
Abstract
Background
This study focuses on the identification of conserved genes involved in myocardial infarction (MI), and then analyzed the differentially expressed genes (DEGs) between the incident and recurrent events to identify MI-recurrent biomarkers.
Methods
Gene expression data of MI peripheral blood were downloaded from GSE97320 and GSE66360 datasets. We identified the common DEGs in these two datasets by functional enrichment analysis and protein–protein interaction (PPI) network analysis. GSE48060 was further analyzed to validate the conserved genes in MI and to compare the DEGs between the incident and recurrent MI.
Results
A total of 477 conserved genes were identified in the comparison between MI and control. Protein–protein interaction (PPI) network showed hub genes, such as MAPK14, STAT3, and MAPKAPK2. Part of those conserved genes was validated in the analysis of GSE48060. The DEGs in the incident and recurrent MI showed significant differences, including RNASE2 and A2M-AS1 as the potential biomarkers of MI recurrence.
Conclusions
The conserved genes in the pathogenesis of MI were identified, benefit for target therapy. Meanwhile, some specific genes may be used as markers for the prediction of recurrent MI.
Similar content being viewed by others


Background
Myocardial infarction (MI) is defined as myocardial cell death due to prolonged ischemia [1]. Worldwide, about 15.9 million MI occurred in 2015. An MI was one of the top five most expensive conditions during inpatient hospitalizations in the US, with a cost of about $11.5 billion for 612,000 hospital stays as estimated in 2011 [2]. The main treatment strategy of MI is myocardial revascularization by the percutaneous coronary intervention (PCI) combined with management of cardiovascular risk factors [3]. Biomarkers are measurable and quantifiable biological parameters which serve as indices for health and physiology assessments. Diagnosis of MI is generally made by combining observation changes in a surface electrocardiogram (ECG) and blood levels of sensitive and specific biomarkers. Overall, the preferred biomarker for each specific category of MI is cTn (I or T) due to its high myocardial tissue specificity as well as high clinical sensitivity [1]. If a cTn assay is not available, the best alternative is MB (muscle/brain) fraction of creatine kinase (CKMB). Elevation of cTn or CKMB in the blood reflects injury leading to necrosis of myocardial cells [1]. In addition, myoglobin, N-terminal proBrain natriuretic peptide, and lactate dehydrogenase have also been considered as clinical diagnosis biomarkers of MI [4]. However, how these biomarkers function myocardial cells injury and necrosis are unclear.
In this study, we identified the conserved genes to investigate the molecular mechanism underlying MI development. Incident MI is defined as the first MI for patients, and it is considered to be a recurrent MI if characteristics of MI occur after 28 days following an incident MI [1, 5]. Differences between first and recurrent events on gene expression profiling are poorly described. Thus, we studied potential differences in the gene expression between patients with an incident and recurrent MI. In addition, little is known of the risk factors of recurrent MI at the transcriptome level. To address this issue, we further detected the potential biomarkers associated with recurrent MI occurrence.
Methods
Datasets
We searched the keywords “myocardial infarction”, “peripheral blood”, “GPL570” in the GEO datasets, and obtained 3 GEO datasets-GSE97320, GSE66360 and GSE48060.
GSE97320 and GSE66360 included gene expression profiles of peripheral blood from patients with MI and normal controls. GSE48060 contained gene expression profiling of patients with incident MI and that with recurrent events as well as normal controls. The platform used in these three datasets is GPL570 HG-U133_Plus_2 Affymetrix Human Genome U133 Plus 2.0 Array.
Differentially expressed gene (DEGs) screen
Gene expression data were first downloaded from each dataset, and the expression levels of genes in each sample were extracted from Series Matrix File(s). And then, R was used to pre-process the downloaded raw data via background correction and quantile normalization. Using Perl [6] probes were transformed into genes. Subsequently, “impute” package [7] was applied to complement the missing expression with its adjacent value.
To screen DEGs between the MI group and the control group, Limma [8] package in R was used. DEGs were screened with |log2(fold change)| > 0.45 and P < 0.05.
Functional enrichment analysis
To obtain the biological function and signaling pathways of conserved genes, GOstats and clusterProfiler [9] packages were used to detect gene ontology categories and KEGG pathways. The threshold of GO function and KEGG pathway of DEGs was all set as P < 0.05.
Protein–protein interaction (PPI) network analysis
To gain insights into the interaction between proteins encoded by DEGs, the database of HPRD [10], BIOGRID [11], and PIP [12] were used to retrieve the predicted interactions of the conserved genes. Then, the PPI network was visualized by the Cytoscape 3.2.1 [13]. A node in the PPI network denotes protein, and the edge denotes the interactions. Cytocluster was further performed to identify the sub-modules.
Statistical analysis
Data were expressed as mean ± SD. A value of P < 0.05 was considered significant.
Results
Identification of conserved genes in MI
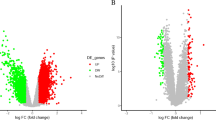
To identify conserved genes involved in MI, comparisons between patients with MI and normal individuals were performed to identify differentially expressed genes (DEGs)in two datasets (GSE97320 and GSE66360), which included gene expression profiles in peripheral blood of patients with MI. A total of 2723 DEGs were identified as the fold change > 1.5 and P value < 0.05 in GSE97320, consisting of 1568 upregulated and 1137 downregulated genes (Fig. 1). In GSE66360, 2486 genes including 1141 upregulated genes and 1345 downregulated genes were differentially expressed between patients with MI and healthy individuals (Fig. 2). The genes regulated consistently in GSE97320 and GSE66360 were defined as the conserved genes. A total of 477 conserved genes were differentially expressed in both datasets, including 289 upregulated genes and 188 downregulated genes with the same consistently changed direction (Table 1). These conserved genes may play an important role in the development of MI.

Heat maps for the DEGs in the microarray of the MI patients and healthy controls from dataset GSE97320. The x-axis represents the samples and y-axis indicates the DEGs

Heat maps for the DEGs in the microarray of the MI patients and healthy controls from dataset GSE66360. The x-axis represents the samples and y-axis indicates the DEGs
Functional enrichment analysis and biological network analysis of the conserved genes
To study the biological function of the 477 conserved genes identified, GO enrichment and KEGG pathway analysis were performed. The GO enrichment analysis revealed 211 GO biological processes (Table 2). Response to lipopolysaccharide, response to molecule of bacterial origin and immune system process were the most significantly enriched biological processes. In addition, 23 KEGG pathways were identified through analyzing the conserved genes, among which osteoclast differentiation was considered as the most remarkably enriched pathway (Table 2).
To investigate the interaction between the proteins encoded by the conserved genes, protein–protein interaction (PPI) network was employed (Fig. 3). Then, further analysis of critical modules by Cytocluster was carried out. 16 key genes such as MAPK14, STAT3, and MAPKAPK2, were found according to the frequency of genes in critical modules their regulation, which was as follow.
Genes | GSE97320 (LogFC) | GSE66360 (LogFC) |
|---|---|---|
MAPK14 | 1.349924696 | 0.536455945 |
STAT3 | 1.780855138 | 1.024539916 |
MAPKAPK2 | 0.765865504 | 0.778311524 |

PPI biological network of the conserved genes
Validation of the conserved genes using dataset GSE48060
GSE48060 dataset included gene expression profiles of the incident and recurrent MI. Comparison between incident MI and normal control (Comparison 1) revealed 89 DEGs, whereas comparison between recurrent MI and normal control (Comparison 2) showed 392 DEGs (Additional file 1: Table S1 and Table 2). To validate the conserved genes, we overlapped the DEGs of the incident and recurrent MI in GSE48060 and the 477 conserved genes gotten in Comparison 1 and Comparison 2. A total of 29 conserved genes was identified in the overlapping analysis.
Identification of the potential genes related to recurrent MI
To study the differences between primary and recurrent events of MI on gene expression profiling, we overlapped the DEGs in the incident and recurrent MI. In incident MI, 58 specific DEGs were identified (Table 3), accounting for 65% of the whole DEGs. And they were mainly enriched in 104 GO biological processes and 8 KEGG pathways (Additional file 1: Table S1). In recurrent MI, 361 specific genes were identified (Table 4) as 93% of the whole DEGs, and the functional enrichment analysis revealed 108 GO processes and 21 KEGG pathways (Additional file 2: Table S2). We further overlapped the specific genes in recurrent MI and conserved genes and found that RNASE2 and A2M-AS1 were potential genes associated with MI recurrence, the regulation of RNASE2 and A2M-AS1 were 0.629609108 and − 0.936691259.
Discussion
The present study not only identifies conserved genes and dysregulated pathways in MI but also reveals several hub genes, such as MAPK14, STAT3, and MAPKAPK2. Gene expression alterations of the incident and recurrent MI reveals significant differences. RNASE2 and A2M-AS1 were identified as potential genes associated with MI recurrence. Those genes could serve as potential biomarkers for MI occurrence or recurrence prediction and diagnosis.
MAPK14, also known as p38α, is one of the four p38 MAPKs, including α, β, δ, and ε isoforms and is the most abundant isoform in human cardiac tissue [14, 15]. P38 MAPK was first reported to be activated by ischemia/reperfusion (I/R) injury [16]. During myocardial ischemia, MAPK14 is found to contribute to infarction, and short-term intraischemic inhibition of this MAPK14 in the intact heart reduces infarction [17]. However, the effect of p38 MAPK on MI is controversial. Mitra et al. has demonstrated that p38 MAPK actually decreases ischemic load during MI, and plays a dual role in pro-survival as well as cardioprotective during ischemia in the absence of reperfusion [18]. The presented study showed MAPK14 upregulation in MI compared with normal tissue. MAPKAPK2 (MAPK-activated protein kinase 2) gene encodes a member of the Ser/Thr protein kinase family. This kinase is regulated through direct phosphorylation by p38 MAP kinase [19]. Inhibition of p38 MAPK leads to a significant decrease in the phosphorylation status of MAPKAPK2 [18]. In conjunction with p38 MAP kinase, MAPKAPK2 is known to be involved in many cellular processes including stress and inflammatory responses, nuclear export, gene expression regulation and cell proliferation [19]. Heat shock protein HSP27 was shown to be one of the substrates of MAPKAPK2and MAPKAPK2 phosphorylates Akt in neutrophils [20]. The isolated perfused rat heart reveals that global ischemia activates MAPKAPK2, and this activation is maintained during reperfusion [16]. MAPKAPK2 has been regarded as a biomarker in MI early stage and recovery [4]. STAT3 (Signal transducer and activator of transcription 3) is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury [21]. STAT3 deficiency causes enhanced susceptibility to myocardial ischemia/reperfusion injury and infarction with increased cardiac apoptosis, increased infarct sizes, and reduced cardiac function and survival [21]. In addition, knockout of STAT3 in mice treated with lipopolysaccharide leads to more cardiac THF production, and fibrosis [22]. Therefore, MAPK14, STAT3, and MAPKAPK2 might be regarded as biomarkers in MI, and the other hub genes are also deserved to be further studies.
Compared to incident cases of MI, recurrent cases of MI experienced more often heart failure, impaired left ventricular ejection fraction, and multivessel disease [23]. In this study, the gene expression profiling between first and recurrent MI showed significant differences, evidenced by that 93% of the whole DEGs in recurrent MI were its specific genes. RNASE2 and A2M-AS1 were regarded as potential genes associated with MI recurrence. RNASE2 gene encodes an enzyme in humans called eosinophil-derived neurotoxin (EDN) [24, 25]. EDN is one of the four major secretory proteins found in the specific granules of the human eosinophilic leukocyte and has been detected in eosinophils, specifically monocytes, and dendritic cells as well as in basophils and neutrophils [26]. EDN was first identified as a neurotoxin, and recent studies suggest that EDN plays a role in antiviral host defense, as a chemoattractant for human dendritic cells, and most recently, as an endogenous ligand for toll-like receptor (TLR) 2 [27]. TLR2 is reported to regulate myocardial ischemia, and sTLR2 may involve in the innate immune response in the pathogenesis of heart failure after acute MI [28]. Thus, we hypothesize that EDN/RNASE2 is likely to be associated with recurrent MI via its direct interactions with TLR2 and dendritic cells. Though little knowledge is available on A2M antisense RNA 1 (A2M-AS1), the relationship between A2M and MI has been reported. The cardiac isoform of A2M (cardiac A2M) is considered as an early marker in cardiac hypertrophy and left ventricular mass in humans. And the further study reveals that cardiac A2M is a valuable marker for the diagnosis of MI diabetic patients and differentiating them from diabetic patients without MI [29]. Thus, the role of A2M-AS1 in recurrent MI need to be further investigated in the future study.
Conclusions
Lacking animal models and cell culture experiment validation are limitations to our study. As an alternative way of validation, here we used GSE48060 dataset to validate the conserved genes. However, further functional experiments are needed to investigate the role of these candidate genes in myocardial infarction though we have reviewed their related functions reported in the previous publication. Meanwhile, the single-nucleotide polymorphism of these candidates may be associated with the risk of heart disease, also deserving for the future investigation. In addition, though myocardial tissues well reflect the characteristics of the injury areas, the blood samples may facilitate clinical diagnosis and treatment via the target genes in the future.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- MI:
-
myocardial infarction
- DEGs:
-
differentially expressed genes
- PPI:
-
protein–protein-interaction
- PCI:
-
percutaneous coronary intervention
- ECG:
-
electrocardiogram
- CKMB:
-
MB (muscle/brain) fraction of creatine kinase
- GO:
-
gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- GEO:
-
gene expression omnibus
- MAPK:
-
mitogen-activated protein kinase
- MAPKAPK2:
-
MAPK-activated protein kinase 2
- STAT3:
-
signal transducer and activator of transcription 3
- EDN:
-
eosinophil-derived neurotoxin
- TLR2:
-
toll-like receptor 2
- RNASE2:
-
human ribonuclease 2
- A2M-AS1:
-
A2M antisense RNA 1
References
Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD, et al. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551–67.
GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet (London, England). 2015;388(10053):1545–602.
Itier R, Roncalli J. New therapies for acute myocardial infarction: current state of research and future promise. Future Cardiol. 2018;14(4):329–42.
Wu K, Zhao Q, Li Z, Li N, Xiao Q, Li X, et al. Bioinformatic screening for key miRNAs and genes associated with myocardial infarction. FEBS Open Bio. 2018;8(6):897–913.
Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, et al. Fourth universal definition of myocardial infarction. J Am Coll Cardiol. 2018;72(18):2231–64.
Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, et al. The Bioperl toolkit: perl modules for the life sciences. Genome Res. 2002;12(10):1611–8.
Hastie T, Tibshirani R, Narasimhan B, Chu G. Impute: imputation for microarray data. Oral History Rev. 2011;1:128–30.
Smyth GK. Limma: linear models for microarray data. In: Bioinformatics and computational biology solutions using R and Bioconductor. 2005. p. 397–420.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, et al. Human protein reference database–2009 update. Nucleic Acids Res. 2009;37(Database issue):D767–72.
Chatr-Aryamontri A, Breitkreutz BJ, Heinicke S, Boucher L, Winter A, Stark C, et al. The BioGRID interaction database: 2013 update. Nucleic Acids Res. 2013;41(Database issue):D816–23.
McDowall MD, Scott MS, Barton GJ. PIPs: human protein-protein interaction prediction database. Nucleic Acids Res. 2009;37(Database issue):D651–6.
Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–2.
Ren J, Zhang S, Kovacs A, Wang Y, Muslin AJ. Role of p38alpha MAPK in cardiac apoptosis and remodeling after myocardial infarction. J Mol Cell Cardiol. 2005;38(4):617–23.
Meng W, Veluchamy A, Hebert HL, Campbell A, Colhoun HM, Palmer CNA. A genome-wide association study suggests that MAPK14 is associated with diabetic foot ulcers. Br J Dermatol. 2017;177(6):1664–70.
Bogoyevitch MA, Gillespie-Brown J, Ketterman AJ, Fuller SJ, Ben-Levy R, Ashworth A, et al. Stimulation of the stress-activated mitogen-activated protein kinase subfamilies in perfused heart. p38/RK mitogen-activated protein kinases and c-Jun N-terminal kinases are activated by ischemia/reperfusion. Circ Res. 1996;79(2):162–73.
Kumphune S, Bassi R, Jacquet S, Sicard P, Clark JE, Verma S, et al. A chemical genetic approach reveals that p38alpha MAPK activation by diphosphorylation aggravates myocardial infarction and is prevented by the direct binding of SB203580. J Biol Chem. 2010;285(5):2968–75.
Mitra A, Ray A, Datta R, Sengupta S, Sarkar S. Cardioprotective role of P38 MAPK during myocardial infarction via parallel activation of alpha-crystallin B and Nrf2. J Cell Physiol. 2014;229(9):1272–82.
Hoover HE, Thuerauf DJ, Martindale JJ, Glembotski CC. alpha B-crystallin gene induction and phosphorylation by MKK6-activated p38. A potential role for alpha B-crystallin as a target of the p38 branch of the cardiac stress response. J Biol Chem. 2000;275(31):23825–33.
Rane MJ, Coxon PY, Powell DW, Webster R, Klein JB, Pierce W, et al. p38 Kinase-dependent MAPKAPK-2 activation functions as 3-phosphoinositide-dependent kinase-2 for Akt in human neutrophils. J Biol Chem. 2001;276(5):3517–23.
Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res. 2004;95(2):187–95.
Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ Res. 2009;104(2):e9–18.
Myftiu S, Sulo E, Burazeri G, Daka B, Sharka I, Shkoza A, et al. Clinical profile and management of patients with incident and recurrent acute myocardial infarction in albania—a call for more focus on prevention strategies. Zdr Varst. 2017;56(4):236–43.
Mastrianni DM, Eddy RL, Rosenberg HF, Corrette SE, Shows TB, Tenen DG, et al. Localization of the human eosinophil Charcot–Leyden crystal protein (lysophospholipase) gene (CLC) to chromosome 19 and the human ribonuclease 2 (eosinophil-derived neurotoxin) and ribonuclease 3 (eosinophil cationic protein) genes (RNS2 and RNS3) to chromosome 14. Genomics. 1992;13(1):240–2.
Rosenberg HF, Tenen DG, Ackerman SJ. Molecular cloning of the human eosinophil-derived neurotoxin: a member of the ribonuclease gene family. Proc Natl Acad Sci USA. 1989;86(12):4460–4.
Rosenberg HF. Eosinophil-derived neurotoxin (EDN/RNase 2) and the mouse eosinophil-associated RNases (mEars): expanding roles in promoting host defense. Int J Mol Sci. 2015;16(7):15442–55.
Rosenberg HF. Eosinophil-derived neurotoxin/RNase 2: connecting the past, the present and the future. Curr Pharm Biotechnol. 2008;9(3):135–40.
Zhang S, Liu W, Liu X, Qi J, Deng C. Biomarkers identification for acute myocardial infarction detection via weighted gene co-expression network analysis. Medicine (Baltimore). 2017;96(47):e8375.
Annapoorani P, Dhandapany PS, Sadayappan S, Ramasamy S, Rathinavel A, Selvam GS. Cardiac isoform of alpha-2 macroglobulin—a new biomarker for myocardial infarcted diabetic patients. Atherosclerosis. 2006;186(1):173–6.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
LQ contributed to data analysis and drafting manuscript; XL designed research, drafted and revised the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
The top 50 significant GO biological processes and all KEGG pathways enriched by the specific genes in incident MI.
Additional file 2: Table S2.
The top 50 significant GO biological processes and all KEGG pathways enriched by the specific genes in recurrent MI.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Qiu, L., Liu, X. Identification of key genes involved in myocardial infarction. Eur J Med Res 24, 22 (2019). https://doi.org/10.1186/s40001-019-0381-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40001-019-0381-x