
Abstract
Background
Pycnodysostosis is an autosomal recessive skeletal dysplasia, the prevalence of which is estimated to be low (1 per million). Nevertheless, in recent years we have found 27 affected individuals from 22 families in Ceará State, a region of the Brazilian Northeast, giving a local prevalence of 3 per million. This local prevalence associated with a high parental consanguinity, suggesting a possible founder effect, prompted us to perform a molecular investigation of these families to test this hypothesis.
Methods
The CTSK gene was sequenced by the Sanger method in the patients and their parents. In addition to 18 families from Ceará, this study also included 15 families from other Brazilian regions. We also investigated the origin of each family from the birthplace of the parents and/or grandparents.
Results
We have studied 39 patients, including 33 probands and 6 sibs, from 33 families with pycnodysostosis and identified six mutations, five previously described (c.436G>C, c.580G>A, c.721C>T, c.830C>T and c.953G>A) and one novel frameshift (c.83dupT). This frameshift variant seems to have a single origin in Ceará State, since the haplotype study using the polymorphic markers D1S2344, D1S442, D1S498 and D1S2715 suggested a common origin. Most of the mutations were found in homozygosity in the patients from Ceará (83.3 %) while in other states the mutations were found in homozygosity in half of patients. We have also shown that most of the families currently living outside of Ceará have northeastern ancestors, suggesting a dispersion of these mutations from the Brazilian Northeast.
Conclusions
The high frequency of pycnodysostosis in Ceará State is the consequence of the high inbreeding in that region. Several mutations, probably introduced a long time ago in Ceará, must have spread due to consanguineous marriages and internal population migration. However, the novel mutation seems to have a single origin in Ceará, suggestive of a founder effect.
Similar content being viewed by others
Background
Pycnodysostosis (OMIM 265800) (Online Mendelian Inheritance in Man) is a rare autosomal recessive disorder, classified in the skeletal dysplasias group with increase of bone density [1]. Besides the altered bone density, the radiological findings include parietal and frontal bossing, delayed closure of sutures and fontanels, obtuse mandibular angle, susceptibility to fractures and acroosteolysis of the distal phalanges [2, 3]. This skeletal dysplasia is characterized by disproportionally short stature associated with a typical clinical phenotype that includes a peculiar facial appearance and hand brachydactyly with short distal phalanges [2, 3]. Hypoplasia of the maxilla, dental crowding, groove palate, nail and clavicle hypoplasia and recurrent respiratory infections are other signs seen in many patients [4, 5]. Lifespan, intelligence and sexual development are often normal [3].
Pycnodysostosis was first described in 1962 by Maroteaux and Lamy [2] but its molecular basis was only elucidated in 1996 by Gelb and collaborators [6]. Studying a large consanguineous Israeli Arab family, these authors found a common genomic region homozygous-by-descent in all 16 individuals affected by pycnodysostosis in the chromosome 1 (1q21) [6]. Among the genes located in this region, the cathepsin K (CTSK) was considered a strong candidate because the encoded protein—lysosomal cysteine protease—is highly expressed in osteoclasts [6]. Sequencing of the CTSK gene in several individuals with a suggestive phenotype of pycnodysostosis has provided evidence for mutations in this gene [6–26].
The CTSK gene is approximately 12 kb in size (Transcript ID: ENST00000271651.7) and contains eight exons (GenBank NM_000396.3). The codon for the translation initiator methionine (ATG) is located in exon 2, whereas the termination codon (TGA) is located in the middle of exon 8 [2] (Fig. 1). Due to the potent collagenolytic action in the collagen type I molecule, the major constituent of the bone matrix, the lysosomal cysteine protease, is prominent for its role in the bone remodeling [2, 3].

Structure of cathepsin K gene showing the distribution of the 51 reported mutations. There are 31 Missenses mutations (black), 9 frameshifts and 1 codon deletion mutations (green), 5 nonsense mutations (red), 4 splice mutations (blue) and 1 stop codon mutation (orange). Exon 1 and the final portion of exon 8 are non-coding (black). The exons are colored according to region of protein they encode. Yellow represents the Pre region (15 amino acids), blue indicates the Pro region (99 amino acids) and red represents the mature protein region (215 amino acids). The mutations found in the present study are into the red boxes and the novel mutation is highlighted with a red arrow. For more details about the described mutations see Additional file 2
There is no specific epidemiologic study to estimate the prevalence of pycnodysostosis, but some authors have estimated it to be between 1 and 1.7 per million based on the number of cases described in the literature worldwide [2, 27–29].
In spite of the rarity of pycnodysostosis, in recent years we have found 27 affected individuals from 22 families (59 % consanguineous), all living in Ceará State, a region with a population of 8,904,459 inhabitants located in the Brazilian Northeast, giving a local prevalence of 3 per million. This high frequency of pycnodysostosis prompted us to perform a molecular investigation of the CTSK gene in these families to evaluate the hypothesis of a possible founder effect. For this survey, we were able to study a total of 18 families from Ceará and also 15 families from other Brazilian regions. Surprisingly, we found five previously described mutations (c.436G>C, c.580G>A, c.721C>T, c.830C>T, and c.953G>A) and one novel frameshift mutation (c.83dupT), suggesting that the high inbreeding of the families might be the cause of the high prevalence in that region. The observation of the same mutations in other Brazilian regions, also associated with high inbreeding and with northeastern ancestors, suggests a dispersion of these mutations from the Brazilian Northeast. On the other hand, a founder effect might explain the novel mutation.
Methods
Patients
A total of 33 families were studied and included 39 patients (33 probands and 6 sibs) and 46 parents. In addition to the clinical and radiological data for each patient, information regarding the birthplace of the respective parents and grandparents was collected. This study had the approval of the Ethics Committee of the State University of Campinas (Comitê de Ética em Pesquisa da Faculdade de Ciências Médicas, Universidade Estadual de Campinas), under the number CEP-671.589, and written informed consent was obtained from each patient and/or from their respective relatives.
Sequencing analysis
The genomic DNA was extracted by the standard phenol–chloroform protocol from peripheral blood samples obtained from patients and their relatives. The exons and exon–intron boundaries were amplified by polymerase chain reaction (PCR) using primers designed by us (see Additional file 1). PCRs were performed in the presence of 50 ng of genomic DNA, 1× KCl buffer, 0.15 U Taq Polymerase, 200 μM each deoxynucleotide triphosphate (dNTP, Thermo Scientific), 0.5 pmol each primer and water to final volume of 10 μL. The PCR products were visualized on 1.5 % agarose gels. The amplicons produced by PCR were purified and were submitted to the bidirectional direct sequencing using the BigDye Terminator Cycle Sequencing Ready Reaction Kit version 3.1 (Applied Biosystems, Foster City, CA, USA), and were analyzed with an ABI 3500 XL automatic sequencer (Applied Biosystems®) using standard methods.
Sequencing data were analyzed through the CodonCodeAligner version 4.1.1 software and the mutations found were compared with online data banks (Ensembl, HGMD®, NCBI, dbSNP and LOVD®). The novel mutation was valued by online prediction mutation software (http://www.mutationtaster.org/). In addition, 100 alleles from 50 control individuals were investigated for the novel mutation.
Haplotype study
Four FAM-labeled microsatellite markers (Fluorescein Amidite) (D1S2344, D1S442, D1S498 and D1S2715) previously used in other similar studies [11, 29–31] were selected from the literature. The primer sequences and physical distance of the markers to CTSK gene were obtained from Ensembl (http://www.ensembl.org/index.html). PCR conditions were performed as described in the sequencing analysis and the primers are indicated in Additional file 1. The amplicons were genotyped using Formamida Hi-Di™ (Applied Biosystems®), 600 LIZ®v2.0 size Standard (Applied Biosystems®) and ABI 3500 XL (Applied Biosystems®). Genotyping data were analyzed using GeneMapper4.1® software.
Results
The sequencing analyses from 33 probands, six sibs, and their parents are summarized in Table 1. The parental origin of the allele could be identified, except in the compound heterozygous individuals from cases 25 and 31, since DNA from both parents was unavailable. Recurrence occurred in nine families, but three were unavailable for study.
Although the pycnodysostosis phenotype is well known, the main clinical and radiological findings of two patients here studied are shown in Fig. 2.

Clinical and radiological findings of two patients affected by pycnodysostosis. a–b The typical facial appearance of the pycnodysostosis (large frontal, midfacial hypoplasia, low set ears, and beaked nose), here observed in the patient 27. c–d Cranial X-rays of patient 27 showing increased bone density, opened sutures, parietal bossing and obtuse mandibular angle. e The hands of patient 27 showing brachydactyly. f X-rays of the hands of the patient 2 showing increased bone density, brachydactyly and acroosteolysis of the distal phalanges, mainly observed in both second fingers
Parental consanguinity was mentioned by 13 families (39. 4 %). In three of them (cases 7, 19 and 23), the parents recognized they are related but were unaware of their degree of kinship, and were therefore interpreted as having a distant consanguinity (F < 1/64) (F = coefficient of inbreeding) (Table 1).
Mutations found in patients from Ceará
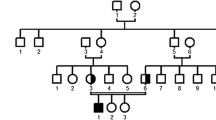
Rather than a single mutation, five different mutations were found in Ceará State, four of which have been previously described: c.436G>C (p.G146R), c.580G>A (p.G194S), c.721C>T (p.R241*), c.953G>A (p.C318Y) and a novel one c.83dupT (p.W29Mfs*10) (Table 1). The geographic distribution of mutations in Ceará State shows that the majority are concentrated in small cities (Fig. 3), especially the novel mutation, that seems to have a single origin in a small region on Ceará’s Northwestern coast.

Distribution of mutations found in patients from Ceará (1–16). Each circle represents a patient carrying one specific mutation (homozygous) or two (compound heterozygous), according to the colors in the caption
The novel mutation (c.83dupT) is a duplication of a thymine in position 83 in the exon 2 of the cDNA (Fig. 4). This frameshift mutation should impact the protein function, since it disrupts ten amino acids at the end of the exon 2 (W29Mfs*10). As expected, analysis in silico predicted this variation as a pathogenic mutation, and moreover, it was absent in 100 alleles from control individuals. A set of all described mutations is shown in Fig. 1 and Additional file 2.

Electropherogram showing a novel frameshift mutation c.83dupT (p.W29Mfs*10) found in Brazilians patients. The red arrow indicates a duplication of a thymine in homozygosity found in a patient with pycnodysostosis and below is the sequencing of a control individual
Mutations found in patients from other States
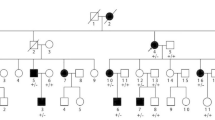
All the mutations found in Ceará, except one (c.580G>A), were also observed in other Brazilian regions (Fig. 5). The c.830C>T mutation was observed in a single family from the South region (Table 1), making a total of six different mutations detected in the complete survey.

Distribution of mutations according to actual address of patients 19–33. Each circle represents a patient carrying one specific mutation (homozygous) or two (compound heterozygous), according to the colors in the caption. AC Acre, AL Alagoas, AM Amazonas, AP Amapá, BA Bahia, CE Ceará, ES Espírito Santo, GO Goiás, MA Maranhão, MG Minas Gerais, MS Mato Grosso do Sul, MT Mato Grosso, PA Pará, PB Paraíba, PE Pernambuco, PI Piauí, PR Paraná, RJ Rio de Janeiro, RN Rio Grande do Norte, RO Rondônia, RR Roraima, RS Rio Grande do Sul, SC Santa Catarina, SE Sergipe, SP São Paulo, TO Tocantins. More information about patients’ origin and precedence is available in Additional file 3
When we analyzed the family background regarding the parents and grandparents birthplaces of patients currently living outside Ceará, we found that all but two (31 and 33) have an ancestor coming from the Northeast region, close to Ceará State (see Additional file 3). Patient 31 is from the backlands of São Paulo State and just recognizes local and European ancestor in his family. Patient 33 is from the hinterland of Rio Grande do Sul State and has Italian ancestors.
Haplotype study
The most frequent mutation in the whole casistic was the c.721C>T (16/33), followed by the c.436G>C (11/33) and the novel mutation c.83dupT (9/33), the latter being concentrated in Ceará state. For this novel mutation, we performed a haplotype study, aiming to test a founder effect hypothesis. Nine patients carrying this novel mutation and their parents were analyzed to identify the haplotype phase. Eight out of 14 chromosomes carrying the c.83dupT mutation also carried a master haplotype of 5.6 cM (according to Genethon), whereas all chromosomes but one from Patient 5, carried a preserved piece of the master haplotype, characterized by markers D1S498 and D1S2715 (Fig. 6a). This preserved region in almost all chromosomes from patients with the same mutations strongly suggests that these patients share a common ancestor. Figure 6b shows the genetic and physical localization from markers and CTSK gene given by Genethon and Ensembl, respectively. In this same figure, we can notice that apart from being different the proportional distance between the markers in the two maps, the order of markers D1S2344 and D1S442 has changed.

a Haplotypes of nine patients with pycnodysostosis carrying the novel frameshift mutation. b Localization and distance of markers and CTSK gene. The markers used in the present study are highlighted. The cathepsin K (CTSK) gene is located between markers D1S442 and D1S498, according to Ensembl. P paternal allele, M maternal allele, cM centiMorgan, Mb mega base pairs, bp base pair
Discussion
Inbreeding population in Ceará State leading to high frequency of pycnodysostosis
Due to the observed high frequency of pycnodysostosis in Ceará State, this survey first focused on the molecular study of patients coming from that region. We have found five different mutations in these patients (c.83dupT, c.436G>C, c.580G>A, c.721C>T, c.953G>A) (Table 1). Although 33.3 % of the families had referred consanguinity, most of the mutations (83.3 %) were found in homozygosity (Table 1), which strongly suggests that this is the real consanguinity rate among these families, probably reflecting a distant consanguinity in some families. Therefore, the initial hypothesis of a founder effect was ruled out and we now propose that the high consanguinity rate is the main cause of the high frequency of pycnodysostosis in this region. The consanguinity rate in the whole Brazilian Northeast is widely recognized as being higher than in other Brazilian regions [32, 33]. While the overall mean value of frequency of consanguineous marriages in Brazil is estimated to be 4.8 %, in Northeastern Brazil this rate varies from 6 to 12 % [34]. According to Freire-Maia, 1957 [33], the mean consanguinity rate in Ceará State is close to 9 %. However, some small and particular areas in the hinterland of the Brazilian Northeast reveal a consanguinity rate as high as 60 % [34].
Consanguineous marriages with the common ancestor(s) placed more distant than F ≤ 1/64, at least in theory, would result in an insignificant probability of homozygosity by descent. However, deleterious autosomal recessive alleles may remain hidden in a heterozygous state within some families for many generations, and then consanguineous marriages between mutation carriers, even with distant consanguinity, could cause them to come to the surface [34]. Since the probability of a carrier finding a partner in the general population who bears the same mutation is very small, this effect is more astonishing for rare diseases [35]. Thus, if we examine a group of families affected by proven autosomal recessive diseases, we must find a higher consanguinity rate than in the original population from which these families come [34].
Novel mutation (c.83dupT, p.W29Mfs*10) and haplotype study
Since the novel mutation was found to be concentrated on the Northwest coast of Ceará State (Fig. 3), we hypothesize this mutation could have been originated or introduced by immigrants in some of those cities and then spread due to consanguineous marriages as a founder effect. The haplotype study showed a master haplotype present in 8 of 14 chromosomes carrying the novel mutation and a preserved region including markers D1S498 and D1S2715 in 13 of 14 chromosomes (Fig. 6a). This reinforces the founder effect hypothesis for this mutation and suggests that the patients carrying the novel mutation have a related common ancestor.
The variations of length found in markers D1S2344 and D1S442 among our patients can be explained by the physical distance of these markers to CTSK gene given by Ensembl. Whereas marker D1S498 is about 0.5 Mb and the marker D1S2715 is 3 Mb far away from cathepsin K gene, the markers D1S2344 and D1S442 are about 5 Mb distant from this gene (Fig. 6b). The more distant the two loci are, the higher the probability of recombination is between them. Although many haplotype studies still use these same markers and consider the genetic localization given by Genethon (Fig. 6b), recombination in the first two was also detected by other authors [11, 29, 31].
Geographic origin and distribution of mutations found in this survey
Unlike the novel mutation, the most frequent mutations c.721C>T and c.436G>C were found more disperse in Ceará State (Fig. 3) and also in other Brazilian regions (Fig. 5), probably suggesting that their dispersion began earlier. The c.721C>T (p.R241*) mutation has already been reported in patients from Portugal [8, 13], Spain [8, 9, 13], Italy [13], Canada [9], and Mexico [31]. The c.436G>C (p.G146R) mutation, however, was identified in patients from Tunisia [25] and Morocco [6]. Moreover, both mutations were already described in other Brazilian patients previously reported, but unfortunately the origin of the patients was not stated [16].
The mutation c.953G>A (C318Y), observed in just one family from Ceará State, was first reported in three Brazilian patients—two in homozygosity and in one case as a compound heterozygous [16]. Recently, it was detected in an African patient [36]. Another mutation, the c.580G>A (p.G194S), described once in a patient living in a region in the South of the Ceará State, was detected in an Italian patient so far [13]. We believe that, unlike the novel mutation c.83dupT, never identified in other patients outside of Brazil, the other mutations (c.436G>C, c.721C>T and c.953G>A) must have been introduced in Northeast Brazil a long time ago, and then spread to other regions due to consanguineous marriages, since most of these patients have ancestors in the Northeast region (see Additional file 3). This is particularly interesting when we look at the novel mutation c.83dupT, which was found in a compound heterozygous state in two patients not living Ceará State (patients 26 and 30). In patient 26, the novel mutation was present in the maternal allele, whose ancestor is from Currais Novos, a city located in Rio Grande do Norte, a neighboring State of Ceará (Fig. 5). In patient 30, the novel mutation was in the paternal allele, whose ancestor is from Ceará State (Additional file 3).
The missense mutation, c.830C>T (p.A277V), observed in homozygosity in a single patient from the southern Brazilian region (Table 1; Fig. 5) was originally described in a patient with Belgian and Algerian background [7] and was then found in Japanese [9, 10, 19] and Pakistani patients [13, 29]. In the family studied here, the ancestors are from Italy. The distant consanguinity (F = 1/256) in this family could support the idea that this mutation arises de novo in an ancestor and remained hidden for generations until a consanguineous marriage revealed it, as expected with rare diseases [35]. Regarding this mutation, it is also worth noting that it is described as a hot spot because of its localization in the CpG dinucleotides regions [2] and near the catalytic domain of the protein [37].
These results suggest that the majority of the mutations found in the families outside Ceará State were spread to other regions by the internal migration of the population. The internal migration in Brazil is usually motivated by economic reasons, when young people leave rural areas or small towns in search of job opportunities in urban centers or larger cities [38]. Bearing in mind that the Northeast region has the lowest socioeconomic level among the five Brazilian regions, the internal migration usually follows the route from Northeast (with a negative migration number) to Southeast (with positive migration), with São Paulo State as the destination of most of migrants [39].
This internal migration from Northeast to Southeast also increases the consanguinity rate in the latter region [32, 40]. In the present study, we have observed that the real consanguinity rate in families outside Ceará State was also high (46.6 %).
Conclusions
The results of the present study suggest that the high consanguinity rate (83.3 %) in Ceará State, based on the homozygosity rate found, could be the main cause of high frequency of pycnodysostosis in that State. We also found a novel mutation (c.83dupT), that seems to have a single origin on the Northwestern coast of Ceará State, may have been spreading as a founder effect, since most of the patients carrying this mutation have the same haplotype, including markers D1S498 and D1S2715. Finally, by investigating the origin of the parental families, we suggest that the mutations found in the patients affected by pycnodysostosis outside Ceará State are derived from those found in Ceará State due to the process of internal migration. In these patients, the consanguinity is also high (46.6 %).
Abbreviations
- A:
-
alanine
- AC:
-
State of Acre
- AL:
-
State of Alagoas
- AM:
-
State of Amazonas
- AP:
-
State of Amapá
- BA:
-
State of Bahia
- bp:
-
base pair
- C or Cys:
-
cysteine
- c.:
-
cDNA localization
- cDNA:
-
complementary DNA, synthesized using a molecule of RNAm as template
- CE:
-
State of Ceará
- CEP:
-
Comitê de Ética em Pesquisa (Ethics Committee)
- cM:
-
centiMorgan
- CTSK:
-
cathepsin K
- D:
-
aspartic acid
- D1S442:
-
polymorphic microsatellite marker
- D1S498:
-
polymorphic microsatellite marker
- D1S2344:
-
polymorphic microsatellite marker
- D1S2715:
-
polymorphic microsatellite marker
- dbSNP:
-
single nucleotide polymorphism database
- del:
-
deletion
- DNA:
-
deoxyribonucleic acid
- dNTP:
-
deoxynucleotide triphosphate
- dup:
-
duplication
- E:
-
glutamic acid
- Ensembl :
-
genome browser for vertebrate that supports research in comparative genomics, evolution, sequence variation and transcriptional regulation
- ES:
-
State of Espírito Santo
- ext:
-
extension
- F:
-
phenilalanine
- FAM:
-
fluorescein amidite—a synthetic equivalent of fluorescein dye
- FCM:
-
Medical Sciences Faculty of Campinas
- FMJ:
-
Medical Sciences Faculty of Juazeiro do Norte
- FMUSP:
-
Medical Sciences Faculty, University of São Paulo
- fs*:
-
frameshift mutation
- G:
-
glycine
- GO:
-
State of Goiás
- H:
-
histidine
- HGMD:
-
Human Gene Mutation Database
- HGVS:
-
Human Genome Variation Society
- I:
-
isoleucine
- IC:
-
inbreeding coefficient, also known as “F” in population genetics
- ICF:
-
Informed consent form
- ins:
-
insertion
- K or Lys:
-
lysine
- Kb:
-
kilobase pairs (103 base pair)
- KCl:
-
potassium chloride
- L:
-
leucine
- LOVD:
-
Leiden Open (source) Variation Database
- M:
-
metionine
- MA:
-
State of Maranhão
- Mb:
-
mega base pairs (106 base pair)
- μL:
-
microliter (10−6 liter)
- μM:
-
micromoles (10−6 mol)
- MG:
-
State of Minas Gerais
- MgCl2 :
-
magnesium chloride
- MS:
-
State of Mato Grosso do Sul
- MT:
-
State of Mato Grosso
- N:
-
asparagine
- NCBI:
-
National Center for Biotechnology Information
- ng:
-
nanogram (10−9 g)
- NM:
-
reference number of human messenger RNA sequencing
- NP:
-
reference number of humam protein sequencing
- OCD:
-
osteochondrodysplasia
- OMIM:
-
Online Mendelian Inheritance in Man
- p.:
-
localization in protein (amino acid number)
- P:
-
proline
- PA:
-
State of Pará
- PB:
-
State of Paraíba
- PCR:
-
polymerase chain reaction
- PE:
-
State of Pernambuco
- PI:
-
State of Piauí
- pMol:
-
picomoles (10−12 mol)
- PR:
-
State of Paraná
- Q:
-
glutamine
- R or Arg:
-
arginine
- r.:
-
RNAm localization
- RJ:
-
State of Rio de Janeiro
- RN:
-
State of Rio Grande do Norte
- RNAm:
-
ribonucleic acid messenger
- RO:
-
State of Rondônia
- RR:
-
State of Roraima
- RS:
-
State of Rio Grande do Sul
- S or Ser:
-
serine
- SC:
-
State of Santa Catarina
- SE:
-
State of Sergipe
- SP:
-
State of São Paulo
- T:
-
threonine
- TO:
-
State of Tocantins
- U:
-
enzyme unit (conversion of 1 micromole of substrate per minute)
- UFSCAR:
-
Federal University of de São Carlos
- UNICAMP:
-
Universidade Estadual de Campinas (State University of Campinas)
- V:
-
valine
- W:
-
tryptofan
- X or *:
-
termination code
- Y:
-
tyrosine
References
Bonafé L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, Nishimura G, Sangiorgi L, Savarirayan R, Sillence D, Spranger J. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet Part A. 2015;9999A:1–24.
Xue Y, Cai T, Shi S, Wang W, Zhang Y, Mao T, Duan X. Clinical and animal research findings in pycnodysostosis and gene mutations of cathepsin K from 1996 to 2011. Orphanet J Rare Dis. 2011;6:20.
Motyckova G, Fisher DE. Pycnodysostosis: role and regulation of cathepsin K in osteoclast function and human disease. Curr Mol Med. 2002;2:407–21.
Alves N, Cantín M. Clinical and radiographic maxillofacial features of pycnodysostosis. Int J Clin Exp Med. 2014;7:492–6.
Testani E, Scarano E, Leoni C, Dittoni S, Losurdo A, Colicchio S, Gnoni V, Vollono C, Zampino G, Paludetti G, Della Marca G. Upper airway surgery of obstructive sleep apnea in pycnodysostosis: case report and literature review. Am J Med Genet A. 2014;164:2029–35.
Gelb B, Shi G, Chapman H, Desnick R. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science. 1996;273:1236–8.
Gelb B, Willner J, Dunn T. Paternal uniparental disomy for chromosome 1 revealed by molecular analysis of a patient with pycnodysostosis. Am J Hum Genet. 1998;62:848–54.
Hou WS, Brömme D, Zhao Y, Mehler E, Dushey C, Weinstein H, Miranda CS, Fraga C, Greig F, Carey J, Rimoin DL, Desnick RJ, Gelb BD. Characterization of novel cathepsin K mutations in the pro and mature polypeptide regions causing pycnodysostosis. J Clin Invest. 1999;103:731–8.
Nishi Y, Atley L, Eyre DE, Edelson JG, Superti-Furga A, Yasud T, Desnick RJ, Gelb BD. Determination of bone markers in pycnodysostosis: effects of cathepsin K deficiency on bone matrix degradation. J Bone Miner Res. 1999;14:1902–8.
Fujita Y, Nakata K, Yasui N, Matsui Y, Kataoka E, Hiroshima K, Shiba RI, Ochi T. Novel mutations of the cathepsin K gene in patients with pycnodysostosis and their characterization. J Clin Endocrinol Metab. 2000;85:425–31.
Haagerup A, Hertz JM, Christensen MF, Binderup H, Kruse TA. Cathepsin K gene mutations and 1q21 haplotypes in at patients with pycnodysostosis in an outbred population. Eur J Hum Genet. 2000;8:431–6.
Fratzl-Zelman N, Valenta A, Roschger P, Nader A, Gelb BD, Fratzl P, Klaushofer K. Decreased bone turnover and deterioration of bone structure in two cases of pycnodysostosis. J Clin Endocrinol Metab. 2004;89:1538–47.
Donnarumma M, Regis S, Tappino B, Rosano C, Assereto S, Corsolini F, Di Rocco M, Filocamo M. Molecular analysis and characterization of nine novel CTSK mutations in twelve patients affected by pycnodysostosis. Mutation in brief #961. Online. Hum Mutat. 2007;28:524.
Schilling AF, Mülhausen C, Lehmann W, Santer R, Schinke T, Rueger JM, Amling M. High bone mineral density in pycnodysostotic patients with a novel mutation in the propeptide of cathepsin K. Osteoporos Int. 2007;18:659–69.
Li H-Y, Ma H-W, Wang H-Q, Ma W-H. Molecular analysis of a novel cathepsin K gene mutation in a Chinese child with pycnodysostosis. J Int Med Res. 2009;37:264–9.
Bertola D, Amaral C, Kim C, Albano L, Aguena M, Passos-Bueno MR. Craniosynostosis in pycnodysostosis: broadening the spectrum of the cranial flat bone abnormalities. Am J Med Genet A. 2010;152A:2599–603.
Khan B, Ahmed Z, Ahmad W. A novel missense mutation in cathepsin K (CTSK) gene in a consanguineous Pakistani family with pycnodysostosis. J Investig Med. 2010;58:720–4.
Toral-López J, Gonzalez-Huerta LM, Sosa B, Orozco S, González HP, Cuevas-Covarrubias SA. Familial pycnodysostosis: identification of a novel mutation in the CTSK gene (cathepsin K). J Investig Med. 2011;59:277–80.
Matsushita M, Kitoh H, Kaneko H, Mishima K, Itoh Y, Hattori T, Ishiguro N. Novel compound heterozygous mutations in the cathepsin K gene in Japanese female siblings with pyknodysostosis. Mol Syndromol. 2012;8550:254–8.
Utokpat P, Panmontha W, Tongkobpetch S, Suphapeetiporn K, Shotelersuk V. Novel CTSK mutation resulting in an entire exon 2 skipping in a Thai girl with pycnodysostosis. Pediatr Int. 2013;55:651–5.
Zheng H, Zhang Z, He JW, Fu WZ, Zhang ZL. A novel mutation (R122Q) in the cathepsin K gene in a Chinese child with pyknodysostosis. Gene. 2013;521:176–9.
Pangrazio A, Puddu A, Oppo M. Exome sequencing identifies CTSK mutations in patients originally diagnosed as intermediate osteopetrosis. Bone. 2014;59:122–6.
Arman A, Bereket A, Coker A, Kiper PÖS, Güran T, Ozkan B, Atay Z, Akçay T, Haliloglu B, Boduroglu K, Alanay Y, Turan S. Cathepsin K analysis in a pycnodysostosis cohort: demographic, genotypic and phenotypic features. Orphanet J Rare Dis. 2014;9:60.
Ozdemir T, Atik T, Karaca E, Onay H, Ozkinay F, Cogulu O. A novel mutation in two families with pycnodysostosis. Clin Dirmophol. 2013;22:102–5.
Tinsa F, Hamouda S, Bellalah M, Bousnina D, Karboul L, Boussetta K, Bousnina S. Unusual feature of pycnodysostosis: pectus carinatum. La Tunisi Médicale. 2014;92:180–1.
Xue Y, Wang L, Xia D, Li Q, Gao S, Dong M, Cai T, Shi S, He L, Hu K, Mao T, Duan X. Dental abnormalities caused by novel compound heterozygous CTSK mutations. J Dent Res. 2015;94:674–81.
Mujawar Q, Naganoor R, Patil H, Thobbi AN, Ukkali S, Malagi N. Pycnodysostosis with unusual findings: a case report. Cases J. 2009;2:6544.
Karakurt L, Yilmaz E, Belhan O, Serin E. Pycnodysostosis associated with bilateral congenital pseudarthrosis of the clavicle. Arch Orthop Trauma Surg. 2003;123:125–7.
Naeem M, Sheikh S, Ahmad W. A mutation in CTSK gene in an autosomal recessive pycnodysostosis family of Pakistani origin. BMC Med Genet. 2009;10:76.
Gelb B, Edelson J, Desnick R. Linkage of pycnodysostosis to chromosome 1q21 by homozygosity mapping. Nat Genet. 1995;173:1236–8.
Johnson M, Polymeropoulos M, Vos H, Ortiz de Luna R, Francomano C. A nonsense mutation in the cathepsin K gene observed in a family with pycnodysostosis. Genome Res. 1996;6:1051–5.
Freire-Maia N. Genetic effects in Brazilian populations due to consanguineous marriages. Am J Med Genet. 1990;35:115–7.
Freire-Maia N. Inbreeding in Brazil. Am J Hum Genet. 1957;9:284–98.
Santos S, Kok F, Weller M. Inbreeding levels in Northeast Brazil: strategies for the prospecting of new genetic disorders. Genet Mol Biol. 2010;223:220–3.
Jaouad IC, Elalaoui SC, Sbiti A, Elkerh F, Belmahi L, Sefiani A. Consanguineous marriages in Morocco and the consequence for the incidence of autosomal recessive disorders. J Biosoc Sci. 2009;41:575–81.
Girbal I, Nunes T, Medeira A, Bandeira T. Pycnodysostosis with novel gene mutation and severe obstructive sleep apnoea: management of a complex case. BMJ Case Rep. 2013;2013:bcr2013200590.
Novinec M, Lenarčič B. Cathepsin K: a unique collagenolytic cysteine peptidase. Biol Chem. 2013;394:1163–79.
Sahota GS. An economic analysis of internal migration in Brazil. J Polit Econ. 1968;76:218–45.
Migration diversity in Brazil : where are the poor people? http://www.abep.nepo.unicamp.br/docs/anais/outros/5EncNacSobreMigracao/public_mig_div_bra.pdf. Accessed 3 Aug 2016.
Moreno CA, Cavalcanti DP: Consanguinidade parental em uma amostra de recém-nascidos do CAISM. In: Anais do XI Congresso Interno de Iniciação Científica da Unicamp. Campinas; 2003.
Authors’ contributions
ATF participated in the design of the study, carried out the molecular genetic studies, analyzed the data and wrote the manuscript. REM and AAP recruited the patients from Ceará. MPFV, MRM, MDG, KCA, DMJR, FTM, FRA, and MCA recruited patients from other regions of Brazil. CDP coordinated the study, participated in the revision of all patients, the molecular results, and in the discussion of the results and revision of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank all the patients and their families. We are also grateful to Thatiane Y Kanazawa, Cynthia Silveira, Karina C Silveira and Luciana Bonadia for the laboratorial support, Maria Dora Lacarrubba Flores for the clinical support, Maynara Cavalcante for her help in recovering the patients’ data from Ceará and the Sector of Educational Support of Faculty of Medical Sciences at the State University of Campinas. We also thank to Dr. Anthony Fensom, who kindly reviewed the text.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Consent for publication
This paper contains images and radiographies from two patients, who have signed the Informed consent form (ICF) agreeing to the use of their images in scientific publications. The head and skull images (6a–d) are from patient 27 and the hand images (6e–f) are from patient 2.
Ethics approval and consent to participate
This study had the approval of the Ethics Committee of the State University of Campinas (Comitê de Ética em Pesquisa da Faculdade de Ciências Médicas, Universidade Estadual de Campinas), under the number CEP-671.589.
Funding
This study was supported by the following Grant sponsors: CNPq 402008/2010-3 and 590148/2011-7. CAPES 3300017023p6. Fapesp 2015/22145-6.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Araujo, T.F., Ribeiro, E.M., Arruda, A.P. et al. Molecular analysis of the CTSK gene in a cohort of 33 Brazilian families with pycnodysostosis from a cluster in a Brazilian Northeast region. Eur J Med Res 21, 33 (2016). https://doi.org/10.1186/s40001-016-0228-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40001-016-0228-7