
Abstract
Cancer poses a significant global health challenge, with predictions of increasing prevalence in the coming years due to limited prevention, late diagnosis, and inadequate success with current therapies. In addition, the high cost of new anti-cancer drugs creates barriers in meeting the medical needs of cancer patients, especially in developing countries. The lengthy and costly process of developing novel drugs further hinders drug discovery and clinical implementation. Therefore, there has been a growing interest in repurposing approved drugs for other diseases to address the urgent need for effective cancer treatments. The aim of this comprehensive review is to provide an overview of the potential of approved non-oncology drugs as therapeutic options for cancer treatment. These drugs come from various chemotherapeutic classes, including antimalarials, antibiotics, antivirals, anti-inflammatory drugs, and antifungals, and have demonstrated significant antiproliferative, pro-apoptotic, immunomodulatory, and antimetastatic properties. A systematic review of the literature was conducted to identify relevant studies on the repurposing of approved non-oncology drugs for cancer therapy. Various electronic databases, such as PubMed, Scopus, and Google Scholar, were searched using appropriate keywords. Studies focusing on the therapeutic potential, mechanisms of action, efficacy, and clinical prospects of repurposed drugs in cancer treatment were included in the analysis. The review highlights the promising outcomes of repurposing approved non-oncology drugs for cancer therapy. Drugs belonging to different therapeutic classes have demonstrated notable antitumor effects, including inhibiting cell proliferation, promoting apoptosis, modulating the immune response, and suppressing metastasis. These findings suggest the potential of these repurposed drugs as effective therapeutic approaches in cancer treatment. Repurposing approved non-oncology drugs provides a promising strategy for addressing the urgent need for effective and accessible cancer treatments. The diverse classes of repurposed drugs, with their demonstrated antiproliferative, pro-apoptotic, immunomodulatory, and antimetastatic properties, offer new avenues for cancer therapy. Further research and clinical trials are warranted to explore the full potential of these repurposed drugs and optimize their use in treating various cancer types. Repurposing approved drugs can significantly expedite the process of identifying effective treatments and improve patient outcomes in a cost-effective manner.
Similar content being viewed by others

Introduction
Cancer is a serious disease that causes high mortality rates worldwide [1,2,3]. The occurrence of different types of cancer and particularly the possibilities of treatment are a big challenge for clinicians [4,5,6,7,8,9]. Malignant tumors such as liver cancer, breast, prostate, pancreas and colorectal cancer are generally difficult to cure at advanced phases with existing conventional treatments [10,11,12,13,14]. Finding new substances for the treatment of cancers was the result of technological development and innovative approaches. Surprisingly, the research on novel agents for cancer treatment lasts long and is a complicated exercise due to the various steps necessary for the isolation, synthesis, and purification of new anticancer substances [13, 15,16,17,18]. After drug discovery, synthesis and selection of appropriate formulation and preclinical pharmacology and toxicology, phase I clinical trials have to be made which begin with the administration of an investigational drug into healthy humans This phase involves the estimation of initial safety and tolerability, pharmacokinetics and assessments of pharmacodynamics. If successful, phase II is performed with the primary objective to explore therapeutic efficacy in patients. After that, for many of them, there is only a small probability to finish successfully phase III clinical trials which are designed to confirm the preliminary evidence obtained in phase II to verify that the potential drug is safe and effective for use in the intended indication and recipient patients. The amount of time and money for the production of new anti-cancer drugs is a big obstacle for the pharma industry for new drugs development, which will have proven therapeutic efficacy [19,20,21,22,23]. Therefore, a novel concept of the use of old FDA-approved drugs is the recent direction to help clinicians and researchers, and, of course, the patients [24]. In this strategy, the advantage is that the assessment of drug safety, such as pharmacodynamics, pharmacokinetics, toxicity, and safety profiles, has been already previously established in Phase I studies. Moreover, old approved drugs can rapidly proceed for further Phase II clinical trials. Therefore, the interest is more and more focused on this strategy because of the lower cost and less time for developing new use of old drugs [19, 25].
Drug repurposing landscape: a brief synopsis
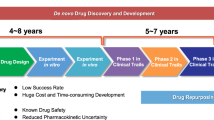
Drug development is a multistep process that includes identifying the therapeutic drug molecule that is clinically effective in the treatment of a disease [26]. This conventional drug discovery strategy implicates the de novo identification of new molecular entities. It has five stages, such as the discovery of the molecule and preclinical study, safety review, clinical studies, FDA review, and FDA post-market safety monitoring [27]. This process involves the identification of candidate molecules, synthesis, characterization, validation, optimization, screening, and assays for therapeutic efficacy [28]. When a product shows favorable outcomes in these studies, then the molecule has to go through a drug development process and subsequently testing in clinical trials [28]. De novo drug development is a time-consuming process and involves significant investments. It usually takes years of work (10–17 years) and costs millions of dollars. Furthermore, it is associated with high failure rates, with roughly 90% of molecules being rejected due to unexpected characteristics, such as safety and efficacy concerns [29, 30]. Despite massive expenditures in drug discovery and tremendous advancements in biological and informational technology over the past several decades, the number of new drugs brought to the phase of clinical trials has not expanded so much and remained relatively constant. Even though total research and development cost for drug discovery has expanded tenfold from 1975 (US $4 billion) to 2009 ($40 billion), there has been no substantial change in the number of new molecules approved since 1975 (in 2013 there were only 6 new drug moieties approved in comparison with the year 1976, where the number of newly approved pharmaceuticals was 27) [31]. The situation has improved in the last decade but not to a large extent. The increasing cost and time in the drug discovery process have resulted in the possibility that if resistance to the available drugs emerges, people with advanced diseases will end up dying before a substitute treatment option could become accessible [32]. Drug development is undoubtedly one of the most complex tasks in pharmaceutical research. In addition to already intimidating complications in pharmacological drug design, numerous obstacles occur due to regulatory, clinical intellectual property, and economic concerns. As a result of these challenging circumstances, the drug development process has become even more prolonged and uncertain. In pursuit of new treatment alternatives for patients with diseases, such as cancer, researchers have turned to drug repurposing tactics [33]. Drug repurposing also termed as drug repositioning, drug rescuing, drug reprofiling, drug recycling, or therapeutic switching, involves identifying and exploring the new therapeutic use of already FDA-approved and in clinical practice used drugs, but used for the treatment of other indications [24]. It is regarded as the most effective strategy in developing drug candidates using novel pharmacological properties and therapeutic characteristics of well-known drugs. Considering that traditional drug discovery is a time-consuming and costly process, the revolutionary strategy of drug repositioning is used to boost the success rate of medication development. Compared to the traditional drug discovery approach, this strategy is more favorable in terms of minimizing the length of time required for drug development while maintaining low costs, high efficiency, and minimal risk of failure [34]. The drug repurposing also offers a significant advantage not only in terms of the availability of preclinical information about the existing drug (a drug to be repurposed) but also provides additional data about clinical aspects such as pharmacokinetic, pharmacodynamics, and toxicity profile of that particular drug [35]. Because of this, these drugs might quickly be tested in Phase II and Phase III clinical trials, and the accompanying development costs could be significantly lower. The risk of failure is reduced, because in-vitro and in-vivo studies, toxicology profiles, chemical optimization, and formulation development have previously been explored. As a result, pharmaceutical companies have directed more and more of their attention to drug repurposing, since it might give a considerable advantage compared to traditional drug development (Fig. 1) [36]. In this context, it is not unexpected that approximately 30% of newly approved drugs in the United States are repurposed drugs [37].

Comparison of drug repurposing with traditional drug discovery
Experimental drug repurposing approach
Binding assay
Techniques such as affinity chromatography, proteomics, and mass spectroscopy are used to identify novel targets for old drugs [38]. The protein target of gefitinib was investigated using HeLa cell extract. Mass spectroscopy results indicated that gefitinib could potentially interact with 20 different protein kinases that might be a target for gefitinib [27].
Phenotypic approaches
The phenotypic drug discovery approach is an experimental strategy that uses the library of accessible drug collections and focuses on finding their biological activities in cells and living organisms. It does not depend on the direct interaction with the target. Changes in in-vitro, in-vivo models and clinical studies can lead to the discovery of new drugs [39]. This is a screening procedure that does not presume the mechanism of action, and the primary output is a change in phenotype or physiological parameters. Cells, physiological systems, and whole organisms can all be used in this process. Depending upon the purpose or phase of drug development, each of these several systems can be used. Cell-based screening provides an increased insight into in vivo processes. On the other hand, in-vivo animal models help to assess the possible use of the existing medications for new phenotypic characteristics [40]. This method led to the discovery that astemizole and its metabolite desmethyl astemizole as effective inhibitors of Plasmodium falciparum growth and development [41].
Drug-centric approach
Drug-centric repurposing strategy is focused on forecasting new use for already approved drugs. This strategy relies heavily on substances that have the potential to interact with a wide range of targets (polypharmacological agents). Even though polypharmacological substances are responsible for triggering undesirable side effects, their activities can be used, because they offer the possibility of additional indications for a specific drug [42]. Polypharmacology seems to become the next major drug development paradigm. A considerable number of drugs are known for their ability to affect many targets simultaneously. Aspirin is used to relieve mild pain, fever and rheumatoid arthritis. It is also used as an anti-inflammatory agent in the treatment of Kawasaki disease and pericarditis. Transient ischemic attacks, ischemic stroke, myocardial infarction and even some types of cancer have all been successfully treated with this drug. Sildenafil, a phosphodiesterase inhibitor, was initially used for the management of erectile dysfunction. However, today, it is widely used for the management of pulmonary hypertension [43]. The majority of kinase inhibitors can inhibit several targets which makes them attractive options for the treatment of some types of cancer. Different multi-targeted tyrosine kinase inhibitors (TKIs) such as imatinib, nilotinib, and vandetanib were approved for clinical use in 2010 to treat solid cancers [44].
Target-based approach
The target-based drug repurposing approach involves the study of candidate drugs with biological targets such as receptors and proteins to different physiological responses to them. According to this method, new indications were discovered by relating a certain drug to a specific disease depending on the protein which it might target [42]. Proteins have many pathophysiological roles both in diseases and in healthy humans; dysfunctional, mutated, or misfolded proteins may trigger pathological responses that cause the development of a disease. The studies of proteins or biomarkers implicated in pathophysiological processes focus on target-based drug repurposing strategy [45]. The target-based strategy involves in silico or virtual high-throughput screening of drugs from various drug libraries or substances databases such as ligand-based screening or molecular docking proceeded by in vitro and in vivo high throughput or high content screening of drugs against a specific protein or biomarker of interest [27]. For instance, a new pharmaceutical molecule N-myristoyl transferase was discovered by this target-based approach for the treatment of filarial nematodes [46].
Knowledge-based
In this drug repurposing approach, models are developed that incorporate drug-related information, such as drug targets, chemical structures, route information, adverse effects, etc. These models are then used to anticipate unknown targets, biomarkers, or disease mechanisms [35].
Pathway or network-based
Pathway-based drug repurposing approach uses information about metabolic pathways, signaling pathways, and protein interaction networks to anticipate the similarity or relationship between a certain drug and a disease [35]. Network or pathway-based drug repurposing uses omics data to understand how drugs interact and communicate with disease targets. As a result, a specialized network with a few targets can be found in a vast network of pathways. Network-based strategies for drug repurposing are more and more in the focus of interest. Network-based computational biology focused on biomolecular interactions and omics data integration seem to be very promising. New drug repurposing research has discovered previously unknown signaling pathways in breast carcinoma subtypes [47, 48]. Kotelnikova et al. described a novel computational algorithm for the treatment planning of glioblastoma. The study analyzed gene expression data using a proprietary algorithm designed for pathway studio called sub-network enrichment analysis (SNEA). This method led to the discovery and FDA approval of fulvestrant (Faslodex1), a drug used to treat hormone receptor-positive metastatic breast carcinoma [49]. A study by Yu et al. suggested an approach for predicting potential drug–disease interactions that may be used for drugs or diseases that have or do not have associated genes. The adverse effects of drugs and disease symptoms were associated with identify drug–module and disease–module pairs using this strategy [50].
Repurposing of non-oncology drugs to treat cancer
Repurposing non-oncology drugs to use against cancer cells is an alternative approach to provide better mitigation possibilities for people with cancer at a lower cost and more quickly. Many methods were used to explore the probable anticancer function of non-cancerous drugs. For drug repurposing of non-oncology medications many in vivo and in vitro trials on pharmacological models and cancer cell lines were performed. Numerous wide-ranging electronic databases, such as the National Institute of Health (NIH) and Molecular Libraries Initiative [51], in which the chemical substances, biological evaluation assays, and genetic relevance of the active chemical substances were used to analyze them as a tool for utilization of drugs repositioning [52]. Repurposing of non-oncology drugs works through many mechanisms, such as cancer monotherapy, inhibiting proliferative signaling, inducing cell death, regulation of cellular metabolism, activation of antitumor immunity, drug combinatorial therapy, reactivating growth suppressors, interfering with replication, decreasing angiogenesis, suppression of invasion, and metastasis [32].
Anthelmintic drugs
Anthelmintic are a class of drugs that are used to treat unicellular protozoa as well as parasite worms in the intestine [53]. Anti-parasitic drugs such as mebendazole, flubendazole, albendazole, ivermectin, and chloroquine are commonly used antiparasitic drugs. Initially, these drugs were used to treat cattle parasites and then subsequently they were recommended for helminthiasis in humans as well. Many studies showed that some anthelmintic drugs have beneficial effects as anticancer agents on pathways, such as activator transcription proteins, signal transducer, and nuclear factor-kappa B (NF-kB) and Wnt/β-catenin (Table 1 and Fig. 2) [53]. Therefore, these anthelmintic drugs might be potential candidates as anticancer drugs.

Illustrative diagram with the relevant mechanisms of repurposing anthelmintic drugs in cancer therapy. Symbols: ↑increase, ↓decrease. Bcl-2 B-cell lymphoma 2, ROS reactive oxygen species, Wnt Wingless-related integration site, XIAP X-linked inhibitor of apoptosis protein
Flubendazole
Flubendazole is a well-known benzimidazole which is an antihelmintic drug that has also antineoplastic effects in different types of malignant diseases, including breast cancer, leukemia, multiple myeloma, and neuroblastoma [54, 55]. Flubendazole initiates significant changes in microtubule targeting sites, induction of apoptosis, induction of reactive oxygen species followed by G2/M phase accretion, and caspases 3, and 7 initiations in malignant cells. Flubendazole acts via different mechanisms such as inhibiting tumor growth, angiogenesis, etc. in pulmonary, liver, and breast cancer [56]. It inhibits trastuzumab resistance by targeting cancer cells, induction of apoptosis, and overexpression of human epidermal growth factor receptor 2 (HER2) in breast cancer [57]. This drug has cytotoxic activities in human colorectal cancer by blocking transcription proteins 3, signal transducer, and autophagy pathway [58]. It also blocks human melanoma cells' growth and metastasis and suppresses programmed cell death protein-1 and myeloid-derived suppressor cell accumulation [59].
Mebendazole
Mebendazole (MZ) is a drug that is frequently prescribed to manage gut parasitic infections. It inhibits tubulin polymerization which has an antiparasitic effect [60]. Mebendazole is a synthetic benzimidazole antihelmintic and a repositioned drug that has already proven its pharmacokinetics and toxicity profile [61]. MZ could be used in combination with temozolomide, which is a drug commonly prescribed in the treatment of malignant gliomas. In both xenograft and syngenic forms of glioma, this combination therapy suppressed tumor development more than temozolomide alone [62]. It also has synergistic effects with docetaxel inhibiting the polymerization of tubulin, mitotic arrest in the G2/M phase, augmenting apoptosis, reducing cell multiplication of prostate cancer, and suppressing tumor growth [63]. It stops mitotic growth in the G2/M phase, double-stranded breaks, and apoptosis in breast cancer which was shown using in vivo and in vitro biological assays [64]. Mebendazole activates the caspase-3 pathway and induces apoptosis. It also inhibits tumor development and stops pulmonary metastases in the later stages of thyroid cancer [65]. Mebendazole has also cytotoxic activity specific for colon cancer, ovarian cancer, endocrine malignancy, and brain tumors [62, 66, 67]. In cholangiocarcinoma, mebendazole induces apoptosis by inhibiting cell multiplication via increasing the expression of caspase-3 [68]. Other studies have found that MZ causes growth suppression in cell lines from many other different types of cancer ex-vivo and in vivo, especially pulmonary cancer [69], colorectal cancer [70], melanoma [71, 72], glioblastoma [62], and medulloblastoma [73]. The effects of MZ in melanoma are achieved by inducing apoptotic cell death, especially by activation of caspases, the pro-apoptotic Bcl-2, and suppression of the repressor of the apoptotic pathway, X-linked blocker of apoptosis (XIAP) [71, 72].
MZ has significant binding interaction potential in colon cancer cells, suggesting that it might be an antagonist of different kinases and oncogenes, such as ABL and BRAF [70], as well as a Hedgehog modulator in medulloblastoma [74]. MZ therapy in rodents caused a reduction in the size of tumors and decreased angiogenesis in comparison with normal animals. Furthermore, the incidence of metastasis in the therapy group was lower [67].
According to several case studies, MZ can also have antitumor effects in a clinical environment when given to patients with cancer [75, 76]. Mukhopadhyay et al. published one of the first studies on the antitumor effects of MZ [67]. It slowed down the growth of lung carcinoma cells but did not affect normal endothelium cells or fibroblasts. After the first- and second-line therapy in refractory tumors, MZ was given to patients with metastatic colorectal carcinoma. The patients had no adverse effects other than an increase in hepatic enzymes, and after that, the dose of the drug was reduced, and therapy efficiently eliminated practically all pulmonary and lymphovascular metastases, and partial recovery of hepatic metastases occurred as well [75]. MZ was prescribed to patients with adrenocortical cancer after the failure of different chemotherapeutics. During approximately one and a half years, there were no adverse effects, the size of the metastases decreased, and the illness was stable [76].
Niclosamide
Niclosamide is a drug of choice [77] and got approval from FDA as an anthelminthic drug. It has also been reported in a series of studies to have cytotoxic effects on ovarian cancer, breast cancer and prostate cancer cells [32, 78]. It targets cancer cells by interfering with the anaerobic metabolism and glucose uptake of these cells [79]. It also has an anticancer effect on multiple signaling pathways, such as metastasis, and signal activation, and as a transducer of transcription proteins Wntβ/-catenin and NF-KB [80, 81]. Some studies showed that it significantly suppressed the development of cancer of breast, liver, and colorectal cancer. Its anti-metastasis effect seems to prevent liver metastasis of colorectal cancer cells. It also has a beneficial effect on pulmonary metastases of breast cancer [82, 83]. However, less bioavailability and the poor solubility of the drug is the biggest obstacle to its clinical development [84]. The intravenous route might help to use the development of this drug as a repositioned drug [85, 86].
Praziquantel
Praziquantel is a broad-spectrum antiparasitic drug, and its mechanism of action is still unclear. Nevertheless, it can intensify the concentration of intracellular Ca2+ and it also causes contractions of muscles [53]. It has been shown that this drug also increases the cytotoxic action of paclitaxel at 20–40 µM. The combination of both drugs, i.e., praziquantel and paclitaxel, acts synergistically reducing the expression of anti-apoptotic protein and X-linked inhibitor of apoptosis protein (XIAP) [53].
Eprinomectin
Eprinomectin has a broad spectrum of effects against different parasitic infections. It showed also a cytotoxic potential against prostate cancer cells. It induces apoptosis in PC-3 cells by affecting reactive oxygen species (ROS). Eprinomectin also causes the stop of mitotic cells at the G1 phase. Moreover, this drug stimulates the translocation of β-catenin and has a significant apoptotic effect and activates caspase-3 and caspase-9. The abovementioned findings might explain why eprinomectin could have cytotoxic effects against advanced prostate cancer [87].
Ivermectin
Ivermectin is an FDA-approved macrocyclic lactone. It has antitumor effect in smaller doses against the Wnt-TCF, and β-catenin, and suppresses the expression of cyclin D in cancer of the colon [88]. Another approach documented the therapeutic effect of this drug in cancer xenografts and melanoma [89]. Reports on neurofibromatosis tumor cells and ovarian cancer cells suggested that this drug might suppress the growth of protein-activated kinases (PAK-1) and thus have a beneficial effect on prostate, breast, and gastric cancers [90]. This drug tends to reduce the growth of a tumor enhancing mitochondrial biogenesis compared to normal cells [91]. Similarly, in another research, it has been shown that this drug also inhibits PAK1 via AKT–mTOR in breast cancer. It reduces yes-associated protein (YAP1) expression and targets connective tissue growth factor (CTGF) in gastric cancer. It also suppresses resistance by dropping p-glycoprotein due to inhibition of ERK/AKT/NF-kB and epidermal growth factor receptor (EGFR) [92]. This drug is a self-renewal marker of cancer stem cells (CSCs) in breast cancer. Therefore, this drug could be a future therapeutic molecule for cancer management [93].
Nitazoxanide
Nitazoxanide is a thiazole-based molecule and it has been reported that it might have antitumor effects due to different mechanisms, such as inhibition of cellular myelocytomatosis (c-MYC), induction of apoptosis, and DNA fragmentation. It also causes autophagy against epithelial tumor cells via mechanistic targeting rapamycin (mTOR) inhibition besides its antihelmintic effects. Similarly, this drug can induce apoptosis by DNA fragmentation condensation of the nucleus along with its anti-parasitic effects [53, 94].
Clioquinol
Clioquinol also seems to have anticancer effects besides its antiparasitic effects. It causes downregulation expression in histone deacetylase (HDACs) in leukemic and malignant myeloma cells. This drug causes apoptosis via mitotic arrest and downregulation of HDAC, resulting in the expression of p53 and p21 [95].
Pyrimethamine
Pyrimethamine is an antiparasitic drug which inhibits tumor growth and metastasis in pulmonary carcinoma by attacking the dihydrofolate reductase (DHFR) and thymidine phosphorylase (TP) [96, 97]. Pyrimethamine, which is a STAT3 antagonist, has chemotherapeutic and immune-stimulatory effects in breast cancer models in mice. Pyrimethamine suppresses STAT3 action in metastatic breast cancer cell lines ex-vivo by reducing tumor growth and invasion and by increasing Lamp1 production in tumour-infiltrating CD8+T lymphocytes [98]. Pyrimethamine also inhibits the growth of ovarian cancer cells both ex-vivo and in-vivo [99].
The most representative mechanisms of anthelmintic drug repurposing in cancer therapy are summarized in Fig. 2.
Antiviral drugs
The combinations of antiviral drugs and conventional chemotherapeutic agents are used to treat different types of malignant diseases such as lymphoma, nasopharyngeal carcinoma, hepatocellular carcinoma, and Kaposi sarcoma using protease inhibitors directed towards the human immune-deficiency virus. Studies on the viruses accompanying cancers can be a useful platform for the development of novel therapeutic approaches not only for treating viral infections but also consequently influencing tumorigeneses [100].
Ritonavir
Ritonavir is a thiazole-based anti-viral drug widely used for the treatment of different viral diseases such as HIV (human immunodeficiency virus) and to increase the effectiveness of protease inhibitors. Several studies have shown antitumor effects of ritonavir. It seems that it can cause apoptosis and inhibit the progression of malignant cells in breast, pancreatic and ovarian carcinoma [101,102,103]. The drug also strengthens the effects of several substances such as temozolomide towards glioma cancer cells [104]. It seems that it could also be used in combination with bortezomib for the treatment of renal cancer [105]. Ritonavir also has anticancer effects against breast cancer by inhibiting Akt phosphorylation and seems to be effective also in lymphocytic leukemia [106].
Nelfinavir
Nelfinavir is a protease inhibitor and is widely used in managing HIV-1 and HIV-2. This drug can decrease phosphorylation of Akt, signal transducer and activator of transcription 3 (STAT-3), and xenografts tumors [107] Several studies reported the beneficial effects of this drug against different types of cancers such as ovarian, breast, lungs, and liposarcoma by inhibiting the signals of Erk 1/2, STAT-3, and Akt [108,109,110,111,112,113]. It has been reported that nelfinavir induces endoplasmic reticulum (ER) stress and also increases the effect of other drugs used for the management of prostate and breast cancers [114, 115]. Numerous studies showed that it inhibits autophagy and cyclooxygenase (COX-2) inhibitors in breast cancers [116]. The mixture of COX-2 and nelfinavir inhibit autophagy and could increase cytotoxic effects directed against cancer of the breast [116]. It also induces pro-apoptotic effects by activating caspase-4 [117, 118]. Some studies found that this drug induces apoptosis mediated by ROS [119, 120]. Other studies also reported antiproliferative effects and limited toxicity against liposarcoma and cystic carcinomas [120, 121].
Acyclovir
Acyclovir was discovered about four decades ago and has become a drug of choice against viral infections, such as Herpes Simplex [122]. A plethora of evidence suggests that this drug is effective in the treatment of different types of cancers [123, 124]. This drug suppresses cell proliferation and induces apoptosis in malignant tumors of of the breast. Another study reported antiproliferative effects of acyclovir in Michigan Cancer Foundation (MCF-7) cell lines by increasing the proteins expression of E-cadherin, reducing the proliferation rate, and increasing apoptosis caspase-3 and wound healing in MCF-7 malignant cells. The results suggested that this drug could be used alone or in combination therapy as a potential candidate for breast cancer treatment. The results of the calorimetric assay indicated the downregulation of aldehyde dehydrogenase (ALDH) activity towards breast cancer cell lines [125].
Ribavirin
Ribavirin is a member of the antiviral drugs class and it seems that it has beneficial effects in different viral diseases, such as polio, hepatitis-C and SARS–coronavirus infection [126,127,128]. It is a guanosine ribonucleoside-based drug. Since it is similar to guanosine it tends to compete for guanylyl transferase and inhibit 5'-mRNA. This drug at micromolecular concentrations binds with eukaryotic translation initiation factor (elF4E) on a purposeful site using a 7-methyl guanosine mRNA cap. This drug suppresses elF4E facilitated oncogenes transformation using 7-methyl guanosine. In another approach, it has been found that it can inhibit elF4E competitive binding to cyclin D1 and thus reduce cyclin D1 protein cells [124]. This drug also causes the translation of VEGF mRNA and other genes [129, 130]. Ribavirin at 2 µm concentration suppresses the progression of human lymphocytes [131].
Cidofovir
Cidofovir is a FDA accepted nucleoside broad-spectrum antiviral drug which is used to treat different viral infections by inhibiting the viral DNA polymerase through its metabolite diphosphate [132,133,134]. Because of this mechanism, the drug can decrease the growth of several types of human cancers [135,136,137,138,139]. This drug also induces apoptosis mitotic arrest in S-phase and causes activation of Poly (ADP-ribose) polymerase (PARP) and caspase in epithelial cells [140,141,142]. It also inhibits DNA synthesis and blocks the growth of cancer cells. It has also been reported that this drug has cytotoxic effects on human glioblastoma cell lines. This drug shows antiproliferative effects both in xenograft models and in vitro studies. It inhibits the gene expression associated with apoptosis in glioblastoma. This drug has antitumor effects because of its different mechanisms, including effects on mitogenic pathways and activation of proapoptotic pathways in glioblastoma [143].
Antibiotics
Sulfisoxazole
Sulfisoxazole is an antibiotic that may inhibit small extracellular vesicular exudation from cancerous mammary cells by interfering with endothelin receptor A. In animal studies of breast cancer xenografts, sulfisoxazole had antitumor and antimetastatic effects (Table 2) [154].
Azithromycin
Azithromycin is a macrolide antibacterial drug. The proliferative potential of cancer cells has been inhibited by this drug. In colon carcinoma cells, ex-vivo and in-vivo, it increases the antineoplastic effectiveness of TNF-α-related apoptosis-inducing ligand (TRAIL) by suppressing autophagy and increasing DR4/5 [155]. Azithromycin also reduces angiogenesis in pulmonary carcinoma by inhibiting vascular endothelial growth factor receptor 2-induced focal adhesion and the PI3K/AKT signalling cascade [156].
Doxycycline
Doxycycline is a tetracycline antibiotic that is used for treating different infections. Some tetracyclines were reported to suppress angiogenesis in the early 1990s [157], and doxycycline was eventually found to have antiproliferative activity in bone and prostate carcinoma and mesothelioma cells [158,159,160]. It has also been shown to induce apoptosis in pancreatic islets [161, 162] and myeloid cells [163]. Matrix metalloproteinases (MMPs) are inhibited by tetracyclines [164]. Doxycycline therapy inhibits penetration by downregulating MMP-2 and MMP-9 levels in myeloid cells [165] and colon carcinoma [166]. Doxycycline has been investigated as a suppressor of tumour progression, because MMPs are assumed to play a crucial role in cancer infiltration and progression. Doxycycline has been shown to reduce different tumours and it seems to be beneficial for breast cancer patients at risk for osteolytic bone metastasis. [167]. The same investigators also demonstrated that zoledronic acid, a medicine used to decrease the incidence of bone fractures in patients with osteosarcoma or osteoporosis, might be useful in combination with doxycycline [168]. MMP-2/9 suppression has also been shown to reduce metastasis in preclinical trials of prostate cancer [169] and squamous skin carcinoma [170]. Doxycycline can reduce EMT-marker transcription in pulmonary and hepatic carcinoma cells, reversing their pro-metastatic character [171, 172]. Doxycycline therapy reduced clonogenic potential and decreased the expression level of stem cell markers in hepatic carcinoma cells enriched with stem-related characteristics. Doxycycline decreased proliferation markers Ki67 and PCNA in the in vivo xenograft mouse model. [173]. Doxycycline was combined with interferon-alpha (IF-α) treatment in a phase II clinical study of renal cancer metastases. VEGF levels were measured to see if there was an antiangiogenic effect. The combined therapy proved to be ineffective in patients with renal carcinoma metastases, despite modest early reduction of VEGF expression in some patients [174]. The efficacy of doxycycline therapy in combination with bone-targeting medicines was evaluated in a recent phase II study in females with breast carcinoma metastases. In this trial, doxycycline was shown to have a negligible effect but it was associated with severe harmful effects [175].
Ionophore antibiotics
Ionophore antibiotics have shown antitumor effects against cancers of the colon, and prostate, as well as endometrial, blood, cerebral, and bone malignancies. Ionophore antibiotics salinomycin and nigericin specifically attack CSCs and it seem that they are slightly more effective than paclitaxel. Cell migration, metastases, and the GTPase K-Ras cascade are all targeted by these drugs. Salinomycin inhibited the hedgehog and WNT/-β catenin pathways, resulting in decreased tumour size of metastatic breast cancer [176]. In lymphomas, salinomycin combined with doxorubicin was reported to have a synergistic effect [176]. Rapamycin, an antimicrobial drug, has an antiproliferative effect by blocking cell cycle progression due to its effect on CDK proteins and mTOR signalling. In vivo investigations have shown that it can also prevent cancer and malignancies caused by the Epstein–Barr virus [177]. Rapamycin has chemotherapeutic potential in different types of human cancers, and it was found to synergize with erlotinib in NSCLC and paediatric glioma [177, 178].
Anthracycline drugs
Anthracycline drugs have also been intensively studied as antimalignant drugs. Doxorubicin, idarubicin, mitoxantrone, daunorubicin and epirubicin are often used anthracycline medicines that have been extensively investigated in solid and blood malignancies [179,180,181]. Garg et al. found that doxorubicin and selinexor promoted the death of thyroid cancer and AML cells when used together [180, 181]. Duocarmycin is an antibiotic that works at low doses. It attaches to the DNA molecule causing DNA to be alkylated, which causes the cell cycle arrest. It is one of the most successful medications used to treat endometrial, breast, bladder, hepatic, thyroid, and pulmonary malignancies [182]. Wang et al. analyzed 124 patients from January 1996 to July 2018 to analyze the effects of chemotherapeutic treatment with gemcitabine and anthracycline (epirubicin and pirarubicin). They analyzed the probability of tumour’s recurrence and therapy failures. Gemcitabine had a lower recurrence percentage and therapy failure rates than anthracycline antimicrobials, suggesting that this approach should be explored for patients who cannot be treated with BCG [183]. Landomycin E is an angucycline antibiotic produced by Streptomyces globisporus which caused cell death in T-cell leukaemia cells by rapidly generating hydrogen peroxide and activating caspases [184]. NAC, in combination with doxorubicin, demonstrated decreased adverse effects on nephrons, somewhat enhanced cytotoxic effects of T-cells, and it slightly enhanced their survivability. NAC, in combination with landomycin, on the other hand, significantly enhanced the lifespan of the rodents while also having some tissue-protective effects. As a result, NAC in combination with landomycin appears to be more effective than doxorubicin. Landomycin seems to be more powerful than doxorubicin at low concentrations in ex-vivo and in-vivo melanoma with fewer adverse effects. However, adverse effects such as cardiovascular events and mucositis suggested the need for other antibacterial drugs. Idarubicin, a doxorubicin analogue, was created. In acute myelogenous leukaemia, idarubicin showed increased lipophilicity and anti-malignant efficacy. In acute myeloid leukaemia clarubicin, an anthracycline antibiotic derived from Streptomyces galilaeus, suppresses RNA production. Amrubicin has chemotherapeutic efficacy against SCLC, lymphoma cells and bladder carcinoma and got marketing authorization in Japan. Zorubicin which is a benzoylhydrazone derivative of the well-known anthracycline antineoplastic antibiotic daunorubicin is at the moment in the confirmatory phase of clinical studies for breast cancer and leukaemia.
Antifungals
Itraconazole
Itraconazole is an antifungal drug that works by inhibiting 14-α-lanosterol demethylase (14-LDM), a crucial enzyme in cholesterol production. Itraconazole also decreases angiogenesis in endothelial cells by reducing endothelial growth [185] and suppresses VEGFR-2 levels and multiple cellular pathways in endothelial cells [186]. Treatment with itraconazole of human vascular endothelial cells (HUVECs) inhibited movement and tube creation by decreasing the phosphorylation of growth factor receptors [187]. Itraconazole therapy of pulmonary carcinoma xenografts reduced angiogenesis and regression in this study. Its antitumor activity may be mediated by different pathways together with its antiangiogenic effects. In glioblastoma cells, removing lipids from the plasma membrane reduced Akt-1 action, which inhibited its downstream target mTOR, causing death and decreased tumor growth [188] (Fig. 3). It has been hypothesized that itraconazole might be a Hedgehog antagonist. In mice treated with itraconazole the proliferation of two hedgehog-based tumor types, a medulloblastoma and a basal cell cancer of the skin, was reduced in vivo [189]. Research on pleural mesothelioma cells came up with similar results [190]. Itraconazole was to a certain extent successful in a randomized clinical study with metastatic castration-resistant prostate carcinoma, with an extended PSA progression-free life, and suppression of Hedgehog signalling [191]. Itraconazole was also used to treat 19 patients with basal cell malignancy in a limited phase II study. Hedgehog signalling was decreased which was followed by slower tumor growth and tumor regression. In a minor phase II trial, combining itraconazole with conventional anticancer therapy for lung cancers improved both progression-free and ultimate survival. The authors of this study speculated that this result could be attributed to the antiangiogenic effects of itraconazole [192]. Nevertheless, there might be some problems with itraconazole treatment of malignant diseases. According to some studies, antifungal medications may affect the effects of other anticancer drugs, particularly antibodies, such as rituximab [193].

Schematic representation of the most representative drug clases used as repurposing drugs in oncology. Symbols: ↑increase, ↓decrease. Akt protein kinase B, Bcl-2 B-cell lymphoma 2, Bcl-xL B-cell lymphoma-extra-large, CDK cyclin-dependent kinases, COX-2 Cyclooxygenase-2, CPX cyclopirox olamine, DR4/5 death receptor 4/5, EGFR epidermal growth factor receptor, EMT epithelial–mesenchymal transition, Erk1/2 extracellular signal-regulated kinase 1/2, HIF-1 hypoxia-inducible factor-1, MMPs matrix metalloproteinase, mTOR mammalian target of rapamycin, NF-KB nuclear factor kappa B, P53 TP53 or tumor protein 53, PARP Poly (ADP-ribose) polymerase, PI3k/Akt phosphoinositide-3-kinase–protein kinase B/Akt, PPAR peroxisome proliferator-activated receptor, ROS reactive oxygen species, SOD2 superoxide dismutase, TNF-α tumor necrosis factor alpha, TRAIL TNF-related apoptosis inducing ligand, VEGFR-2 vascular endothelial growth factor receptor-2, Wnt wingless-related integration site, XIAP X-linked inhibitor of apoptosis protein
Rapamycin
Rapamycin, also called “sirolimus,” is a drug that was first discovered for its potent antifungal effects [194]. Because of its unique immunosuppressive effects, rapamycin has been routinely used to prevent rejection after organ transplantation. Rapamycin has relatively recently been identified as an mTOR inhibitor that may be used to treat Kras Pten endocrine ductal adenocarcinoma, resulting in inhibition of proliferation and tumor size shrinkage [195]. By transcriptional activation of Atg7 and DAPK, rapamycin was also used to activate cellular autophagy and increase the chemotherapeutic effects of dihydro-artemisinin in breast tumor cells [196]. Due to the increased expression of p73, rapamycin improves the susceptibility of ER-positive breast cancer cells to tamoxifen [197].
Griseofulvin
Griseofulvin causes apoptotic cell death in lymphoma and leukemia cells. [198]. It affects microtubule assembly in MCF-7 cells causing apoptotic death and cell cycle arrest, and it has a stimulatory effect together with vinblastine [199]. The same chemotherapeutic effects have been observed in colorectal and cervical cancers [200, 201]. Centrosome clumping has also been associated with the production of micronuclei in prostate carcinoma. In pulmonary and prostate cancer, combining radiation with griseofulvin therapy showed synergistic anticancer effects. The sulfonyl group substitution analogues of griseofulvin have antiproliferative effects on oral cancer cells and cytotoxic activity on breast cancer cells [202].
Clotrimazole
Clotrimazole inhibits glioblastoma cell invasion and metastasis. It inhibited actin polymerization and promoted glycolytic inflow in breast, colon, and pulmonary cancer cells [203]. Kadavakollu et al. [204] evaluated many potential anti-cancer pathways. In prostate and cervical cancers, as well as lymphoid malignancies, ruthenium combined with other drugs, had stronger cytotoxic activity than the monotherapy with individual drugs [205]. A combination of imatinib and clotrimazole inhibited the glycolysis pathway more effectively and boosted NO and VEGF production in breast cancer cells [206].
Ciclopirox
Ciclopirox (CPX), an antifungal drug, induced ageing in p53 deficient HeLa cells by a mechanism unrelated to mTOR [207]. Due to degrading CDC–CDK, downregulating Bcl-xL, and activating the caspase-dependent cascade, it causes apoptotic cell death in breast and colorectal cancer, and rhabdomyosarcoma cells [208, 209]. Prolonged exposure to this drug caused p53-independent caspase stimulation and cell death [207]. It has also been demonstrated that it suppresses HPV genetic mutations. By activating oxygen radicals, caspase-3, and lowering Bcl-xL levels, CPX was significantly more effective than gemcitabine in endocrine carcinoma. Nevertheless, when it came to triggering apoptotic pathways, the combination of these drugs was significantly more efficacious than each of these medicines alone [210]. According to ex-vivo and in-vivo studies in colorectal cancer, CPX causes autophagy through the depletion of DJ-1 and the formation of oxygen radicals [211]. According to Ahmad et al. ethacrynic acid and CPX had an additive anticancer effect in hepatocellular carcinoma. However, low concentrations of CPX were harmful not just to cancer cells but also to healthy cells [212].
Nannocystin A
Nannocystin A showed chemotherapeutic effects on colorectal and breast cancer cells. Nannocystin A has been shown to target eukaryotic elongation factor 1 in proteome studies [213, 214]. These findings support the repositioning of an antifungal drug with anticancer potential. In future, additional clinical testing is needed to confirm this.
NSAIDS/anti-inflammatory drugs
Aspirin
Aspirin is a drug that has effects on cyclooxygenase (COX) isoenzymes 1 and 2 and is widely used in the treatment and prevention of myocardial infarction in patients with coronary heart disease. COX-1 is important for platelet synthesis of thromboxane A2, which results in platelet aggregation and adhesion to cells, including malignant cells. Platelets covering tumor cells prevent the immune system from recognizing these cells, favoring the development and spread of cancer. COX-2 is important in the production of prostaglandin E2, which significantly stimulates the growth of tumor cells [215,216,217]. Recent pharmacological studies have analyzed the potential of aspirin as a therapeutic approach in spontaneous or chemically provoked tumors [218,219,220]. Many clinical trials have found that taking aspirin after a diagnosis is associated with a better prognosis in patients with colorectal cancer [221,222,223,224]. However, because most of these studies were retrospective and the patient recruitment was not homogenous, there is a lot of contradictory information. To determine the function of aspirin as a potential anticancer therapy, prospective trials are required. Several clinical trials are now in progress, the majority of which are looking at the effect of aspirin in preventing relaps of the illness.
Ibuprofen
Ibuprofen is a non-selective cyclooxygenase inhibitor. It slows down the development of prostate carcinoma [225]. It also has a radio-sensitizing effect ex-vivo but at larger doses than those that have been documented to suppress eicosanoid production, indicating that other pathways are included [226, 227]. Anti-angiogenesis, initiation of apoptosis, and decrease of cellular proliferation were found to have antitumor effects on gastric adenocarcinoma cells ex-vivo, along with modulating the expression levels of the cancer-related genes Akt, PCNA, Bax, P53, and Bcl2 [228] (Fig. 3). It has been shown that TNF-α upregulated metastatic melanoma cell migration in vitro and that this could be reduced by ibuprofen both in solution and delivered from a hydrogel. Although this might be attributed to the induction of apoptotic cell death, the mechanism of this is still not completely explained [229, 230]. Chemosensitivity could also be modulated by ibuprofen. Ibuprofen therapy reduced the amounts of Hsp70, a heat shock protein associated with apoptotic tolerance in lung carcinoma cells. Following ibuprofen therapy, blocking Hsp70 and inducing apoptosis improved responsiveness to the anticancer drug, cisplatin [231].
Naproxen
Naproxen is a propionic-acid analogue that reduces cell growth, provokes programmed cell death, and restricts metastasis [232]. This drug is a non-selective cyclooxygenase inhibitor. Ex-vivo and in-vivo chemotherapeutic effects have been documented in breast, leukemic, bladder, colorectal, and osteosarcoma cells. Naproxen causes the death of bladder carcinoma cells ex-vivo by targeting PI3K [233, 234]. A combination of cholesterol-lowering drug atorvastatin and naproxen effectively suppressed colon adenocarcinomas in experimental animals in-vivo [235]. A combination of calcitriol and naproxen has been tested in phase II clinical trials for preventing the relapse of prostate carcinoma. Such a combined therapy has been well-tolerated, with 19% of enrolled participants having a reduction in PSA doubling time (PSADT) and 67% having a prolongation of PSADT when compared with baseline [236].
Diclofenac
Diclofenac is an acetic acid derivative with a modest affinity for cyclooxygenase-2. It has been shown that this drug has beneficial effects on several malignant tumors including fibrosarcoma, hepatoma, colorectal, endometrial, and endocrine carcinoma. The effect of diclofenac (3%) and calcitriol was shown on different carcinoma cancer cell types, such as endometrial, breast, cerebral, colorectal, endocrine, non-small cell lung cancer, hepatic, and showed a higher cytotoxic effect in cells from chronic lymphocytic leukemia than in normal lymphocytes[237, 238]. Furthermore, growth rates and degree of vasculature were significantly decreased in experiments on rats with fibrosarcoma and hepatoma [239]. Diclofenac also has an anticancer effect in colon carcinoma [240, 241]. Diclofenac also reduced tumor growth in a mouse model of pancreatic cancer [242], as well as in ovarian cancer [243]. In vitro evidence suggests that diclofenac therapy induces apoptotic cell death by inhibiting antioxidant SOD2, resulting in a greater proportion of free radicals [244]. Despite the increasing amount of data supporting the anticancer effects of diclofenac, there are at the moment no active clinical studies evaluating the effects of diclofenac as a chemotherapeutic agent. A phase II clinical study for basal cell carcinoma, on the other hand, was just completed. Diclofenac was tested as a stand-alone treatment and in combination with calcitriol. The study found that diclofenac applied topically was more successful than combination therapy, with complete histologic tumor regression in 64.3% [245].
Celecoxib
Celecoxib, a specific COX-2 blocker, has chemotherapeutic effects in different types of cancer. In randomized controlled studies celecoxib combined with chemotherapeutic treatment has beneficial effects on breast cancer, progressive pulmonary carcinoma, and transitory bladder cancer [246,247,248,249]. Through miRNA-145/TGFBR2/Smad3 axis, celecoxib suppresses the epithelial-to-mesenchymal shift in cancer bladder cells [250]. Celecoxib decreases liver cancer cells proliferation and metastasis by addressing PNO1 and reduces AKT/c-Met-induced hepatocarcinogenesis by inhibiting COX-2/Akt/FASN pathway [249, 251, 252]. Celecoxib has effects on proline metabolism, generating an upregulation in proapoptotic markers (PRODH/POX, PPAR), lowering HIF-1 levels, and triggering squamous skin carcinoma programmed cell death. In human oral cancer cells, combination therapy with celecoxib and calyculin-A suppresses epithelial–mesenchymal shift [253, 254].
Indomethacin
Indomethacin is an antinociceptive non-steroidal anti-inflammatory drug (NSAID) widely used in the management of rheumatoid diseases [255]. It has been noted that patients who were treated for a long period with NSAIDs had a decreased chance of acquiring cancer which was confirmed in several clinical studies [255]. Furthermore, there is a growing number of publications suggesting that indomethacin and indomethacin-dependent prodrugs have chemo-preventive effects against different malignant diseases by inhibiting COX-1/2-associated angiogenesis [256, 257]. Indomethacin inhibits cancer cell progression by competing with calcium-related signalling and the creation of focal interactions [258]. A COX-independent mode of activity for indomethacin's antiproliferative effect has been discovered in several studies, as indicated by cell growth suppression in indomethacin-treated colonic cancer cells that would not exhibit COX-1/2. [259,260,261]. Lin et al. relatively recently proposed that the chemotherapeutic activity of indomethacin might be due to MAPK-associated pathway suppression [262]. They used computational scanning to perform this drug repurposing using a current drug repository. Indomethacin was proven to have a stronger association with ShcPTB by interacting with the phosphotyrosine binding (PTB) region of adaptor protein Shc (ShcPTB) as a readout. It seems that indomethacin competes against active EGFR by interacting with ShcPTB without disrupting the ERK-binding region, preventing EGFR from recruiting Shc and inducing abnormal signalling as a consequence of ERK production. Indomethacin suppresses cancer cell proliferation by disturbing PKC–p38–DRP1 axis-based mitochondrial dynamics or downregulating Wnt/β-catenin signalling to effectively target MAPK mechanisms [263]. NSAIDs, such as indomethacin, are currently seriously being considered as potential anti-carcinogenic medicines [264,265,266,267]. The therapeutic use of indomethacin offers a lot of potentials particularly if data would be collected enabling a better knowledge of the processes concerning its antiproliferative effects.
Antimalarial drugs
Artemisinins
A drug development initiative for the management of malaria led to the innovation of artemisinins. Artemisinins are plant extracts that have been used in Chinese traditional medicine for centuries.
When used as a treatment for malaria, artemisinins trigger the production of free radicals in infected erythrocytes, eradicating the plasmodium parasite [268]. The effects of artemisinins are attributed to the interaction of endoperoxide moiety with the Fe-containing heme groups in the affected RBCs [269]. Artesunate is by far the most researched artemisinin which could be used for cancer medication repurposing. Ex-vivo and in-vivo data indicate that it might have antiangiogenic effects, the effects on the formation of free radicals, and the modification of antimicrobial resistance in a wide range of cancers [270]. Artesunate has antiproliferative and pro-apoptotic effects on lymphoma and myeloma cells [271], as well as on hepatocellular cancer cells [272, 273]. An increase in the expression of pro-apoptotic proteins, such as caspase-3, a reduction in the MYC oncogene, and a reduction of many anti-apoptotic proteins are all possible explanations for why this drug might have anti-malignant effects. The chemotherapeutic efficacy of artesunate in hepatocellular cancer is increased when combined with sorafenib [272] and gemcitabine [273]. Antiangiogenic effects of artesunate have been documented in renal cancer and hepatocellular carcinoma, with decreased tumor development in vivo, lower vessel number, and decreased vascular endothelial growth factor [274].
Dihydroartemisinin (DHA) is an analogue of artemisinin that inhibits leukemia cell proliferation by inducing autophagy and programmed cell death which is reactive oxygen species-dependent [275]. Decreased expression of the protein transferrin receptor 1 (TfR1) and cell cycle arrest were two factors that contributed to the effect of DHA. DHA therapy showed a significant reduction in iron in hepatoma and breast cancer cells due to the downregulation of TfR1, a protein that plays a significant role in iron absorption [276]. In leukemic cells, the TfR1 level was reduced [277]. TfR1 level was required for DHA responsiveness in different papillomavirus-infected cells and cervical cancer cells. In an in-vivo papilloma model, DHA therapy also suppressed tumour development [278].
Chloroquine
Chloroquine (CQ) is an antimalarial drug which was developed in the 1930s became the most widely used synthetic antimalarial drug during the 1960s and 1970s until the development of newer antimalarials in the whole world. Chloroquine has relatively recently been found to have potential anticancer effects. Chloroquine triggers tumor apoptosis by p53 and Rab8b-dependent Par-4 release, resulting in Par-4-dependent suppression of metastatic tumor development [279]. By reverting tumour-based macrophages to the M1 phenotype, chloroquine has effects on anticancer immune system response [280]. Chloroquine inhibits extrinsic neutrophil entrapment which decreases hypercoagulability in pancreatic malignant tumors [281]. Chloroquine suppresses tumour-associated Kv10.1 gates and reduces MDAMB-231 breast carcinoma cell motility ex-vivo [282]. In colon cancer, the combination of temsirolimus and chloroquine improves radiosensitivity [283]. Chloroquine sensitizes human T-cell blood cancer with oncogenic NOTCH1 mutations to secretase reduction [284]. Chloroquine is a derivative of quinoline similar to that of clioquinol. Many studies have reported its antitumor effects both in-vivo and in vitro. It has a synergistic effect in combination with paclitaxel and can stop the growth of breast malignant tumors [285]. Chloroquine, in combination with gemcitabine, seems to be able to eradicate tumor cells in xenograft models [149].
Hydroxychloroquine
Hydroxychloroquine is an analogue of chloroquine which has the same therapeutic effects as chloroquine although with less systemic toxicity. Hydroxychloroquine blocks intracellular lysosomal activities and improves the anticancer activities of breast cancer and glioblastoma [286, 287]. Hydroxychloroquine is an autophagy blocker that significantly increases the chemotherapeutic activity of bevacizumab on glioblastoma by suppressing of autophagy [288]. Ex-vivo and in-vivo, hydroxychloroquine increase the chemotherapeutic potential of the anti-angiogenesis drug BC001, suppressing the development of gastric carcinoma [289]. Pulmonary cancer cells are suppressed by hydroxychloroquine due to the increased chemo-sensitizing effect and effects on the change of M2-TAMs to M1-related macrophages, which improves the CD8+ T cell immunological reaction [290].
Quinacrine
Quinacrine was first identified in the 1920s as an antimalarial drug [291]. It is also used as an antibiotic, and a pleural sclerosing substance to manage giardiasis—a protozoal infection of the intestinal tract, certain types of lupus erythematosus and rheumatoid arthritis [292,293,294]. Quinacrine has also been used in clinical studies for the management of Creutzfeldt–Jakob disease including a new variant of CJD which is linked to contamination of food by the bovine spongiform encephalopathy (BSE). Quinacrine seems to be very suitable for repurposing for cancer therapy [295,296,297]. Earlier studies have shown that quinacrine has beneficial effects on different malignancies and that these effects are mediated by p53 activation. Quinacrine promotes p53 expression in renal cancer using suggesting that simultaneous inhibition of NF-kappaB and activation of p53 by a single small molecule can have anti-cancer effects [298]. It seems that quinacrine's cytotoxicity is associated with elevated p53 levels. It also seems that the facilitates chromatin transcription (FACT) protein complex, which seems to be trapped on chromatin and induces CK2-induced phosphorylation of p53, is responsible for quinacrine-mediated p53 transcription [299, 300]. Conversely, there are indications that the down-regulation of p53 increases the quinacrine effect in MCF-7 cells when compared with normal cells [301]. Nonetheless, the results of some studies suggest that quinacrine cytotoxicity on cancer cells is influenced by the p53 expression, at least to a certain extent [302, 303]. Studies with quinacrine as a chemotherapeutic drug are published recently quite often [32]. For example, in a Phase, I and Phase II clinical study, Fox Chase Cancer Center researchers combined quinacrine with capecitabine to treat colon carcinoma (NCT01844076). In the Phase I clinical study, quinacrine was combined with erlotinib for the management of recurrent or delayed pulmonary cancer (NCT01839955). Overall, quinacrine is very promising as an anticancer treatment, and the effects of this drug might be associated with the stimulation of p53, a crucial growth inhibitor that is dysregulated in many malignant diseases [32].
Atovaquone
In recent years, Atovaquone, a well-studied molecule known for its role as a non-oncological and anti-malarial drug, has garnered significant attention in the field of cancer therapy [304]. With its established safety profile and extensive clinical use, Atovaquone has emerged as a potential candidate for repurposing in the treatment of cancer [305]. Atovaquone, primarily used as an antiparasitic agent against malaria, has demonstrated promising anticancer properties in preclinical studies. Multiple investigations have shown its ability to inhibit cancer cell growth and induce apoptosis in various cancer types, including lung, breast, colon, and prostate cancers. These findings highlight the potential of Atovaquone as an effective anticancer agent [305].
Mechanistically, Atovaquone exerts its anticancer effects through multiple pathways. It has been shown to disrupt mitochondrial function, leading to energy depletion and apoptosis in cancer cells [304]. In addition, Atovaquone has demonstrated the ability to inhibit specific signaling pathways involved in cancer cell proliferation and survival, such as the PI3K/AKT pathway [306]. Moreover, the favorable safety profile of Atovaquone, established through its extensive use as an anti-malarial agent, further supports its potential for repurposing in cancer therapy. The well-tolerated nature of Atovaquone could potentially minimize adverse effects commonly associated with traditional chemotherapy agents, offering a more favorable treatment option for cancer patients [304, 306]. Although the repurposing of Atovaquone for cancer therapy is still in its early stages, ongoing preclinical and clinical studies are actively investigating its efficacy and safety in cancer treatment [304]. These studies aim to evaluate the optimal dosage, combination strategies, and patient selection criteria for Atovaquone-based therapies. Preliminary results from these studies have shown promising outcomes, warranting further investigation and clinical trials. Atovaquone, an anti-malarial drug, has emerged as a well-studied molecule with a high safety profile; its potential as an anticancer agent has been supported by pharmacological preclinical evidence demonstrating its ability to inhibit cancer cell growth and induce apoptosis [304, 306]. Ongoing research and clinical studies will shed more light on the efficacy and safety of Atovaquone in cancer therapy, paving the way for its potential inclusion as a repurposed drug in the treatment armamentarium against cancer.
Myorelaxant agents
Thiocolchicoside
Thiocolchicoside is a chemically synthesized colchicoside produced from Gloriosa superba (Liliaceae) that has been authorized in Europe (EMA) only as an add-on treatment for painful muscle contractures (permanent tightening of the muscle tissue) [307]. Surprisingly, thiocolchicoside has lately been mentioned in several publications to have anticancer effects. Reuter et al., for instance, found that thiocolchicoside has anticancer effects in different malignant diseases including leukemia, lymphoma, and squamous cell carcinoma. Thiocolchicoside inhibited NF-B and COX-2 stimulation by promoting ubiquitination deterioration of IB, a major suppressor of the NF-B signalling cascade controlling IKK status and p65 nuclear translocation [308]. It seems that thiocolchicoside can block the receptor stimulator of the NF-kB ligand and the NF-B signalling cascade, which inhibits cancer-induced bone metastasis [309]. Different pharmaceutical firms market thiocolchicoside as a myorelaxant with anti-inflammatory and analgesic effects and advertise its use as a nociceptive medication [310]. The effects of thiocolchicoside, when used in the treatment of lower back pain, have been validated in several clinical trials [311, 312]. Despite the limited clinical trials exploring its anticancer effects, thiocolchicoside, a half-century-old medication, might be useful in cancer treatment through drug repurposing. However, this has to be proven in clinical trials, because until now no such evidence does exist.
Conclusion
Human cancers are different diseases and, therefore, need different treatment approaches and options. There has been a significant development in discovering new drugs for different types of malignant diseases during the last two decades. However, due to acquired resistance to existing medicines, a considerable number of cancer patients are incurable, which causes frustration for scientists and physicians. The budgets of many countries for treating human cancers are limited, and they cannot afford the current chemotherapy treatments which are more and more expensive. As a result, drug repurposing has been identified as one of the most promising ways to find novel anticancer therapies that are cheaper and can be faster to obtain marketing authorization. Academics, scientists, and pharmaceutical businesses all recognize the value of drug repurposing in dealing with the rising burden of human cancers. Different types of drugs that can have anti-cancer cell effects have been discussed in this review. By targeting the well-studied mechanisms implicated in carcinogenesis, these drugs have been shown to limit cellular growth, metastasis, and invasion or induce cell cycle arrest and apoptosis. Clinical trials are currently being performed with repurposed drugs based on their preclinical anti-cancer efficacy. Some of them have been approved by the FDA for the treatment of human malignant diseases, such as raloxifene which has been approved for breast cancer, and thalidomide, which is used to treat multiple myeloma. A meta-analysis of medications including metformin, statins, and aspirin demonstrated their association with a lower risk of cancer, and these treatments may be licensed for cancer treatment in the near future. Scientists can now predict a treatment's efficacy, mode of action, and safety in different diseases, including cancer, thanks to advances in pharmacogenomics and high-throughput drug screening methods. Drug repurposing brings up a whole new world of research into existing drugs, potentially allowing more prompt and cheaper therapy for malignant diseases.
In conclusion, this comprehensive review explores the repurposing of non-oncology drugs for cancer therapy, focusing on elucidating mechanisms, evaluating efficacy, and exploring clinical prospects. One notable finding from our analysis is the observation of higher IC50 values associated with the repurposed compounds during in vitro studies. The higher IC50 values suggest that the repurposed compounds may exhibit reduced potency in inhibiting cancer cell growth compared to traditional anticancer agents. While this finding poses challenges, it also presents opportunities for future investigations. Understanding the underlying reasons for these elevated IC50 values is crucial for optimizing the therapeutic potential of repurposed compounds in cancer treatment. Several factors could contribute to the observed higher IC50 values, including differences in target specificity, altered pharmacokinetics, or complex interactions within the cancer microenvironment. Addressing these issues requires a multi-faceted approach involving in-depth mechanistic studies, refinement of drug formulations, and innovative combination strategies. To overcome the limitations posed by higher IC50 values, further research should focus on enhancing the effectiveness of repurposed compounds through various strategies. These may include identifying synergistic drug combinations, optimizing drug delivery systems to enhance bioavailability, or exploring novel formulations to improve target specificity. In addition, the use of advanced preclinical models that better recapitulate the complexities of human cancer biology could provide valuable insights into the efficacy of repurposed compounds. While higher IC50 values observed in vitro pose challenges, it is important to consider that repurposed drugs have the advantage of established safety profiles, known pharmacokinetics, and potentially reduced development timelines. By addressing the issue of higher IC50 values and leveraging the strengths of repurposed compounds, we can advance the field of cancer therapy by potentially identifying new treatment options that are both effective and safe. In summary, the observation of higher IC50 values for repurposed compounds during in vitro studies highlights the need for further investigation and optimization. By addressing these challenges head-on and capitalizing on the unique opportunities provided by repurposed drugs, we can accelerate the development of innovative cancer therapies and improve patient outcomes. This discrepancy raises important considerations for further research and clinical translation.
Availability of data and materials
Not applicable.
Abbreviations
- Akt:
-
Protein kinase B
- Bax:
-
Bcl-2-associated X protein
- Bcl2:
-
B-cell lymphoma 2
- BSE:
-
Bovine spongiform encephalopathy
- c-myc:
-
Cellular myelocytomatosis
- COX-2:
-
Cyclooxygenase-2
- CSCs:
-
Cancer stem cells
- CSCs:
-
Cancer stem cells
- CTGF:
-
Connective tissue growth factor
- DAPK:
-
Death-associated protein kinase
- DHFR:
-
Dihydrofolate reductase
- EGFR:
-
Epidermal growth factor receptor
- eIF4E:
-
Eukaryotic translation initiation factor 4E
- ER:
-
Endoplasmic reticulum
- FACT:
-
Facilitates chromatin transcription
- FDA:
-
Food and drug administration
- HDACs:
-
Histone deacetylase
- HER2:
-
Human epidermal growth factor receptor-2
- HIV:
-
Human immunodeficiency virus
- HUVEC:
-
Human vascular endothelial cells
- MCF-7:
-
Michigan Cancer Foundation-7 cell line
- MDA-MB-231:
-
MD Anderson-Metastatic Brest-231 cell line
- MMPs:
-
Matrix metalloproteinases
- mTOR:
-
Mammalian target of rapamycin
- MZ:
-
Mebendazole
- NF-Kb:
-
Nuclear factor-kB
- NIH:
-
National Institute of Health
- NO:
-
Nitric oxide
- NSAID:
-
Non-steroidal anti-inflammatory drug
- P53:
-
Tumor protein TP53
- PAK-1:
-
Protein-activated kinases-1
- PARP:
-
Poly (ADP-ribose) polymerase
- PCNA:
-
Proliferating cell nuclear antigen
- PSADT:
-
PSA doubling time
- PTB:
-
Phosphotyrosine binding
- ROS:
-
Reactive oxygen species
- Shc-PTB:
-
Shc-Phosphotyrosine binding
- SNEA:
-
Sub-network enrichment analysis
- SOD2:
-
Superoxide dismutase 2
- STAT3:
-
Signal transducer and activator of transcription-3
- TfR1:
-
Transferrin receptor protein-1
- TKIs:
-
Tyrosine kinase inhibitors
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- VEGF:
-
Vascular endothelial growth factor
- XIAP:
-
X-linked blocker of apoptosis
- YAP1:
-
Yes-associated protein-1
References
Ozkan G, Günal-Köroğlu D, Karadag A, Capanoglu E, Cardoso SM, Al-Omari B, Calina D, Sharifi-Rad J, Cho WC. A mechanistic updated overview on lycopene as potential anticancer agent. Biomed Pharmacother. 2023;1(161):114428.
Sharma R, Abbasi-Kangevari M, Abd-Rabu R, Abidi H, Abu-Gharbieh E, Acuna JM, Adhikari S, Advani SM, Afzal MS, Meybodi MA, Ahinkorah BO. Global, regional, and national burden of colorectal cancer and its risk factors, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Gastroenterol Hepatol. 2022;7(7):627–47.
Mohi-Ud-Din R, Mir RH, Wani TU, Alsharif KF, Alam W, Albrakati A, Saso L, Khan H. The regulation of endoplasmic reticulum stress in Cancer: special focuses on luteolin patents. Molecules. 2022;27(8):2471.
Iqbal MJ, Javed Z, Herrera-Bravo J, Sadia H, Anum F, Raza S, Tahir A, Shahwani MN, Sharifi-Rad J, Calina D, Cho WC. Biosensing chips for cancer diagnosis and treatment: a new wave towards clinical innovation. Cancer Cell Int. 2022;22(1):1–6.
Ianoși SL, Batani A, Ilie MA, Tampa M, Georgescu SR, Zurac S, Boda D, Ianosi NG, Neagoe D, Calina D, Tutunaru C. Non-invasive imaging techniques for the in vivo diagnosis of Bowen’s disease: three case reports. Oncol Lett. 2019;17(5):4094–101.
Jain D, Chaudhary P, Varshney N, Bin Razzak KS, Verma D, Khan Zahra TR, Janmeda P, Sharifi-Rad J, Daştan SD, Mahmud S, Docea AO. Tobacco smoking and liver cancer risk: potential avenues for carcinogenesis. J Oncol. 2021;10:2021.
Mir RH, Mir PA, Mohi-Ud-Din R, Sabreen S, Maqbool M, Shah AJ, Shenmar K, Raza SN, Pottoo FH. A comprehensive review on journey of pyrrole scaffold against multiple therapeutic targets. Anti-Cancer Agent Med Chem. 2022;22(19):3291–303.
Mir RH, Mohi-ud-din R, Wani TU, Dar MO, Shah AJ, Lone B, Pooja C, Masoodi MH. Indole: a privileged heterocyclic moiety in the management of cancer. Curr Org Chem. 2021;25(6):724–36.
Mohi-Ud-Din R, Mir RH, Sawhney G, Dar MA, Bhat ZA. Possible pathways of hepatotoxicity caused by chemical agents. Curr Drug Metab. 2019;20(11):867–79.
Dhyani P, Quispe C, Sharma E, Bahukhandi A, Sati P, Attri DC, Szopa A, Sharifi-Rad J, Docea AO, Mardare I, Calina D. Anticancer potential of alkaloids: a key emphasis to colchicine, vinblastine, vincristine, vindesine, vinorelbine and vincamine. Cancer Cell Int. 2022;22(1):1–20.
Garzoli S, Alarcón-Zapata P, Seitimova G, Alarcón-Zapata B, Martorell M, Sharopov F, Fokou PV, Dize D, Yamthe LR, Les F, Cásedas G. Natural essential oils as a new therapeutic tool in colorectal cancer. Cancer Cell Int. 2022;22(1):407.
Mir PA, Mohi-Ud-Din R, Banday N, Maqbool M, Raza SN, Farooq S, Afzal S, Mir RH. Anticancer potential of thymoquinone: a novel bioactive natural compound from Nigella sativa L. Anti-Cancer Agent Med Chem. 2022;22(20):3401–15.
Mohi-Ud-Din R, Mir RH, Sabreen S, Jan R, Pottoo FH, Singh IP. Recent insights into therapeutic potential of plant-derived flavonoids against cancer. Anti-Cancer Agent Med Chem. 2022;22(20):3343–69.
Bhat IA, Kabeer SW, Reza MI, Mir RH, Dar MO. AdipoRon: a novel insulin sensitizer in various complications and the underlying mechanisms: a review. Curr Mol Pharmacol. 2020;13(2):94–107.
Almatroodi SA, Alsahli MA, Almatroudi A, Rahmani AH. Garlic and its active compounds: a potential candidate in the prevention of cancer by modulating various cell signalling pathways. Anti-Cancer Agent Med Chem. 2019;19(11):1314–24.
Wani TU, Mohi-Ud-Din R, Mir RH, Itoo AM, Mir KB, Fazli AA, Pottoo FH. Exosomes harnessed as nanocarriers for cancer therapy-current status and potential for future clinical applications. Curr Mol Med. 2021;21(9):707–23.
Hassan R, Mohi-Ud-Din R, Dar MO, Shah AJ, Mir PA, Shaikh M, Pottoo FH. Bioactive heterocyclic compounds as potential therapeutics in the treatment of gliomas: a review. Anti-Cancer Agent Med Chem. 2022;22(3):551–65.
Mohi-ud-Din R, Mir RH, Mir PA, Banday N, Shah AJ, Sawhney G, Bhat MM, Batiha GE, Pottoo FH. Dysfunction of ABC transporters at the surface of BBB: potential implications in intractable epilepsy and applications of nanotechnology enabled drug delivery. Curr Drug Metab. 2022;23(9):735–56.
Bertolini F, Sukhatme VP, Bouche G. Drug repurposing in oncology—patient and health systems opportunities. Nat Rev Clin Oncol. 2015;12(12):732–42.
Xue H, Li J, Xie H, Wang Y. Review of drug repositioning approaches and resources. Int J Biol Sci. 2018;14(10):1232.
Taheri Y, Joković N, Vitorović J, Grundmann O, Maroyi A, Calina D. The burden of the serious and difficult-to-treat infections and a new antibiotic available: cefiderocol. Front Pharmacol. 2021;14(11):578823.
Mir RH, Mir PA, Shah AJ, Banday N, Sabreen S, Maqbool M, Jan R, Shafi N, Masoodi MH. Curcumin as a privileged scaffold molecule for various biological targets in drug development. Stud Nat Prod Chem. 2022;1(73):405–34.
Mir RH, Mir PA, Uppal J, Chawla A, Patel M, Bardakci F, Adnan M, Mohi-Ud-Din R. Evolution of natural product scaffolds as potential proteasome inhibitors in developing cancer therapeutics. Metabolites. 2023;13(4):509.
Sadeghi HM, Adeli I, Calina D, Docea AO, Mousavi T, Daniali M, Nikfar S, Tsatsakis A, Abdollahi M. Polycystic ovary syndrome: a comprehensive review of pathogenesis, management, and drug repurposing. Int J Mol Sci. 2022;23(2):583.
Maxmen A. Busting the billion-dollar myth: how to slash the cost of drug development. Nature. 2016;536(7617):388.
Reichel A, Lienau P. Pharmacokinetics in drug discovery: an exposure-centred approach to optimising and predicting drug efficacy and safety. In: Nielsch U, Fuhrmann U, Jaroch S, editors. New approaches to drug discovery. Berlin: Springer; 2016. p. 235–60.
Rudrapal M, Khairnar SJ, Jadhav AG. Drug repurposing (DR): an emerging approach in drug discovery. Drug Repurp-Hypothesis Mol Aspect Ther Appl. 2020;13:10.
Deore AB, Dhumane JR, Wagh R, Sonawane R. The stages of drug discovery and development process. Asian J Pharm Res Dev. 2019;7(6):62–7.
Ávalos-Moreno M, López-Tejada A, Blaya-Cánovas JL, Cara-Lupiañez FE, González-González A, Lorente JA, Sánchez-Rovira P, Granados-Principal S. Drug repurposing for triple-negative breast cancer. J Personal Med. 2020;10(4):200.
Fogel DB. Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: a review. Contempor Clin Trials Commun. 2018;1(11):156–64.
Shim JS, Liu JO. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int J Biol Sci. 2014;10(7):654.
Zhang Z, Zhou L, Xie N, Nice EC, Zhang T, Cui Y, Huang C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct Target Ther. 2020;5(1):113.
Hernández-Lemus E, Martínez-García M. Pathway-based drug-repurposing schemes in cancer: the role of translational bioinformatics. Front Oncol. 2021;14(10):605680.
Sahoo BM, Ravi Kumar BV, Sruti J, Mahapatra MK, Banik BK, Borah P. Drug repurposing strategy (DRS): emerging approach to identify potential therapeutics for treatment of novel coronavirus infection. Front Mol Biosci. 2021;26(8):628144.
Park K. A review of computational drug repurposing. Transl Clin Pharmacol. 2019;27(2):59–63.
Ko Y. Computational drug repositioning: current progress and challenges. Appl Sci. 2020;10(15):5076.
Plenge RM, Scolnick EM, Altshuler D. Validating therapeutic targets through human genetics. Nat Rev Drug Discov. 2013;12(8):581–94.
Sonaye HV, Sheikh RY, Doifode CA. Drug repurposing: iron in the fire for older drugs. Biomed Pharmacother. 2021;1(141):111638.
Kim TW. Drug repositioning approaches for the discovery of new therapeutics for Alzheimer’s disease. Neurotherapeutics. 2015;12:132–42.
Ciallella JR, Reaume AG. In vivo phenotypic screening: clinical proof of concept for a drug repositioning approach. Drug Discov Today Technol. 2017;1(23):45–52.
Ayyar P, Subramanian U. Repurposing–second life for drugs. Pharmacia. 2022;69(1):51–9.
Parvathaneni V, Kulkarni NS, Muth A, Gupta V. Drug repurposing: a promising tool to accelerate the drug discovery process. Drug Discov Today. 2019;24(10):2076–85.
Reddy AS, Zhang S. Polypharmacology: drug discovery for the future. Expert Rev Clin Pharmacol. 2013;6(1):41–7.
Zhang W, Bai Y, Wang Y, Xiao W. Polypharmacology in drug discovery: a review from systems pharmacology perspective. Curr Pharm Des. 2016;22(21):3171–81.
Gns HS, Saraswathy GR, Murahari M, Krishnamurthy M. An update on drug repurposing: re-written saga of the drug’s fate. Biomed Pharmacother. 2019;1(110):700–16.
Galvin BD, Li Z, Villemaine E, Poole CB, Chapman MS, Pollastri MP, Wyatt PG, Carlow CK. A target repurposing approach identifies N-myristoyltransferase as a new candidate drug target in filarial nematodes. PLoS Negl Trop Dis. 2014;8(9):e3145.
Mejía-Pedroza RA, Espinal-Enríquez J, Hernández-Lemus E. Pathway-based drug repositioning for breast cancer molecular subtypes. Front Pharmacol. 2018;15(9):905.
Jin G, Wong ST. Toward better drug repositioning: prioritizing and integrating existing methods into efficient pipelines. Drug Discov Today. 2014;19(5):637–44.
Kotelnikova E, Yuryev A, Mazo I, Daraselia N. Computational approaches for drug repositioning and combination therapy design. J Bioinform Comput Biol. 2010;8(03):593–606.
Iorio F, Saez-Rodriguez J, Bernardo DD. Network based elucidation of drug response: from modulators to targets. BMC Syst Biol. 2013;7(1):1–9.
Austin CP, Brady LS, Insel TR, Collins FS. NIH molecular libraries initiative. Science. 2004;306(5699):1138–9.
Skrabanek L, Saini HK, Bader GD, Enright AJ. Computational prediction of protein–protein interactions. Mol Biotechnol. 2008;38:1–7.
Laudisi F, Marônek M, Di Grazia A, Monteleone G, Stolfi C. Repositioning of anthelmintic drugs for the treatment of cancers of the digestive system. Int J Mol Sci. 2020;21(14):4957.
Armando RG, Mengual Gómez DL, Gomez DE. New drugs are not enough-drug repositioning in oncology: An update. Int J Oncol. 2020;56(3):651–84.
Hou ZJ, Luo X, Zhang W, Peng F, Cui B, Wu SJ, Zheng FM, Xu J, Xu LZ, Long ZJ, Wang XT. Flubendazole, FDA-approved anthelmintic, targets breast cancer stem-like cells. Oncotarget. 2015;6(8):6326.
Oh E, Kim YJ, An H, Sung D, Cho TM, Farrand L, Jang S, Seo JH, Kim JY. Flubendazole elicits anti-metastatic effects in triple-negative breast cancer via STAT3 inhibition. Int J Cancer. 2018;143(8):1978–93.
Kim YJ, Sung D, Oh E, Cho Y, Cho TM, Farrand L, Seo JH, Kim JY. Flubendazole overcomes trastuzumab resistance by targeting cancer stem-like properties and HER2 signaling in HER2-positive breast cancer. Cancer Lett. 2018;1(412):118–30.
Lin S, Yang L, Yao Y, Xu L, Xiang Y, Zhao H, Wang L, Zuo Z, Huang X, Zhao C. Flubendazole demonstrates valid antitumor effects by inhibiting STAT3 and activating autophagy. J Exp Clin Cancer Res. 2019;38:1–3.
Li Y, Acharya G, Elahy M, Xin H, Khachigian LM. The anthelmintic flubendazole blocks human melanoma growth and metastasis and suppresses programmed cell death protein-1 and myeloid-derived suppressor cell accumulation. Cancer Lett. 2019;10(459):268–76.
Laclette JP, Guerra G, Zetina C. Inhibition of tubulin polymerization by mebendazole. Biochem Biophys Res Commun. 1980;92(2):417–23.
Guerini AE, Triggiani L, Maddalo M, Bonù ML, Frassine F, Baiguini A, Alghisi A, Tomasini D, Borghetti P, Pasinetti N, Bresciani R. Mebendazole as a candidate for drug repurposing in oncology: An extensive review of current literature. Cancers. 2019;11(9):1284.
Bai RY, Staedtke V, Aprhys CM, Gallia GL, Riggins GJ. Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro Oncol. 2011;13(9):974–82.
Rushworth LK, Hewit K, Munnings-Tomes S, Somani S, James D, Shanks E, Dufès C, Straube A, Patel R, Leung HY. Repurposing screen identifies mebendazole as a clinical candidate to synergise with docetaxel for prostate cancer treatment. Br J Cancer. 2020;122(4):517–27.
Zhang Z, Ji J, Liu H. Drug repurposing in oncology: Current evidence and future direction. Curr Med Chem. 2021;28(11):2175–94.
Williamson T, Mendes TB, Joe N, Cerutti JM, Riggins GJ. Mebendazole inhibits tumor growth and prevents lung metastasis in models of advanced thyroid cancer. Endocr Relat Cancer. 2020;27(3):123–36.
Pantziarka P, Bouche G, Meheus L, Sukhatme V, Sukhatme VP. Repurposing Drugs in Oncology (ReDO)—mebendazole as an anti-cancer agent. Ecancermedicalscience. 2014;8:443.
Mukhopadhyay T, Sasaki JI, Ramesh R, Roth JA. Mebendazole elicits a potent antitumor effect on human cancer cell lines both in vitro and in vivo. Clin Cancer Res. 2002;8(9):2963–9.
Sawanyawisuth K, Williamson T, Wongkham S, Riggins GJ. Effect of the antiparasitic drug mebendazole on cholangiocarcinoma growth. Southeast Asian J Trop Med Public Health. 2014;45(6):1264.
Sasaki JI, Ramesh R, Chada S, Gomyo Y, Roth JA, Mukhopadhyay T. The anthelmintic drug mebendazole induces mitotic arrest and apoptosis by depolymerizing tubulin in non-small cell lung cancer cells. Mol Cancer Ther. 2002;1(13):1201–9.
Nygren P, Fryknäs M, Ågerup B, Larsson R. Repositioning of the anthelmintic drug mebendazole for the treatment for colon cancer. J Cancer Res Clin Oncol. 2013;139:2133–40.
Doudican N, Rodriguez A, Osman I, Orlow SJ. Mebendazole induces apoptosis via Bcl-2 inactivation in chemoresistant melanoma cells. Mol Cancer Res. 2008;6(8):1308–15.
Doudican NA, Byron SA, Pollock PM, Orlow SJ. XIAP downregulation accompanies mebendazole growth inhibition in melanoma xenografts. Anticancer Drugs. 2013;24(2):181–8.
Bodhinayake I, Symons M, Boockvar JA. Repurposing mebendazole for the treatment of medulloblastoma. Neurosurgery. 2015;76(2):N15–6.
Larsen AR, Bai RY, Chung JH, Borodovsky A, Rudin CM, Riggins GJ, Bunz F. Repurposing the antihelmintic mebendazole as a hedgehog inhibitor. Mol Cancer Ther. 2015;14(1):3–13.
Nygren P, Larsson R. Drug repositioning from bench to bedside: tumour remission by the antihelmintic drug mebendazole in refractory metastatic colon cancer. Acta Oncol. 2014;53(3):427–8.
Dobrosotskaya IY, Hammer GD, Schteingart DE, Maturen KE, Worden FP. Mebendazole monotherapy and long-term disease control in metastatic adrenocortical carcinoma. Endocr Pract. 2011;17(3):e59-62.
Giovanelli A, Silva CL, Medeiros L, Vasconcellos MC. The molluscicidal activity of niclosamide (Bayluscide WP70®) on Melanoides tuberculata (Thiaridae), a snail associated with habitats of Biomphalaria glabrata (Planorbidae). Mem Inst Oswaldo Cruz. 2002;97:743–5.
Lu L, Dong J, Wang L, Xia Q, Zhang D, Kim H, Yin T, Fan S, Shen Q. Activation of STAT3 and Bcl-2 and reduction of reactive oxygen species (ROS) promote radioresistance in breast cancer and overcome of radioresistance with niclosamide. Oncogene. 2018;37(39):5292–304.
Pampori NA, Singh G, Srivastava VM. Cotugnia digonopora: carbohydrate metabolism and effect of anthelmintics on immature worms. J Helminthol. 1984;58(1):39–47.
Liao Z, Nan G, Yan Z, Zeng L, Deng Y, Ye J, Zhang Z, Qiao M, Li R, Denduluri S, Wang J. The anthelmintic drug niclosamide inhibits the proliferative activity of human osteosarcoma cells by targeting multiple signal pathways. Curr Cancer Drug Targets. 2015;15(8):726–38.
Pan JX, Ding K, Wang CY. Niclosamide, an old antihelminthic agent, demonstrates antitumor activity by blocking multiple signaling pathways of cancer stem cells. Chin J Cancer. 2012;31(4):178.
Sack U, Walther W, Scudiero D, Selby M, Kobelt D, Lemm M, Fichtner I, Schlag PM, Shoemaker RH, Stein U. Novel effect of antihelminthic Niclosamide on S100A4-mediated metastatic progression in colon cancer. J Natl Cancer Inst. 2011;103(13):1018–36.
Ye T, Xiong Y, Yan Y, Xia Y, Song X, Liu L, Li D, Wang N, Zhang L, Zhu Y, Zeng J. The anthelmintic drug niclosamide induces apoptosis, impairs metastasis and reduces immunosuppressive cells in breast cancer model. PLoS ONE. 2014;9(1):e85887.
Chen H, Yang Z, Ding C, Chu L, Zhang Y, Terry K, Liu H, Shen Q, Zhou J. Discovery of O-alkylamino-tethered niclosamide derivatives as potent and orally bioavailable anticancer agents. ACS Med Chem Lett. 2013;4(2):180–5.
Ma R, Ma ZG, Gao JL, Tai Y, Li LJ, Zhu HB, Li L, Dong DL, Sun ZJ. Injectable pegylated niclosamide (polyethylene glycol-modified niclosamide) for cancer therapy. J Biomed Mater Res, Part A. 2020;108(1):30–8.
Lin CK, Bai MY, Hu TM, Wang YC, Chao TK, Weng SJ, Huang RL, Su PH, Lai HC. Preclinical evaluation of a nanoformulated antihelminthic, niclosamide, in ovarian cancer. Oncotarget. 2016;7(8):8993.
Samy AL, Bakthavachalam V, Vudutha M, Vinjamuri S, Chinnapaka S, Munirathinam G. Eprinomectin, a novel semi-synthetic macrocylic lactone is cytotoxic to PC3 metastatic prostate cancer cells via inducing apoptosis. Toxicol Appl Pharmacol. 2020;15(401):115071.
Melotti A, Mas C, Kuciak M, Lorente-Trigos A, Ruiz i Altaba A. The river blindness drug I vermectin and related macrocyclic lactones inhibit WNT-TCF pathway responses in human cancer. EMBO Mol Med. 2014;6(10):1263–78.
Drinyaev VA, Mosin VA, Kruglyak EB, Novik TS, Sterlina TS, Ermakova NV, Kublik LN, Levitman MK, Shaposhnikova VV, Korystov YN. Antitumor effect of avermectins. Eur J Pharmacol. 2004;501(1–3):19–23.
Kirtonia A, Gala K, Fernandes SG, Pandya G, Pandey AK, Sethi G, Khattar E, Garg M. Repurposing of drugs: An attractive pharmacological strategy for cancer therapeutics. Semin Cancer Biol. 2021;68:258–78.
Wang K, Gao W, Dou Q, Chen H, Li Q, Nice EC, Huang C. Ivermectin induces PAK1-mediated cytostatic autophagy in breast cancer. Autophagy. 2016;12(12):2498–9.
Nambara S, Masuda T, Nishio M, Kuramitsu S, Tobo T, Ogawa Y, Hu Q, Iguchi T, Kuroda Y, Ito S, Eguchi H. Antitumor effects of the antiparasitic agent ivermectin via inhibition of Yes-associated protein 1 expression in gastric cancer. Oncotarget. 2017;8(64):107666.
Dominguez-Gomez G, Chavez-Blanco A, Medina-Franco JL, Saldivar-Gonzalez F, Flores-Torrontegui Y, Juarez M, Díaz-Chávez J, Gonzalez-Fierro A, Dueñas-González A. Ivermectin as an inhibitor of cancer stem-like cells. Mol Med Rep. 2018;17(2):3397–403.
Alavi SE, Shahmabadi HE. Anthelmintics for drug repurposing: Opportunities and challenges. Saudi Pharm J. 2021;29(5):434–45.
Cao B, Li J, Zhu J, Shen M, Han K, Zhang Z, Yu Y, Wang Y, Wu D, Chen S, Sun A. The antiparasitic clioquinol induces apoptosis in leukemia and myeloma cells by inhibiting histone deacetylase activity. J Biol Chem. 2013;288(47):34181–9.
Lin MX, Lin SH, Lin CC, Yang CC, Yuan SY. In vitro and in vivo antitumor effects of pyrimethamine on non-small cell lung cancers. Anticancer Res. 2018;38(6):3435–45.
Liu H, Qin Y, Zhai D, Zhang Q, Gu J, Tang Y, Yang J, Li K, Yang L, Chen S, Zhong W. Antimalarial drug pyrimethamine plays a dual role in antitumor proliferation and metastasis through targeting DHFR and TP. Mol Cancer Ther. 2019;18(3):541–55.
Khan MW, Saadalla A, Ewida AH, Al-Katranji K, Al-Saoudi G, Giaccone ZT, Gounari F, Zhang M, Frank DA, Khazaie K. The STAT3 inhibitor pyrimethamine displays anti-cancer and immune stimulatory effects in murine models of breast cancer. Cancer Immunol Immunother. 2018;67:13–23.
Liu Y, Zhou H, Yi T, Wang H. Pyrimethamine exerts significant antitumor effects on human ovarian cancer cells both in vitro and in vivo. Anticancer Drugs. 2019;30(6):571–8.
Seaberg EC, Wiley D, Martínez-Maza O, Chmiel JS, Kingsley L, Tang Y, Margolick JB, Jacobson LP, Multicenter AIDS Cohort Study (MACS). Cancer incidence in the multicenter AIDS Cohort Study before and during the HAART era: 1984 to 2007. Cancer. 2010;116(23):5507–16.
Kumar S, Bryant CS, Chamala S, Qazi A, Seward S, Pal J, Steffes CP, Weaver DW, Morris R, Malone JM, Shammas MA. Ritonavir blocks AKT signaling, activates apoptosis and inhibits migration and invasion in ovarian cancer cells. Mol Cancer. 2009;8(1):1–2.
Batchu RB, Gruzdyn OV, Bryant CS, Qazi AM, Kumar S, Chamala S, Kung ST, Sanka RS, Puttagunta US, Weaver DW, Gruber SA. Ritonavir-mediated induction of apoptosis in pancreatic cancer occurs via the RB/E2F-1 and AKT pathways. Pharmaceuticals. 2014;7(1):46–57.
Sleire L, Førde HE, Netland IA, Leiss L, Skeie BS, Enger PØ. Drug repurposing in cancer. Pharmacol Res. 2017;1(124):74–91.
Kast RE, Ramiro S, Lladó S, Toro S, Coveñas R, Muñoz M. Antitumor action of temozolomide, ritonavir and aprepitant against human glioma cells. J Neurooncol. 2016;126:425–31.
Sato A, Asano T, Ito K, Asano T. Ritonavir interacts with bortezomib to enhance protein ubiquitination and histone acetylation synergistically in renal cancer cells. Urology. 2012;79(4):966-e13.
Adekola KU, Dalva Aydemir S, Ma S, Zhou Z, Rosen ST, Shanmugam M. Investigating and targeting chronic lymphocytic leukemia metabolism with the human immunodeficiency virus protease inhibitor ritonavir and metformin. Leuk Lymphoma. 2015;56(2):450–9.
Popović-Djordjević J, Quispe C, Giordo R, Kostić A, Stanković JS, Fokou PV, Carbone K, Martorell M, Kumar M, Pintus G, Sharifi-Rad J. Natural products and synthetic analogues against HIV: a perspective to develop new potential anti-HIV drugs. Eur J Med Chem. 2022;5(233):114217.
Bono C, Karlin L, Harel S, Mouly E, Labaume S, Galicier L, Apcher S, Sauvageon H, Fermand JP, Bories JC, Arnulf B. The human immunodeficiency virus-1 protease inhibitor nelfinavir impairs proteasome activity and inhibits the proliferation of multiple myeloma cells in vitro and in vivo. Haematologica. 2012;97(7):1101.
Pore N, Gupta AK, Cerniglia GJ, Jiang Z, Bernhard EJ, Evans SM, Koch CJ, Hahn SM, Maity A. Nelfinavir down-regulates hypoxia-inducible factor 1α and VEGF expression and increases tumor oxygenation: implications for radiotherapy. Can Res. 2006;66(18):9252–9.
Gills JJ, LoPiccolo J, Tsurutani J, Shoemaker RH, Best CJ, Abu-Asab MS, Borojerdi J, Warfel NA, Gardner ER, Danish M, Hollander MC. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res. 2007;13(17):5183–94.
Brüning A, Burger P, Vogel M, Rahmeh M, Gingelmaier A, Friese K, Lenhard M, Burges A. Nelfinavir induces the unfolded protein response in ovarian cancer cells, resulting in ER vacuolization, cell cycle retardation and apoptosis. Cancer Biol Ther. 2009;8(3):226–32.
Guan M, Fousek K, Jiang C, Guo S, Synold T, Xi B, Shih CC, Chow WA. Nelfinavir induces liposarcoma apoptosis through inhibition of regulated intramembrane proteolysis of SREBP-1 and ATF6. Clin Cancer Res. 2011;17(7):1796–806.
Cho HY, Thomas S, Golden EB, Gaffney KJ, Hofman FM, Chen TC, Louie SG, Petasis NA, Schönthal AH. Enhanced killing of chemo-resistant breast cancer cells via controlled aggravation of ER stress. Cancer Lett. 2009;282(1):87–97.
Guan M, Fousek K, Chow WA. Nelfinavir inhibits regulated intramembrane proteolysis of sterol regulatory element binding protein-1 and activating transcription factor 6 in castration-resistant prostate cancer. FEBS J. 2012;279(13):2399–411.
Guan M, Su L, Yuan YC, Li H, Chow WA. Nelfinavir and nelfinavir analogs block site-2 protease cleavage to inhibit castration-resistant prostate cancer. Sci Rep. 2015;5(1):9698.
Thomas S, Sharma N, Golden EB, Cho H, Agarwal P, Gaffney KJ, Petasis NA, Chen TC, Hofman FM, Louie SG, Schönthal AH. Preferential killing of triple-negative breast cancer cells in vitro and in vivo when pharmacological aggravators of endoplasmic reticulum stress are combined with autophagy inhibitors. Cancer Lett. 2012;325(1):63–71.
Pyrko P, Kardosh A, Wang W, Xiong W, Schönthal AH, Chen TC. HIV-1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Can Res. 2007;67(22):10920–8.
Tian X, Ye J, Alonso-Basanta M, Hahn SM, Koumenis C, Dorsey JF. Modulation of CCAAT/enhancer binding protein homologous protein (CHOP)-dependent DR5 expression by nelfinavir sensitizes glioblastoma multiforme cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J Biol Chem. 2011;286(33):29408–16.
Xiang T, Du L, Pham P, Zhu B, Jiang S. Nelfinavir, an HIV protease inhibitor, induces apoptosis and cell cycle arrest in human cervical cancer cells via the ROS-dependent mitochondrial pathway. Cancer Lett. 2015;364(1):79–88.
Pan J, Mott M, Xi B, Hepner E, Guan M, Fousek K, Magnusson R, Tinsley R, Valdes F, Frankel P, Synold T. Phase I study of nelfinavir in liposarcoma. Cancer Chemother Pharmacol. 2012;70:791–9.
Hoover AC, Milhem MM, Anderson CM, Sun W, Smith BJ, Hoffman HT, Buatti JM. Efficacy of nelfinavir as monotherapy in refractory adenoid cystic carcinoma: results of a phase II clinical trial. Head Neck. 2015;37(5):722–6.
Elion GB. Mechanism of action and selectivity of acyclovir. Am J Med. 1982;73(1):7–13.
Assouline S, Culjkovic B, Cocolakis E, Rousseau C, Beslu N, Amri A, Caplan S, Leber B, Roy DC, Miller WH Jr, Borden KL. Molecular targeting of the oncogene eIF4E in acute myeloid leukemia (AML): a proof-of-principle clinical trial with ribavirin. Blood. 2009;114(2):257–60.
Kentsis A, Topisirovic I, Culjkovic B, Shao L, Borden KL. Ribavirin suppresses eIF4E-mediated oncogenic transformation by physical mimicry of the 7-methyl guanosine mRNA cap. Proc Natl Acad Sci. 2004;101(52):18105–10.
Shaimerdenova M, Karapina O, Mektepbayeva D, Alibek K, Akilbekova D. The effects of antiviral treatment on breast cancer cell line. Infect Agent Cancer. 2017;12:1.
Sidwell RW, Huffman JH, Khare GP, Allen LB, Witkowski JT, Robins RK. Broad-spectrum antiviral activity of virazole: 1-β-D-ribofuranosyl-1, 2, 4-triazole-3-carboxamide. Science. 1972;177(4050):705–6.
Crotty S, Maag D, Arnold JJ, Zhong W, Lau JY, Hong Z, Andino R, Cameron CE. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat Med. 2000;6(12):1375–9.
Tam RC, Lau JY, Hong Z. Mechanisms of action of ribavirin in antiviral therapies. Antiviral Chem Chemother. 2001;12(5):261–72.
Von der Haar T, Gross JD, Wagner G, McCarthy JE. The mRNA cap-binding protein eIF4E in post-transcriptional gene expression. Nat Struct Mol Biol. 2004;11(6):503–11.
Graff JR, Zimmer SG. Translational control and metastatic progression: enhanced activity of the mRNA cap-binding protein eIF-4E selectively enhances translation of metastasis-related mRNAs. Clin Exp Metas. 2003;20:265–73.
Drach JC, Thomas MA, Barnett JW, Smith SH, Shipman C Jr. Tritiated thymidine incorporation does not measure DNA synthesis in ribavirin-treated human cells. Science. 1981;212(4494):549–51.
Zimmermann T, Stingele K, Hartmann M, Haas J, von Einsiedel R, Wildemann B. Successful treatment of aids related PML with HAART and cidofovir. Eur J Med Res. 2001;6(5):190–62.
Bronson JJ, Ho HT, De Boeck HI, Woods K, Ghazzouli IS, Martin JC, Hitchcock MJ. Biochemical pharmacology of acyclic nucleotide analogues. Ann NY Acad Sci. 1990;1(616):398–407.
De Clercq E. Therapeutic potential of Cidofovir (HPMPC, Vistide) for the treatment of DNA virus (ie herpes-, papova-, pox-and adenovirus) infections. Verhandelingen-Koninklijke Academie voor Geneeskunde Van Belgie. 1996;58(1):19–47.
Andrei G, Snoeck R, Piette J, Delvenne P, De Clercq E. Inhibiting effects of cidofovir (HPMPC) on the growth of the human cervical carcinoma (SiHa) xenografts in athymic nude mice. Oncol Res Featur Preclin Clin Cancer Ther. 1998;10(10):533–9.
Liekens S, Andrei G, Vandeputte M, De Clercq E, Neyts J. Potent inhibition of hemangioma formation in rats by the acyclic nucleoside phosphonate analogue cidofovir. Can Res. 1998;58(12):2562–7.
Liekens S, Verbeken E, De Clercq E, Neyts J. Potent inhibition of hemangiosarcoma development in mice by cidofovir. Int J Cancer. 2001;92(2):161–7.
Neyts J, Sadler R, De Clercq E, Raab-Traub N, Pagano JS. The antiviral agent cidofovir [(S)-1-(3-hydroxy-2-phosphonyl-methoxypropyl) cytosine] has pronounced activity against nasopharyngeal carcinoma grown in nude mice. Can Res. 1998;58(3):384–8.
Murono S, Raab-Traub N, Pagano JS. Prevention and inhibition of nasopharyngeal carcinoma growth by antiviral phosphonated nucleoside analogs. Can Res. 2001;61(21):7875–7.
Liekens S, Gijsbers S, Vanstreels E, Daelemans D, De Clercq E, Hatse S. The nucleotide analog cidofovir suppresses basic fibroblast growth factor (FGF2) expression and signaling and induces apoptosis in FGF2-overexpressing endothelial cells. Mol Pharmacol. 2007;71(3):695–703.
Liekens S, Neyts J, De Clercq E, Verbeken E, Ribatti D, Presta M. Inhibition of fibroblast growth factor-2-induced vascular tumor formation by the acyclic nucleoside phosphonate cidofovir. Can Res. 2001;61(13):5057–64.
Yan K, He LJ, Cheng W, Ji ZZ, Zhao BX, Hui XL, Cao SS, Chen B, He L, Lang SH, Miao Y. Inhibiting gastric cancer-associated angiogenesis by CIAPIN1 siRNA. Cancer Biol Ther. 2009;8(11):1058–63.
Hadaczek P, Ozawa T, Soroceanu L, Yoshida Y, Matlaf L, Singer E, Fiallos E, James CD, Cobbs CS. Cidofovir: a novel antitumor agent for glioblastoma. Clin Cancer Res. 2013;19(23):6473–83.
Zhang L, Dratver MB, Yazal T, Dong K, Nguyen A, Yu G, Dao A, Dratver MB, Duhachek-Muggy S, Bhat K, Alli C. Mebendazole potentiates radiation therapy in triple-negative breast cancer. Int J Radiat Oncol Biol Phys. 2019;103(1):195–207.
Chen L, Wang L, Shen H, Lin H, Li D. Anthelminthic drug niclosamide sensitizes the responsiveness of cervical cancer cells to paclitaxel via oxidative stress-mediated mTOR inhibition. Biochem Biophys Res Commun. 2017;484(2):416–21.
King ML, Lindberg ME, Stodden GR, Okuda H, Ebers SD, Johnson A, Montag A, Lengyel E, MacLean Ii JA, Hayashi K. WNT7A/β-catenin signaling induces FGF1 and influences sensitivity to niclosamide in ovarian cancer. Oncogene. 2015;34(26):3452–62.
Tang M, Hu X, Wang Y, Yao X, Zhang W, Yu C, Cheng F, Li J, Fang Q. Ivermectin, a potential anticancer drug derived from an antiparasitic drug. Pharmacol Res. 2021;1(163):105207.
Darwish WM, Bayoumi NA, El-Kolaly MT. Laser-responsive liposome for selective tumor targeting of nitazoxanide nanoparticles. Eur J Pharm Sci. 2018;1(111):526–33.
Balic A, Sørensen MD, Trabulo SM, Sainz B Jr, Cioffi M, Vieira CR, Miranda-Lorenzo I, Hidalgo M, Kleeff J, Erkan M, Heeschen C. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol Cancer Ther. 2014;13(7):1758–71.
Dalva-Aydemir S, Bajpai R, Martinez M, Adekola KU, Kandela I, Wei C, Singhal S, Koblinski JE, Raje NS, Rosen ST, Shanmugam M. Targeting the metabolic plasticity of multiple myeloma with FDA-approved ritonavir and metformin. Clin Cancer Res. 2015;21(5):1161–71.
Johnson CE, Hunt DK, Wiltshire M, Herbert TP, Sampson JR, Errington RJ, Davies DM, Tee AR. Endoplasmic reticulum stress and cell death in mTORC1-overactive cells is induced by nelfinavir and enhanced by chloroquine. Mol Oncol. 2015;9(3):675–88.
Tan H, He L, Cheng Z. Inhibition of eIF4E signaling by ribavirin selectively targets lung cancer and angiogenesis. Biochem Biophys Res Commun. 2020;529(3):519–25.
Verhees F, Legemaate D, Demers I, Jacobs R, Haakma WE, Rousch M, Kremer B, Speel EJ. The antiviral agent cidofovir induces DNA damage and mitotic catastrophe in HPV-positive and-negative head and neck squamous cell carcinomas in vitro. Cancers. 2019;11(7):919.
Im EJ, Lee CH, Moon PG, Rangaswamy GG, Lee B, Lee JM, Lee JC, Jee JG, Bae JS, Kwon TK, Kang KW. Sulfisoxazole inhibits the secretion of small extracellular vesicles by targeting the endothelin receptor A. Nat Commun. 2019;10(1):1387.
Qiao X, Wang X, Shang Y, Li Y, Chen SZ. Azithromycin enhances anticancer activity of TRAIL by inhibiting autophagy and up-regulating the protein levels of DR4/5 in colon cancer cells in vitro and in vivo. Cancer Commun. 2018;38:1–3.
Li F, Huang J, Ji D, Meng Q, Wang C, Chen S, Wang X, Zhu Z, Jiang C, Shi Y, Liu S. Azithromycin effectively inhibits tumor angiogenesis by suppressing vascular endothelial growth factor receptor 2-mediated signaling pathways in lung cancer. Oncol Lett. 2017;14(1):89–96.
Tamargo RJ, Bok RA, Brem H. Angiogenesis inhibition by minocycline. Can Res. 1991;51(2):672–5.
Fife RS, Rougraff BT, Proctor C, Sledge GW Jr. Inhibition of proliferation and induction of apoptosis by doxycycline in cultured human osteosarcoma cells. J Lab Clin Med. 1997;130(5):530–4.
Fife RS, Sledge GW Jr, Roth BJ, Proctor C. Effects of doxycycline on human prostate cancer cells in vitro. Cancer Lett. 1998;127(1–2):37–41.
Rubins JB, Charboneau D, Alter MD, Bitterman PB, Kratzke RA. Inhibition of mesothelioma cell growth in vitro by doxycycline. J Lab Clin Med. 2001;138(2):101–6.
Mouratidis PX, Colston KW, Dalgleish AG. Doxycycline induces caspase-dependent apoptosis in human pancreatic cancer cells. Int J Cancer. 2007;120(4):743–52.
Son K, Fujioka S, Iida T, Furukawa K, Fujita T, Yamada H, Chiao PJ, Yanaga K. Doxycycline induces apoptosis in PANC-1 pancreatic cancer cells. Anticancer Res. 2009;29(10):3995–4003.
Song H, Fares M, Maguire KR, Sidén Å, Potacova Z. Cytotoxic effects of tetracycline analogues (doxycycline, minocycline and COL-3) in acute myeloid leukemia HL-60 cells. PLoS ONE. 2014;9(12):e114457.
Golub LM, Ramamurthy NS, McNamara TF, Greenwald RA, Rifkin BR. Tetracyclines inhibit connective tissue breakdown: new therapeutic implications for an old family of drugs. Crit Rev Oral Biol Med. 1991;2(3):297–321.
Iwasaki H, Inoue H, Mitsuke Y, Badran A, Ikegaya S, Ueda T. Doxycycline induces apoptosis by way of caspase-3 activation with inhibition of matrix metalloproteinase in human T-lymphoblastic leukemia CCRF-CEM cells. J Lab Clin Med. 2002;140(6):382–6.
Onoda T, Ono T, Dhar DK, Yamanoi A, Fujii T, Nagasue N. Doxycycline inhibits cell proliferation and invasive potential: combination therapy with cyclooxygenase-2 inhibitor in human colorectal cancer cells. J Lab Clin Med. 2004;143(4):207–16.
Duivenvoorden WC, Popovic SV, Lhoták S, Seidlitz E, Hirte HW, Tozer RG, Singh G. Doxycycline decreases tumor burden in a bone metastasis model of human breast cancer. Can Res. 2002;62(6):1588–91.
Duivenvoorden WC, Vukmirović-Popović S, Kalina M, Seidlitz E, Singh G. Effect of zoledronic acid on the doxycycline-induced decrease in tumour burden in a bone metastasis model of human breast cancer. Br J Cancer. 2007;96(10):1526–31.
Lokeshwar BL, Selzer MG, Zhu BQ, Block NL, Golub LM. Inhibition of cell proliferation, invasion, tumor growth and metastasis by an oral non-antimicrobial tetracycline analog (COL-3) in a metastatic prostate cancer model. Int J Cancer. 2002;98(2):297–309.
Shen LC, Chen YK, Lin LM, Shaw SY. Anti-invasion and anti-tumor growth effect of doxycycline treatment for human oral squamous-cell carcinoma–in vitro and in vivo studies. Oral Oncol. 2010;46(3):178–84.
Qin Y, Zhang Q, Lee S, Zhong WL, Liu YR, Liu HJ, Zhao D, Chen S, Xiao T, Meng J, Jing XS. Doxycycline reverses epithelial-to-mesenchymal transition and suppresses the proliferation and metastasis of lung cancer cells. Oncotarget. 2015;6(38):40667.
Meng J, Sun B, Zhao X, Zhang D, Zhao X, Gu Q, Dong X, Zhao N, Liu P, Liu Y. Doxycycline as an inhibitor of the epithelial-to-mesenchymal transition and vasculogenic mimicry in hepatocellular carcinoma. Mol Cancer Ther. 2014;13(12):3107–22.
Yang B, Lu Y, Zhang AI, Zhou A, Zhang L, Zhang L, Gao L, Zang Y, Tang X, Sun L. Doxycycline induces apoptosis and inhibits proliferation and invasion of human cervical carcinoma stem cells. PLoS ONE. 2015;10(6):e0129138.
Huie M, Oettel K, Van Ummersen L, Kim KM, Zhang Y, Staab MJ, Horvath D, Marnocha R, Douglas J, Drezen A, Alberti D. Phase II study of interferon-alpha and doxycycline for advanced renal cell carcinoma. Invest New Drugs. 2006;24:255–60.
Addison CL, Simos D, Wang Z, Pond G, Smith S, Robertson S, Mazzarello S, Singh G, Vandermeer L, Fernandes R, Iyengar A. A phase 2 trial exploring the clinical and correlative effects of combining doxycycline with bone-targeted therapy in patients with metastatic breast cancer. J Bone Oncol. 2016;5(4):173–9.
Boesch M, Sopper S, Wolf D. Ionophore antibiotics as cancer stem cell-selective drugs: open questions. Oncologist. 2016;21(11):1291–3.
Vaysberg M, Balatoni CE, Nepomuceno RR, Krams SM, Martinez OM. Rapamycin inhibits proliferation of epstein-barr virus-positive B-cell lymphomas through modulation of cell-cycle protein expression. Transplantation. 2007;83(8):1114–21.
Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab. 2014;19(3):373–9.
Young RC, Ozols RF, Myers CE. The anthracycline antineoplastic drugs. N Engl J Med. 1981;305(3):139–53.
Garg M, Kanojia D, Mayakonda A, Ganesan TS, Sadhanandhan B, Suresh S, Nagare RP, Said JW, Doan NB, Ding LW, Baloglu E. Selinexor (KPT-330) has antitumor activity against anaplastic thyroid carcinoma in vitro and in vivo and enhances sensitivity to doxorubicin. Sci Rep. 2017;7(1):9749.
Garg M, Nagata Y, Kanojia D, Mayakonda A, Yoshida K, Haridas Keloth S, Zang ZJ, Okuno Y, Shiraishi Y, Chiba K, Tanaka H. Profiling of somatic mutations in acute myeloid leukemia with FLT3-ITD at diagnosis and relapse. Blood J Am Soc Hematol. 2015;126(22):2491–501.
Boyle KE, Boger DL, Wroe A, Vazquez M. Duocarmycin SA, a potent antitumor antibiotic, sensitizes glioblastoma cells to proton radiation. Bioorg Med Chem Lett. 2018;28(16):2688–92.
Wang TW, Yuan H, Diao WL, Yang R, Zhao XZ, Guo HQ. Comparison of gemcitabine and anthracycline antibiotics in prevention of superficial bladder cancer recurrence. BMC Urol. 2019;19:1–5.
Panchuk RR, Lehka LV, Terenzi A, Matselyukh BP, Rohr J, Jha AK, Downey T, Kril IJ, Herbacek I, van Schoonhoven S, Heffeter P. Rapid generation of hydrogen peroxide contributes to the complex cell death induction by the angucycline antibiotic landomycin E. Free Radical Biol Med. 2017;1(106):134–47.
Chong CR, Xu J, Lu J, Bhat S, Sullivan DJ Jr, Liu JO. Inhibition of angiogenesis by the antifungal drug itraconazole. ACS Chem Biol. 2007;2(4):263–70.
Nacev BA, Grassi P, Dell A, Haslam SM, Liu JO. The antifungal drug itraconazole inhibits vascular endothelial growth factor receptor 2 (VEGFR2) glycosylation, trafficking, and signaling in endothelial cells. J Biol Chem. 2011;286(51):44045–56.
Aftab BT, Dobromilskaya I, Liu JO, Rudin CM. Itraconazole inhibits angiogenesis and tumor growth in non–small cell lung cancer. Can Res. 2011;71(21):6764–72.
Liu R, Li J, Zhang T, Zou L, Chen Y, Wang K, Lei Y, Yuan K, Li Y, Lan J, Cheng L. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: involvement of abnormal cholesterol trafficking. Autophagy. 2014;10(7):1241–55.
Kim J, Tang JY, Gong R, Kim J, Lee JJ, Clemons KV, Chong CR, Chang KS, Fereshteh M, Gardner D, Reya T. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17(4):388–99.
You M, Varona-Santos J, Singh S, Robbins DJ, Savaraj N, Nguyen DM. Targeting of the Hedgehog signal transduction pathway suppresses survival of malignant pleural mesothelioma cells in vitro. J Thorac Cardiovasc Surg. 2014;147(1):508–16.
Antonarakis ES, Heath EI, Smith DC, Rathkopf D, Blackford AL, Danila DC, King S, Frost A, Ajiboye AS, Zhao M, Mendonca J. Repurposing itraconazole as a treatment for advanced prostate cancer: a noncomparative randomized phase II trial in men with metastatic castration-resistant prostate cancer. Oncologist. 2013;18(2):163–73.
Rudin CM, Brahmer JR, Juergens RA, Hann CL, Ettinger DS, Sebree R, Smith R, Aftab BT, Huang P, Liu JO. Phase 2 study of pemetrexed and itraconazole as second-line therapy for metastatic nonsquamous non–small-cell lung cancer. J Thorac Oncol. 2013;8(5):619–23.
Ringshausen I, Feuerstacke Y, Krainz P, den Hollander J, Hermann K, Buck A, Peschel C, zum Meyer Bueschenfelde C. Antifungal therapy with itraconazole impairs the anti-lymphoma effects of rituximab by inhibiting recruitment of CD20 to cell surface lipid rafts. Cancer Res. 2010;70(11):4292–6.
Ali ES, Mitra K, Akter S, Ramproshad S, Mondal B, Khan IN, Islam MT, Sharifi-Rad J, Calina D, Cho WC. Recent advances and limitations of mTOR inhibitors in the treatment of cancer. Cancer Cell Int. 2022;22(1):1–6.
Morran DC, Wu J, Jamieson NB, Mrowinska A, Kalna G, Karim SA, Au AY, Scarlett CJ, Chang DK, Pajak MZ, Oien KA. Targeting mTOR dependency in pancreatic cancer. Gut. 2014;63(9):1481–9.
Liu Q, Zhou X, Li C, Zhang X, Li CL. Rapamycin promotes the anticancer action of dihydroartemisinin in breast cancer MDA-MB-231 cells by regulating expression of Atg7 and DAPK. Oncol Lett. 2018;15(4):5781–6.
Zhu L, Li XX, Shi L, Wu J, Qian JY, Xia TS, Zhou WB, Sun X, Zhou XJ, Wei JF, Ding Q. Rapamycin enhances the sensitivity of ER-positive breast cancer cells to tamoxifen by upregulating p73 expression. Oncol Rep. 2019;41(1):455–64.
Schmeel LC, Schmeel FC, Kim Y, Blaum-Feder S, Schmidt-Wolf IG. Griseofulvin efficiently induces apoptosis in in vitro treatment of lymphoma and multiple myeloma. Anticancer Res. 2017;37(5):2289–95.
Rathinasamy K, Jindal B, Asthana J, Singh P, Balaji PV, Panda D. Griseofulvin stabilizes microtubule dynamics, activates p53 and inhibits the proliferation of MCF-7 cells synergistically with vinblastine. BMC Cancer. 2010;10:1–3.
Ho YS, Duh JS, Jeng JH, Wang YJ, Liang YC, Lin CH, Tseng CJ, Yu CF, Chen RJ, Lin JK. Griseofulvin potentiates antitumorigenesis effects of nocodazole through induction of apoptosis and G2/M cell cycle arrest in human colorectal cancer cells. Int J Cancer. 2001;91(3):393–401.
Panda D, Rathinasamy K, Santra MK, Wilson L. Kinetic suppression of microtubule dynamic instability by griseofulvin: implications for its possible use in the treatment of cancer. Proc Natl Acad Sci. 2005;102(28):9878–83.
Liéby-Muller F, Le Baliner QH, Grisoni S, Fournier E, Guilbaud N, Marion F. Synthesis and activities towards resistant cancer cells of sulfone and sulfoxide griseofulvin derivatives. Bioorg Med Chem Lett. 2015;25(10):2078–81.
Furtado CM, Marcondes MC, Sola-Penna M, de Souza ML, Zancan P. Clotrimazole preferentially inhibits human breast cancer cell proliferation, viability and glycolysis. PLoS ONE. 2012;7(2):e30462.
Kadavakollu S, Stailey C, Kunapareddy CS, White S. Clotrimazole as a cancer drug: a short review. Med Chem. 2014;4(11):722.
Robles-Escajeda E, Martínez A, Varela-Ramirez A, Sánchez-Delgado RA, Aguilera RJ. Analysis of the cytotoxic effects of ruthenium–ketoconazole and ruthenium–clotrimazole complexes on cancer cells. Cell Biol Toxicol. 2013;29:431–43.
Motawi TM, Sadik NA, Fahim SA, Shouman SA. Combination of imatinib and clotrimazole enhances cell growth inhibition in T47D breast cancer cells. Chem Biol Interact. 2015;25(233):147–56.
Braun JA, Herrmann AL, Blase JI, Frensemeier K, Bulkescher J, Scheffner M, Galy B, Hoppe-Seyler K, Hoppe-Seyler F. Effects of the antifungal agent ciclopirox in HPV-positive cancer cells: Repression of viral E6/E7 oncogene expression and induction of senescence and apoptosis. Int J Cancer. 2020;146(2):461–74.
Zhou H, Shen T, Luo Y, Liu L, Chen W, Xu B, Han X, Pang J, Rivera CA, Huang S. The antitumor activity of the fungicide ciclopirox. Int J Cancer. 2010;127(10):2467–77.
Shen T, Shang C, Zhou H, Luo Y, Barzegar M, Odaka Y, Wu Y, Huang S. Ciclopirox inhibits cancer cell proliferation by suppression of Cdc25A. Genes Cancer. 2017;8(3–4):505.
Mihailidou C, Papakotoulas P, Papavassiliou AG, Karamouzis MV. Superior efficacy of the antifungal agent ciclopirox olamine over gemcitabine in pancreatic cancer models. Oncotarget. 2018;9(12):10360.
Zhou J, Zhang L, Wang M, Zhou L, Feng X, Yu L, Lan J, Gao W, Zhang C, Bu Y, Huang C. CPX targeting DJ-1 triggers ROS-induced cell death and protective autophagy in colorectal cancer. Theranostics. 2019;9(19):5577.
Al-Dali AM, Weiher H, Schmidt-Wolf IG. Utilizing ethacrynic acid and ciclopirox olamine in liver cancer. Oncol Lett. 2018;16(5):6854–60.
Hoffmann H, Kogler H, Heyse W, Matter H, Caspers M, Schummer D, Klemke-Jahn C, Bauer A, Penarier G, Debussche L, Brönstrup M. Discovery, structure elucidation, and biological characterization of nannocystin A, a macrocyclic myxobacterial metabolite with potent antiproliferative properties. Angew Chem Int Ed. 2015;54(35):10145–8.
Krastel P, Roggo S, Schirle M, Ross NT, Perruccio F, Aspesi P Jr, Aust T, Buntin K, Estoppey D, Liechty B, Mapa F. Nannocystin A: an elongation factor 1 inhibitor from myxobacteria with differential anti-cancer properties. Angew Chem Int Ed. 2015;54(35):10149–54.
Ishida J, Konishi M, Ebner N, Springer J. Repurposing of approved cardiovascular drugs. J Transl Med. 2016;14:1–5.
Mir RH, Shah AJ, Mohi-Ud-Din R, Pottoo FH, Dar M, Jachak SM, Masoodi MH. Natural anti-inflammatory compounds as drug candidates in Alzheimer’s disease. Curr Med Chem. 2021;28(23):4799–825.
Mir RH, Masoodi MH. Anti-inflammatory plant polyphenolics and cellular action mechanisms. Curr Bioact Compd. 2020;16(6):809–17.
Gasic G, Gasic T, Murphy S. Anti-metastatic effect of aspirin. Lancet. 1972;300(7783):932–3.
Kolenich J, Mansour E, Flynn A. Haematological effects of aspirin. Lancet. 1972;300(7779):714.
Mir RH, Sawhney G, Verma R, Ahmad B, Kumar P, Ranjana S, Bhagat A, Madishetti S, Ahmed Z, Jachak SM, Choi S. Origanum vulgare L.: in vitro assessment of cytotoxicity, molecular docking studies, antioxidant and anti-inflammatory activity in LPS stimulated RAW 264.7 cells. Med Chem. 2021;17(9):983–93.
Reimers MS, Bastiaannet E, van Herk-Sukel MP, Lemmens VE, van den Broek CB, van de Velde CJ, de Craen AJ, Liefers GJ. Aspirin use after diagnosis improves survival in older adults with colon cancer: a retrospective cohort study. J Am Geriatr Soc. 2012;60(12):2232–6.
McCowan C, Munro AJ, Donnan PT, Steele RJ. Use of aspirin post-diagnosis in a cohort of patients with colorectal cancer and its association with all-cause and colorectal cancer specific mortality. Eur J Cancer. 2013;49(5):1049–57.
Goh HH, Leong WQ, Chew MH, Pan YS, Tony LK, Chew L, Tan IB, Toh HC, Tang CL, Fu WP, Chia WK. Post-operative aspirin use and colorectal cancer-specific survival in patients with stage I-III colorectal cancer. Anticancer Res. 2014;34(12):7407–14.
Ahmad G, Hassan R, Dhiman N, Ali A. Assessment of anti-inflammatory activity of 3-acetylmyricadiol in LPSStimulated raw 264.7 macrophages. Comb Chem High Throughput Screen. 2022;25(1):204–10.
Zhou Q, Zhao S, Gan L, Wang Z, Peng S, Li Q, Liu H, Liu X, Wang Z, Shi Q, Estill J. Use of non-steroidal anti-inflammatory drugs and adverse outcomes during the COVID-19 pandemic: a systematic review and meta-analysis. EClinicalMedicine. 2022;1:46.
Palayoor ST, Bump EA, Calderwood SK, Bartol S, Coleman CN. Combined antitumor effect of radiation and ibuprofen in human prostate carcinoma cells. Clin Cancer Res. 1998;4(3):763–71.
Hassan Mir R, Godavari G, Siddiqui NA, Ahmad B, Mothana RA, Ullah R, Almarfadi OM, Jachak SM, Masoodi MH. Design, synthesis, molecular modelling, and biological evaluation of oleanolic acid-arylidene derivatives as potential anti-inflammatory agents. Drug Des Dev Ther. 2021;4:385–97.
Akrami H, Aminzadeh S, Fallahi H. Inhibitory effect of ibuprofen on tumor survival and angiogenesis in gastric cancer cell. Tumor Biol. 2015;36:3237–43.
Redpath M, Marques CM, Dibden C, Waddon A, Lalla R, MacNeil S. Ibuprofen and hydrogel-released ibuprofen in the reduction of inflammation-induced migration in melanoma cells. Br J Dermatol. 2009;161(1):25–33.
Mir RH, Wani TU, Jan R, Shah AJ, Sabreen S, Mir PA, Rasool S, Masoodi MH, Bhat ZA. Nigella sativa as a therapeutic candidate for arthritis and related disorders. In: Khan A, Rehman M, editors. Black seeds (Nigella sativa). Amsterdam: Elsevier; 2022. p. 295–312.
Endo H, Yano M, Okumura Y, Kido H. Ibuprofen enhances the anticancer activity of cisplatin in lung cancer cells by inhibiting the heat shock protein 70. Cell Death Dis. 2014;5(1):e1027.
Kolawole OR, Kashfi K. NSAIDs and cancer resolution: new paradigms beyond cyclooxygenase. Int J Mol Sci. 2022;23(3):1432.
Kim MS, Kim JE, Lim DY, Huang Z, Chen H, Langfald A, Lubet RA, Grubbs CJ, Dong Z, Bode AM. Naproxen induces cell-cycle arrest and apoptosis in human urinary bladder cancer cell lines and chemically induced cancers by targeting PI3K. Cancer Prev Res. 2014;7(2):236–45.
Mir RH, Mir PA, Maqbool M, Banday N, Farooq S, Raza SN, Chawla PA. Therapeutic potential of plant-derived flavonoids against inflammation. In: Prasher P, Zacconi F, Dua K, Rathbone M, Withey J, editors. Recent developments in anti-inflammatory therapy. Cambridge: Academic Press; 2023. p. 279–93.
Suh N, Reddy BS, DeCastro A, Paul S, Lee HJ, Smolarek AK, So JY, Simi B, Wang CX, Janakiram NB, Steele V. Combination of atorvastatin with sulindac or naproxen profoundly inhibits colonic adenocarcinomas by suppressing the p65/β-catenin/cyclin D1 signaling pathway in rats. Cancer Prev Res. 2011;4(11):1895–902.
Srinivas S, Feldman D. A phase II trial of calcitriol and naproxen in recurrent prostate cancer. Anticancer Res. 2009;29(9):3605–10.
Wickström M, Danielsson K, Rickardson L, Gullbo J, Nygren P, Isaksson A, Larsson R, Lövborg H. Pharmacological profiling of disulfiram using human tumor cell lines and human tumor cells from patients. Biochem Pharmacol. 2007;73(1):25–33.
Triscott J, Rose Pambid M, Dunn SE. Concise review: bullseye: targeting cancer stem cells to improve the treatment of gliomas by repurposing disulfiram. Stem Cells. 2015;33(4):1042–6.
Peterson HI. Effects of prostaglandin synthesis inhibitors on tumor growth and vascularization: experimental studies in the rat. Invasion Metastasis. 1983;3(3):151–9.
Hixson LJ, Alberts DS, Krutzsch M, Einsphar J, Brendel K, Gross PH, Paranka NS, Baier M, Emerson S, Pamukcu R. Antiproliferative effect of nonsteroidal antiinflammatory drugs against human colon cancer cells. Cancer Epidemiol Biomark Prev. 1994;3(5):433–8.
Dar MO, Mir RH, Mohiuddin R, Masoodi MH, Sofi FA. Metal complexes of xanthine and its derivatives: Synthesis and biological activity. J Inorg Biochem. 2023;10:112290.
Mayorek N, Naftali-Shani N, Grunewald M. Diclofenac inhibits tumor growth in a murine model of pancreatic cancer by modulation of VEGF levels and arginase activity. PLoS ONE. 2010;5(9):e12715.
Valle BL, D’Souza T, Becker KG, Wood WH III, Zhang Y, Wersto RP, Morin PJ. Non-steroidal anti-inflammatory drugs decrease E2F1 expression and inhibit cell growth in ovarian cancer cells. PLoS ONE. 2013;8(4):e61836.
Cecere F, Iuliano A, Albano F, Zappelli C, Castellano I, Grimaldi P, Masullo M, De Vendittis E, Ruocco MR. Diclofenac-induced apoptosis in the neuroblastoma cell line SH-SY5Y: possible involvement of the mitochondrial superoxide dismutase. Biomed Res Int. 2010;1:2010.
Brinkhuizen T, Frencken KJ, Nelemans PJ, Hoff ML, Kelleners-Smeets NW, Zur Hausen A, van der Horst MP, Rennspiess D, Winnepenninckx VJ, van Steensel MA, Mosterd K. The effect of topical diclofenac 3% and calcitriol 3 μg/g on superficial basal cell carcinoma (sBCC) and nodular basal cell carcinoma (nBCC): a phase II, randomized controlled trial. J Am Acad Dermatol. 2016;75(1):126–34.
Hamy AS, Tury S, Wang X, Gao J, Pierga JY, Giacchetti S, Brain E, Pistilli B, Marty M, Espié M, Benchimol G. Celecoxib with neoadjuvant chemotherapy for breast cancer might worsen outcomes differentially by COX-2 expression and ER status: exploratory analysis of the REMAGUS02 trial. J Clin Oncol. 2019;37(8):624.
Edelman MJ, Wang X, Hodgson L, Cheney RT, Baggstrom MQ, Thomas SP, Gajra A, Bertino E, Reckamp KL, Molina J, Schiller JH. Phase III randomized, placebo-controlled, double-blind trial of celecoxib in addition to standard chemotherapy for advanced non–small-cell lung cancer with cyclooxygenase-2 overexpression: CALGB 30801 (Alliance). J Clin Oncol. 2017;35(19):2184.
Kelly JD, Tan WS, Porta N, Mostafid H, Huddart R, Protheroe A, Bogle R, Blazeby J, Palmer A, Cresswell J, Johnson M. BOXIT—a randomised phase III placebo-controlled trial evaluating the addition of celecoxib to standard treatment of transitional cell carcinoma of the bladder (CRUK/07/004). Eur Urol. 2019;75(4):593–601.
Mir RH, Banday N, Sabreen S, Shah AJ, Jan R, Wani TU, Farooq S, Bhat ZA. Resveratrol: a potential drug candidate with multispectrum therapeutic application. Stud Nat Prod Chem. 2022;1(73):99–137.
Liu X, Wu Y, Zhou Z, Huang M, Deng W, Wang Y, Zhou X, Chen L, Li Y, Zeng T, Wang G. Celecoxib inhibits the epithelial-to-mesenchymal transition in bladder cancer via the miRNA-145/TGFBR2/Smad3 axis. Int J Mol Med. 2019;44(2):683–93.
Dai H, Zhang S, Ma R, Pan L. Celecoxib inhibits hepatocellular carcinoma cell growth and migration by targeting PNO1. Med Sci Monit. 2019;25:7351.
Qiu Z, Zhang C, Zhou J, Hu J, Sheng L, Li X, Chen L, Li X, Deng X, Zheng G. Celecoxib alleviates AKT/c-Met-triggered rapid hepatocarcinogenesis by suppressing a novel COX-2/AKT/FASN cascade. Mol Carcinog. 2019;58(1):31–41.
Tołoczko-Iwaniuk N, Dziemiańczyk-Pakieła D, Celińska-Janowicz K, Zaręba I, Klupczyńska A, Kokot ZJ, Nowaszewska BK, Reszeć J, Borys J, Miltyk W. Proline-dependent induction of apoptosis in oral squamous cell carcinoma (OSCC)—the effect of celecoxib. Cancers. 2020;12(1):136.
Velmurugan BK, Hua CH, Tsai MH, Lee CP, Chung CM, Ko YC. Combination of celecoxib and calyculin-A inhibits epithelial-mesenchymal transition in human oral cancer cells. Biotech Histochem. 2020;95(5):341–8.
Aboelella NS, Brandle C, Okoko O, Gazi MY, Ding ZC, Xu H, Gorman G, Bollag R, Davila ML, Bryan LJ, Munn DH. Indomethacin-induced oxidative stress enhances death receptor 5 signaling and sensitizes tumor cells to adoptive T-cell therapy. J Immunother Cancer. 2022;10(7):e004938.
Chennamaneni S, Zhong B, Lama R, Su B. COX inhibitors Indomethacin and Sulindac derivatives as antiproliferative agents: synthesis, biological evaluation, and mechanism investigation. Eur J Med Chem. 2012;1(56):17–29.
Touhey S, O’Connor R, Plunkett S, Maguire A, Clynes M. Structure–activity relationship of indomethacin analogues for MRP-1, COX-1 and COX-2 inhibition: identification of novel chemotherapeutic drug resistance modulators. Eur J Cancer. 2002;38(12):1661–70.
Guo YC, Chang CM, Hsu WL, Chiu SJ, Tsai YT, Chou YH, Hou MF, Wang JY, Lee MH, Tsai KL, Chang WC. Indomethacin inhibits cancer cell migration via attenuation of cellular calcium mobilization. Molecules. 2013;18(6):6584–96.
Zhang YJ, Bao YJ, Dai Q, Yang WY, Cheng P, Zhu LM, Wang BJ, Jiang FH. mTOR signaling is involved in indomethacin and nimesulide suppression of colorectal cancer cell growth via a COX-2 independent pathway. Ann Surg Oncol. 2011;18:580–8.
Brunelli C, Amici C, Angelini M, Fracassi C, Belardo G, Santoro MG. The non-steroidal anti-inflammatory drug indomethacin activates the eIF2α kinase PKR, causing a translational block in human colorectal cancer cells. Biochemical journal. 2012;443(2):379–86.
Cheng YL, Zhang GY, Li C, Lin J. Screening for novel protein targets of indomethacin in HCT116 human colon cancer cells using proteomics. Oncol Lett. 2013;6(5):1222–8.
Lin CC, Suen KM, Stainthorp A, Wieteska L, Biggs GS, Leitão A, Montanari CA, Ladbury JE. Targeting the Shc-EGFR interaction with indomethacin inhibits MAP kinase pathway signalling. Cancer Lett. 2019;10(457):86–97.
Mazumder S, De R, Debsharma S, Bindu S, Maity P, Sarkar S, Saha SJ, Siddiqui AA, Banerjee C, Nag S, Saha D. Indomethacin impairs mitochondrial dynamics by activating the PKCζ–p38–DRP1 pathway and inducing apoptosis in gastric cancer and normal mucosal cells. J Biol Chem. 2019;294(20):8238–58.
Cuzick J. Preventive therapy for cancer. Lancet Oncol. 2017;18(8):e472-82.
Thun MJ, Jacobs EJ, Patrono C. The role of aspirin in cancer prevention. Nat Rev Clin Oncol. 2012;9(5):259–67.
Rostom A, Dubé C, Lewin G, Tsertsvadze A, Barrowman N, Code C, Sampson M, Moher D. Nonsteroidal anti-inflammatory drugs and cyclooxygenase-2 inhibitors for primary prevention of colorectal cancer: a systematic review prepared for the US Preventive Services Task Force. Ann Intern Med. 2007;146(5):376–89.
Nagaraj AB, Wang QQ, Joseph P, Zheng C, Chen Y, Kovalenko O, Singh S, Armstrong A, Resnick K, Zanotti K, Waggoner S. Using a novel computational drug-repositioning approach (DrugPredict) to rapidly identify potent drug candidates for cancer treatment. Oncogene. 2018;37(3):403–14.
Kundu CN, Das S, Nayak A, Satapathy SR, Das D, Siddharth S. Anti-malarials are anti-cancers and vice versa–one arrow two sparrows. Acta Trop. 2015;1(149):113–27.
Cui L, Su XZ. Discovery, mechanisms of action and combination therapy of artemisinin. Expert Rev Anti Infect Ther. 2009;7(8):999–1013.
Augustin Y, Krishna S, Kumar D, Pantziarka P. The wisdom of crowds and the repurposing of artesunate as an anticancer drug. Ecancermedicalscience. 2015;9:ed50.
Holien T, Olsen OE, Misund K, Hella H, Waage A, Rø TB, Sundan A. Lymphoma and myeloma cells are highly sensitive to growth arrest and apoptosis induced by artesunate. Eur J Haematol. 2013;91(4):339–46.
Vandewynckel YP, Laukens D, Geerts A, Vanhove C, Descamps B, Colle I, Devisscher L, Bogaerts E, Paridaens A, Verhelst X, Van Steenkiste C. Therapeutic effects of artesunate in hepatocellular carcinoma: repurposing an ancient antimalarial agent. Eur J Gastroenterol Hepatol. 2014;26(8):861–70.
Hou J, Wang D, Zhang R, Wang H. Experimental therapy of hepatoma with artemisinin and its derivatives: in vitro and in vivo activity, chemosensitization, and mechanisms of action. Clin Cancer Res. 2008;14(17):5519–30.
Zhou HJ, Wang WQ, Wu GD, Lee J, Li A. Artesunate inhibits angiogenesis and downregulates vascular endothelial growth factor expression in chronic myeloid leukemia K562 cells. Vascul Pharmacol. 2007;47(2–3):131–8.
Wang Z, Hu W, Zhang JL, Wu XH, Zhou HJ. Dihydroartemisinin induces autophagy and inhibits the growth of iron-loaded human myeloid leukemia K562 cells via ROS toxicity. FEBS Open Bio. 2012;1(2):103–12.
Ba Q, Zhou N, Duan J, Chen T, Hao M, Yang X, Li J, Yin J, Chu R, Wang H. Dihydroartemisinin exerts its anticancer activity through depleting cellular iron via transferrin receptor-1. PLoS ONE. 2012. https://doi.org/10.1371/journal.pone.0042703.
Zhou HJ, Wang Z, Li A. Dihydroartemisinin induces apoptosis in human leukemia cells HL60 via downregulation of transferrin receptor expression. Anticancer Drugs. 2008;19(3):247–55.
Disbrow GL, Baege AC, Kierpiec KA, Yuan H, Centeno JA, Thibodeaux CA, Hartmann D, Schlegel R. Dihydroartemisinin is cytotoxic to papillomavirus-expressing epithelial cells in vitro and in vivo. Can Res. 2005;65(23):10854–61.
Burikhanov R, Hebbar N, Noothi SK, Shukla N, Sledziona J, Araujo N, Kudrimoti M, Wang QJ, Watt DS, Welch DR, Maranchie J. Chloroquine-inducible Par-4 secretion is essential for tumor cell apoptosis and inhibition of metastasis. Cell Rep. 2017;18(2):508–19.
Chen D, Xie J, Fiskesund R, Dong W, Liang X, Lv J, Jin X, Liu J, Mo S, Zhang T, Cheng F. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat Commun. 2018;9(1):873.
Boone BA, Murthy P, Miller-Ocuin J, Doerfler WR, Ellis JT, Liang X, Ross MA, Wallace CT, Sperry JL, Lotze MT, Neal MD. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer. 2018;18:1–2.
Valdés-Abadía B, Morán-Zendejas R, Rangel-Flores JM, Rodríguez-Menchaca AA. Chloroquine inhibits tumor-related Kv10.1 channel and decreases migration of MDA-MB-231 breast cancer cells in vitro. Eur J Pharmacol. 2019;855:262–6.
Shiratori H, Kawai K, Hata K, Tanaka T, Nishikawa T, Otani K, Sasaki K, Kaneko M, Murono K, Emoto S, Sonoda H. The combination of temsirolimus and chloroquine increases radiosensitivity in colorectal cancer cells. Oncol Rep. 2019;42(1):377–85.
Hounjet J, Habets R, Schaaf MB, Hendrickx TC, Barbeau LM, Yahyanejad S, Rouschop KM, Groot AJ, Vooijs M. The anti-malarial drug chloroquine sensitizes oncogenic NOTCH1 driven human T-ALL to γ-secretase inhibition. Oncogene. 2019;38(27):5457–68.
Choi DS, Blanco E, Kim YS, Rodriguez AA, Zhao H, Huang TH, Chen CL, Jin G, Landis MD, Burey LA, Qian W. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells. 2014;32(9):2309–23.
Cook KL, Wärri A, Soto-Pantoja DR, Clarke PA, Cruz MI, Zwart A, Clarke R. Chloroquine inhibits autophagy to potentiate antiestrogen responsiveness in ER+ breast cancer. Clin Cancer Res. 2014;20(12):3222–32.
Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, McAfee Q. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy. 2014;10(8):1359–68.
Liu LQ, Wang SB, Shao YF, Shi JN, Wang W, Chen WY, Ye ZQ, Jiang JY, Fang QX, Zhang GB, Xuan ZX. Hydroxychloroquine potentiates the anti-cancer effect of bevacizumab on glioblastoma via the inhibition of autophagy. Biomed Pharmacother. 2019;1(118):109339.
Wang W, Liu L, Zhou Y, Ye Q, Yang X, Jiang J, Ye Z, Gao F, Tan X, Zhang G, Fang Q. Hydroxychloroquine enhances the antitumor effects of BC001 in gastric cancer. Int J Oncol. 2019;55(2):405–14.
Li Y, Cao F, Li M, Li P, Yu Y, Xiang L, Xu T, Lei J, Tai YY, Zhu J, Yang B. Hydroxychloroquine induced lung cancer suppression by enhancing chemo-sensitization and promoting the transition of M2-TAMs to M1-like macrophages. J Exp Clin Cancer Res. 2018;37:1–6.
Van Dyke K, Lantz C, Szustkiewicz C. Quinacrine: mechanisms of antimalarial action. Science. 1970;169(3944):492–3.
Requena-Méndez A, Goñi P, Rubio E, Pou D, Fumadó V, Lóbez S, Aldasoro E, Cabezos J, Valls ME, Treviño B, Martínez Montseny AF. The use of quinacrine in nitroimidazole-resistant Giardia duodenalis: an old drug for an emerging problem. J Infect Dis. 2017;215(6):946–53.
Egorin MJ, Trump DL, Wainwright CW. Quinacrine ochronosis and rheumatoid arthritis. JAMA. 1976;236(4):385–6.
Larrieu AJ, Tyers GF, Williams EH, O’Neill MJ, Derrick JR. Intrapleural instillation of quinacrine for treatment of recurrent spontaneous pneumothorax. Ann Thorac Surg. 1979;28(2):146–50.
Geschwind MD, Kuo AL, Wong KS, Haman A, Devereux G, Raudabaugh BJ, Johnson DY, Torres-Chae CC, Finley R, Garcia P, Thai JN. Quinacrine treatment trial for sporadic Creutzfeldt-Jakob disease. Neurology. 2013;81(23):2015–23.
Collinge J, Gorham M, Hudson F, Kennedy A, Keogh G, Pal S, Rossor M, Rudge P, Siddique D, Spyer M, Thomas D. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol. 2009;8(4):334–44.
Oien DB, Pathoulas CL, Ray U, Thirusangu P, Kalogera E, Shridhar V. Repurposing quinacrine for treatment-refractory cancer. Semin Cancer Biol. 2021;68:21–30.
Gurova KV, Hill JE, Guo C, Prokvolit A, Burdelya LG, Samoylova E, Khodyakova AV, Ganapathi R, Ganapathi M, Tararova ND, Bosykh D. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-κB-dependent mechanism of p53 suppression in tumors. Proc Natl Acad Sci. 2005;102(48):17448–53.
Nesher E, Safina A, Aljahdali I, Portwood S, Wang ES, Koman I, Wang J, Gurova KV. Role of chromatin damage and chromatin trapping of FACT in mediating the anticancer cytotoxicity of DNA-binding small-molecule drugs. Can Res. 2018;78(6):1431–43.
Gasparian AV, Burkhart CA, Purmal AA, Brodsky L, Pal M, Saranadasa M, Bosykh DA, Commane M, Guryanova OA, Pal S, Safina A. Curaxins: anticancer compounds that simultaneously suppress NF-κB and activate p53 by targeting FACT. Sci Transl Med. 2011;3(95):95ra74.
Park S, Oh AY, Cho JH, Yoon MH, Woo TG, Kang SM, Lee HY, Jung YJ, Park BJ. Therapeutic effect of quinacrine, an antiprotozoan drug, by selective suppression of p-CHK1/2 in p53-negative malignant cancers. Mol Cancer Res. 2018;16(6):935–46.
Preet R, Siddharth S, Satapathy SR, Das S, Nayak A, Das D, Wyatt MD, Kundu CN. Chk1 inhibitor synergizes quinacrine mediated apoptosis in breast cancer cells by compromising the base excision repair cascade. Biochem Pharmacol. 2016;1(105):23–33.
Mohapatra P, Preet R, Das D, Satapathy SR, Choudhuri T, Wyatt MD, Kundu CN. Quinacrine-mediated autophagy and apoptosis in colon cancer cells is through a p53-and p21-dependent mechanism. Oncol Res Featur Preclin Clin Cancer Ther. 2012;20(2–3):81–91.
Gupta N, Srivastava SK. Atovaquone: an antiprotozoal drug suppresses primary and resistant breast tumor growth by inhibiting HER2/β-catenin signaling. Mol Cancer Ther. 2019;18(10):1708–20.
Gupta N, Gaikwad S, Kaushik I, Wright SE, Markiewski MM, Srivastava SK. Atovaquone suppresses triple-negative breast tumor growth by reducing immune-suppressive cells. Int J Mol Sci. 2021;22(10):5150.
Fiorillo M, Lamb R, Tanowitz HB, Mutti L, Krstic-Demonacos M, Cappello AR, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Repurposing atovaquone: targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget. 2016;7(23):34084.
Tüzün F, Ünalan H, Öner N, Özgüzel H, Kirazli Y, İçağasioğlu A, Kuran B, Tüzün Ş, Başar G. Multicenter, randomized, double-blinded, placebo-controlled trial of thiocolchicoside in acute low back pain. Joint Bone Spine. 2003;70(5):356–61.
Reuter S, Prasad S, Phromnoi K, Ravindran J, Sung B, Yadav VR, Kannappan R, Chaturvedi MM, Aggarwal BB. Thiocolchicoside exhibits anticancer effects through downregulation of NF-κB pathway and its regulated gene products linked to inflammation and cancer. Cancer Prev Res. 2010;3(11):1462–72.
Reuter S, Gupta SC, Phromnoi K, Aggarwal BB. Thiocolchicoside suppresses osteoclastogenesis induced by RANKL and cancer cells through inhibition of inflammatory pathways: a new use for an old drug. Br J Pharmacol. 2012;165(7):2127–39.
Micheau O, Dufour F, Walczak H. Thiocolchicoside a semi-synthetic derivative of the Glory Lily: a new weapon to fight metastatic bone resorption. Br J Pharmacol. 2012;165(7):2124–6.
Sproviero E, Albamonte E, Costantino C, Giossi A, Mancuso M, Rigamonti A, Tornari P, Caggiano G. Efficacy and safety of a fixed combination of intramuscular diclofenac 75 mg+ thiocolchicoside 4 mg in the treatment of acute low back pain: a phase III, randomized, double blind, controlled trial. Eur J Phys Rehabil Med. 2018;54(5):654–62.
Rao R, Panghate A, Chandanwale A, Sardar I, Ghosh M, Roy M, Banerjee B, Goswami A, Kotwal pp. Clinical comparative study: efficacy and tolerability of tolperisone and thiocolchicoside in acute low back pain and spinal muscle spasticity. Asian Spine J. 2012;6(2):115.
Wang T, Fu X, Jin T, Zhang L, Liu B, Wu Y, Xu F, Wang X, Ye K, Zhang W, Ye L. Aspirin targets P4HA2 through inhibiting NF-κB and LMCD1-AS1/let-7g to inhibit tumour growth and collagen deposition in hepatocellular carcinoma. EBioMedicine. 2019;1(45):168–80.
Fujiwara N, Singal AG, Hoshida Y. Dose and duration of aspirin use to reduce incidental hepatocellular carcinoma. Hepatology. 2019;70(6):2216.
Yao Y, Guo Q, Cao Y, Qiu Y, Tan R, Yu Z, Zhou Y, Lu N. Artemisinin derivatives inactivate cancer-associated fibroblasts through suppressing TGF-β signaling in breast cancer. J Exp Clin Cancer Res. 2018;37(1):1–4.
Li X, Ba Q, Liu Y, Yue Q, Chen P, Li J, Zhang H, Ying H, Ding Q, Song H, Liu H. Dihydroartemisinin selectively inhibits PDGFRα-positive ovarian cancer growth and metastasis through inducing degradation of PDGFRα protein. Cell Discov. 2017;3(1):1–3.
Acknowledgements
The authors acknowledge University of Kashmir for providing necessary facilities to complete this work.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
All authors contributed equally and made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas. That is, revising or critically reviewing the article; giving final approval of the version to be published; agreeing on the journal to which the article has been submitted; and, confirming to be accountable for all aspects of the work.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors wish to confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mohi-ud-din, R., Chawla, A., Sharma, P. et al. Repurposing approved non-oncology drugs for cancer therapy: a comprehensive review of mechanisms, efficacy, and clinical prospects. Eur J Med Res 28, 345 (2023). https://doi.org/10.1186/s40001-023-01275-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40001-023-01275-4