. Glucosamine inhibits angiotensin II-induced cytoplasmic Ca2+ elevation in neonatal cardiomyocytes via protein-associated O-linked N-acetylglucosamine. Am J Physiol Cell Physiol. 2006;290:C57-65.
Hegyi B, Borst JM, Bailey LRJ, Shen EY, Lucena AJ, Navedo MF, Bossuyt J, Bers DM. Hyperglycemia regulates cardiac K(+) channels via O-GlcNAc-CaMKII and NOX2-ROS-PKC pathways. Basic Res Cardiol. 2020;115:71.
Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502:372–6.
Kronlage M, Dewenter M, Grosso J, Fleming T, Oehl U, Lehmann LH, Falcao-Pires I, Leite-Moreira AF, Volk N, Grone HJ, Muller OJ, Sickmann A, Katus HA, Backs J. O-GlcNAcylation of histone deacetylase 4 protects the diabetic heart from failure. Circulation. 2019;140:580–94.
Gorlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: a mutual interplay. Redox Biol. 2015;6:260–71.
Ryu IH, Lee KY, Do SI. Abeta-affected pathogenic induction of S-nitrosylation of OGT and identification of Cys-NO linkage triplet. Biochim Biophys Acta. 2016;1864:609–21.
Yoon CK, Yoon SY, Hwang JS, Shin YJ. O-GlcNAc signaling augmentation protects human corneal endothelial cells from oxidative stress via AKT pathway activation. Curr Eye Res. 2020;45:556–62.
Hu Y, Suarez J, Fricovsky E, Wang H, Scott BT, Trauger SA, Han W, Hu Y, Oyeleye MO, Dillmann WH. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J Biol Chem. 2009;284:547–55.
Gawlowski T, Suarez J, Scott B, Torres-Gonzalez M, Wang H, Schwappacher R, Han X, Yates JR 3rd, Hoshijima M, Dillmann W. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-beta-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem. 2012;287:30024–34.
Cividini F, Scott BT, Dai A, Han W, Suarez J, Diaz-Juarez J, Diemer T, Casteel DE, Dillmann WH. O-GlcNAcylation of 8-oxoguanine DNA glycosylase (Ogg1) impairs oxidative mitochondrial DNA lesion repair in diabetic hearts. J Biol Chem. 2016;291:26515–28.
Banerjee PS, Ma J, Hart GW. Diabetes-associated dysregulation of O-GlcNAcylation in rat cardiac mitochondria. Proc Natl Acad Sci U S A. 2015;112:6050–5.
Xue Q, Yan R, Ji S, Yu S. Regulation of mitochondrial network homeostasis by O-GlcNAcylation. Mitochondrion. 2022;65:45–55.
Li T, Zhang Z, Kolwicz SC Jr, Abell L, Roe ND, Kim M, Zhou B, Cao Y, Ritterhoff J, Gu H, Raftery D, Sun H, Tian R. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab. 2017;25:374–85.
Ma J, Liu T, Wei AC, Banerjee P, O’Rourke B, Hart GW. O-GlcNAcomic profiling identifies widespread O-Linked beta-N-Acetylglucosamine modification (O-GlcNAcylation) in oxidative phosphorylation system regulating cardiac mitochondrial function. J Biol Chem. 2015;290:29141–53.
Becker T, Wagner R. Mitochondrial outer membrane channels: emerging diversity in transport processes. BioEssays. 2018;40: e1800013.
McCommis KS, Baines CP. The role of VDAC in cell death: friend or foe? Biochim Biophys Acta. 2012;1818:1444–50.
Naumova N, Sachl R. Regulation of cell death by mitochondrial transport systems of calcium and Bcl-2 proteins. Membranes (Basel). 2020;10:299.
Siddiqui WA, Ahad A, Ahsan H. The mystery of BCL2 family: Bcl-2 proteins and apoptosis: an update. Arch Toxicol. 2015;89:289–317.
Rajamani U, Essop MF. Hyperglycemia-mediated activation of the hexosamine biosynthetic pathway results in myocardial apoptosis. Am J Physiol Cell Physiol. 2010;299:C139–47.
Ren J, Bi Y, Sowers JR, Hetz C, Zhang Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat Rev Cardiol. 2021;18:499–521.
Watson LJ, Long BW, DeMartino AM, Brittian KR, Readnower RD, Brainard RE, Cummins TD, Annamalai L, Hill BG, Jones SP. Cardiomyocyte Ogt is essential for postnatal viability. Am J Physiol Heart Circ Physiol. 2014;306:H142–53.
Zhang X, Hu C, Ma ZG, Hu M, Yuan XP, Yuan YP, Wang SS, Kong CY, Teng T, Tang QZ. Tisp40 prevents cardiac ischemia/reperfusion injury through the hexosamine biosynthetic pathway in male mice. Nat Commun. 2023;14:3383.
Jang I, Kim HB, Seo H, Kim JY, Choi H, Yoo JS, Kim JW, Cho JW. O-GlcNAcylation of eIF2alpha regulates the phospho-eIF2alpha-mediated ER stress response. Biochim Biophys Acta. 2015;1853:1860–9.
Herrero-Beaumont G, Largo R. Glucosamine and O-GlcNAcylation: a novel immunometabolic therapeutic target for OA and chronic, low-grade systemic inflammation? Ann Rheum Dis. 2020;79:1261–3.
Zhu X, Sang L, Wu D, Rong J, Jiang L. Effectiveness and safety of glucosamine and chondroitin for the treatment of osteoarthritis: a meta-analysis of randomized controlled trials. J Orthop Surg Res. 2018;13:170.
Not LG, Marchase RB, Fulop N, Brocks CA, Chatham JC. Glucosamine administration improves survival rate after severe hemorrhagic shock combined with trauma in rats. Shock. 2007;28:345–52.
Ju Y, Hua J, Sakamoto K, Ogawa H, Nagaoka I. Modulation of TNF-alpha-induced endothelial cell activation by glucosamine, a naturally occurring amino monosaccharide. Int J Mol Med. 2008;22:809–15.
Xing D, Gong K, Feng W, Nozell SE, Chen YF, Chatham JC, Oparil S. O-GlcNAc modification of NFkappaB p65 inhibits TNF-alpha-induced inflammatory mediator expression in rat aortic smooth muscle cells. PLoS ONE. 2011;6: e24021.
Li Y, Liu H, Xu QS, Du YG, Xu J. Chitosan oligosaccharides block LPS-induced O-GlcNAcylation of NF-kappaB and endothelial inflammatory response. Carbohydr Polym. 2014;99:568–78.
Kregel KC. Heat shock proteins: modifying factors in physiological stress responses and acquired thermotolerance. J Appl Physiol. 1985;2002(92):2177–86.
Kazemi Z, Chang H, Haserodt S, McKen C, Zachara NE. O-linked beta-N-acetylglucosamine (O-GlcNAc) regulates stress-induced heat shock protein expression in a GSK-3beta-dependent manner. J Biol Chem. 2010;285:39096–107.
Tanimoto T, Parseghian MH, Nakahara T, Kawai H, Narula N, Kim D, Nishimura R, Weisbart RH, Chan G, Richieri RA, Haider N, Chaudhry F, Reynolds GT, Billimek J, Blankenberg FG, Sengupta PP, Petrov AD, Akasaka T, Strauss HW, Narula J. Cardioprotective effects of HSP72 administration on ischemia-reperfusion injury. J Am Coll Cardiol. 2017;70:1479–92.
Fernandez-Fernandez MR, Gragera M, Ochoa-Ibarrola L, Quintana-Gallardo L, Valpuesta JM. Hsp70 - a master regulator in protein degradation. FEBS Lett. 2017;591:2648–60.
Song YJ, Zhong CB, Wang XB. Heat shock protein 70: a promising therapeutic target for myocardial ischemia-reperfusion injury. J Cell Physiol. 2019;234:1190–207.
Gong J, Jing L. Glutamine induces heat shock protein 70 expression via O-GlcNAc modification and subsequent increased expression and transcriptional activity of heat shock factor-1. Minerva Anestesiol. 2011;77:488–95.
Singleton KD, Wischmeyer PE. Glutamine induces heat shock protein expression via O-glycosylation and phosphorylation of HSF-1 and Sp1. JPEN J Parenter Enteral Nutr. 2008;32:371–6.
Lefebvre T, Cieniewski C, Lemoine J, Guerardel Y, Leroy Y, Zanetta JP, Michalski JC. Identification of N-acetyl-d-glucosamine-specific lectins from rat liver cytosolic and nuclear compartments as heat-shock proteins. Biochem J. 2001;360:179–88.
Guinez C, Losfeld ME, Cacan R, Michalski JC, Lefebvre T. Modulation of HSP70 GlcNAc-directed lectin activity by glucose availability and utilization. Glycobiology. 2006;16:22–8.
Guinez C, Mir AM, Leroy Y, Cacan R, Michalski JC, Lefebvre T. Hsp70-GlcNAc-binding activity is released by stress, proteasome inhibition, and protein misfolding. Biochem Biophys Res Commun. 2007;361:414–20.
Guinez C, Lemoine J, Michalski JC, Lefebvre T. 70-kDa-heat shock protein presents an adjustable lectinic activity towards O-linked N-acetylglucosamine. Biochem Biophys Res Commun. 2004;319:21–6.
Overath T, Kuckelkorn U, Henklein P, Strehl B, Bonar D, Kloss A, Siele D, Kloetzel PM, Janek K. Mapping of O-GlcNAc sites of 20 S proteasome subunits and Hsp90 by a novel biotin-cystamine tag. Mol Cell Proteomics. 2012;11:467–77.
Roquemore EP, Chevrier MR, Cotter RJ, Hart GW. Dynamic O-GlcNAcylation of the small heat shock protein alpha B-crystallin. Biochemistry. 1996;35:3578–86.
Hoter A, Amiri M, Prince A, Amer H, Warda M, Naim HY. Differential glycosylation and modulation of camel and human HSP isoforms in response to thermal and hypoxic stresses. Int J Mol Sci. 2018;19:402.
Park JH, Nakamura Y, Li W, Hamanaka G, Arai K, Lo EH, Hayakawa K. Effects of O-GlcNAcylation on functional mitochondrial transfer from astrocytes. J Cereb Blood Flow Metab. 2021;41:1523–35.
Xu H, Gu H, Yang Y, Cai E, Ding F, Yu S. 2-(4-Methoxyphenyl)Ethyl-2-acetamido-2-Deoxy-beta-D-Pyranoside exerts a neuroprotective effect through regulation of energy homeostasis and O-GlcNAcylation. J Mol Neurosci. 2019;69:177–87.
Xu H, Du M, Shen Y, Yang Y, Ding F, Yu S. Enhancement of O-GlcNAcylation on mitochondrial proteins with 2-(4-Methoxyphenyl)ethyl-2-acetamido-2-deoxy-beta-d-pyranoside, contributes to the mitochondrial network, cellular bioenergetics and stress response in neuronal cells under ischemic-like conditions. Molecules. 2021;26:5883.
Wu K, Chen L, Qiu Z, Zhao B, Hou J, Lei S, Jiang M, Xia Z. Protective effect and mechanism of Xbp1s regulating HBP/O-GlcNAcylation through GFAT1 on brain injury after SAH. Biomedicines. 2023;11:1259.
Hwang SY, Shin JH, Hwang JS, Kim SY, Shin JA, Oh ES, Oh S, Kim JB, Lee JK, Han IO. Glucosamine exerts a neuroprotective effect via suppression of inflammation in rat brain ischemia/reperfusion injury. Glia. 2010;58:1881–92.
Cha MY, Cho HJ, Kim C, Jung YO, Kang MJ, Murray ME, Hong HS, Choi YJ, Choi H, Kim DK, Choi H, Kim J, Dickson DW, Song HK, Cho JW, Yi EC, Kim J, Jin SM, Mook-Jung I. Mitochondrial ATP synthase activity is impaired by suppressed O-GlcNAcylation in Alzheimer’s disease. Hum Mol Genet. 2015;24:6492–504.
Zhu L, Tao T, Zhang D, Liu X, Ke K, Shen A. NOS1AP O-GlcNAc modification involved in neuron apoptosis induced by excitotoxicity. Int J Mol Sci. 2015;16:16560–75.
Wang Z, Li X, Spasojevic I, Lu L, Shen Y, Qu X, Hoffmann U, Warner DS, Paschen W, Sheng H, Yang W. Increasing O-GlcNAcylation is neuroprotective in young and aged brains after ischemic stroke. Exp Neurol. 2021;339: 113646.
Jiang M, Yu S, Yu Z, Sheng H, Li Y, Liu S, Warner DS, Paschen W, Yang W. XBP1 (X-Box-Binding Protein-1)-dependent O-GlcNAcylation is neuroprotective in ischemic stroke in young mice and its impairment in aged mice is rescued by thiamet-G. Stroke. 2017;48:1646–54.
Liu S, Sheng H, Yu Z, Paschen W, Yang W. O-linked beta-N-acetylglucosamine modification of proteins is activated in post-ischemic brains of young but not aged mice: Implications for impaired functional recovery from ischemic stress. J Cereb Blood Flow Metab. 2016;36:393–8.
Zhao J, Dong L, Huo T, Cheng J, Li X, Huangfu X, Sun S, Wang H, Li L. O-GlcNAc Transferase (OGT) protects cerebral neurons from death during ischemia/reperfusion (I/R) injury by modulating Drp1 in mice. Neuromolecular Med. 2022;24:299–310.
Luo Y, Chen P, Yang L, Duan X. Metabolomic analysis and pharmacological validation of the cerebral protective effect of 3,4-dihydroxybenzaldehyde on cerebral ischemia-reperfusion injury. Mol Med Rep. 2023;27:9.
Luo Y, Chen P, Yang LP. Duan XH [Quality control mechanism of mitochondria by 3,4-dihydroxybenzaldehyde through OGT-PINK1 pathway]. Zhongguo Zhong Yao Za Zhi. 2023;48:3308–16.
Li R, Shen Y, Li X, Lu L, Wang Z, Sheng H, Hoffmann U, Yang W. Activation of the XBP1s/O-GlcNAcylation pathway improves functional outcome after cardiac arrest and resuscitation in young and aged mice. Shock. 2021;56:755–61.
He QQ, Yang M, Huang J, Wu W, Tang K, Zhang Y, Zhou J, Ou W, Xie M, Liang Y, Lu P, Zuo Y, Yu H, Li T. Hypoxia-triggered O-GlcNAcylation in the brain drives the glutamate-glutamine cycle and reduces sensitivity to sevoflurane in mice. Br J Anaesth. 2022;129:703–15.
Park J, Jung S, Kim SM, Park IY, Bui NA, Hwang GS, Han IO. Repeated hypoxia exposure induces cognitive dysfunction, brain inflammation, and amyloidbeta/p-Tau accumulation through reduced brain O-GlcNAcylation in zebrafish. J Cereb Blood Flow Metab. 2021;41:3111–26.
Roy-O’Reilly M, McCullough LD. Age and sex are critical factors in ischemic stroke pathology. Endocrinology. 2018;159:3120–31.
Park SJ, Bae JE, Jo DS, Kim JB, Park NY, Fang J, Jung YK, Jo DG, Cho DH. Increased O-GlcNAcylation of Drp1 by amyloid-beta promotes mitochondrial fission and dysfunction in neuronal cells. Mol Brain. 2021;14:6.
Hwang JS, Kwon MY, Kim KH, Lee Y, Lyoo IK, Kim JE, Oh ES, Han IO. Lipopolysaccharide (LPS)-stimulated iNOS induction is increased by glucosamine under normal glucose conditions but is inhibited by glucosamine under high glucose conditions in macrophage cells. J Biol Chem. 2017;292:1724–36.
Wang AC, Jensen EH, Rexach JE, Vinters HV, Hsieh-Wilson LC. Loss of O-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proc Natl Acad Sci U S A. 2016;113:15120–5.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Number [81401094]), the Municipal Health Commission of Nantong (Grant Number [MSZ2022048]), and the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Contributions
QX, SJ, HX and SY wrote the main manuscript text, QX and SJ prepared all figures and tables. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xue, Q., Ji, S., Xu, H. et al. O-GlcNAcylation: a pro-survival response to acute stress in the cardiovascular and central nervous systems. Eur J Med Res 29, 174 (2024). https://doi.org/10.1186/s40001-024-01773-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40001-024-01773-z