
Abstract
Inflammation is an essential defense mechanism in health; however, excessive inflammation contributes to the pathophysiology of several chronic diseases. Although anti-inflammatory drugs are essential for controlling inflammation, they have several side effects. Recent findings suggest that naturally derived compounds possess physiological activities, including anti-inflammatory, antifungal, antiviral, anticancer, and immunomodulatory activities. Therefore, this study aimed to investigate the anti-inflammatory effects and molecular mechanisms of 2,5,6-trimethoxy-p-terphenyl (TP1), extracted from the Antarctic lichen Stereocaulon alpinum, using in vitro models. TP1 treatment decreased the production of nitric oxide (NO) and reactive oxygen species (ROS) in LPS-stimulated Raw264.7 macrophages. Additionally, TP1 treatment significantly decreased the mRNA levels of pro-inflammatory cytokines (IL-1β, TNF-α, IL-6) and the mRNA and protein levels of the pro-inflammatory enzymes (inducible nitric oxide synthase and cyclooxygenase-2). Moreover, TP1 suppressed lipopolysaccharide-induced phosphorylation of the NF-κB and MAPK signaling pathways in Raw264.7 macrophages. Conclusively, these results suggest that TP1 ameliorates inflammation by suppressing the expression of pro-inflammatory cytokines, making it a potential anti-inflammatory drug for the treatment of severe inflammatory diseases.
Similar content being viewed by others


Introduction
Inflammation, the body’s “natural defense system” against injury and disease, has five representative signals: fever, redness, dysfunction, swelling, and ache [1]. Although inflammation is essential to maintain health, excessive inflammation can damage host cells and cause diseases [2]. Pattern recognition receptors (PRRs) recognize pathogen-associated molecular patterns (PAMPs), specialized motif of bacteria, viruses, and fungi, and trigger inflammatory responses through intracellular signaling [3]. Toll-like receptors (TLRs) are the most well-known members of the PRR family [4]. TLR4 recognizes lipopolysaccharides (LPS) [5]. LPS is a major component of the cell wall of gram-negative bacteria and can trigger the release of numerous inflammatory cytokines, resulting in an acute inflammatory response [6]. The binding of LPS to TLR4 induces the formation of myeloid differentiation primary response protein 88 (MyD88), interleukin-1 receptor-associated kinases (IRAKs), and TNF receptor-associated factor 6 (TRAF6) complex [7]. These processes can trigger intracellular signaling cascades by stimulating the AKT/NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways, including ERK1/2, JNKs, and p38 MAPKs [8]. Stimulation of these pathways can induce the expression of pro-inflammatory mediators, such as pro-inflammatory cytokines (interleukin-6 [IL-6], IL-1β, and tumor necrosis factor-alpha [TNF-α]) and pro-inflammatory enzymes, such as inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) [9].
Sepsis, one of the deadliest diseases worldwide, is an acute inflammatory disease caused by an uncontrolled inflammatory response [10]. Over the years, the incidence of sepsis has increased, and LPS is a major cause of sepsis [11, 12]. Although extensive studies have been performed to treat sepsis, no effective treatment has been developed [13]. Currently, sepsis is mainly managed using antibiotics, intravenous fluids, and vasoactive drugs [14]. Although ceftriaxone and vancomycin are commonly used antibiotics for sepsis treatment [15, 16], they have several side effects, including urolithiasis, nephrolithiasis, and maculopapular or erythematous rashes [17, 18]. Therefore, it is crucial to develop novel anti-inflammatory drugs with low toxicity for treating sepsis.
Natural compounds are recognized as sources of therapeutic agents owing to their structural diversity [19]. Moreover, 13 drugs based on natural products were approved for clinical use between 2005 and 2007 [20]. Antarctic plants produce secondary metabolites with various physiological activities as a survival strategy in extreme environments with low temperatures and high ultraviolet rays [21]. For example, lobaric acid, a metabolite of Streocaulon alpinum, inhibits protein tyrosine phosphatase 1B (PTP1B) [22]. Libertellenone G and libertellenone H, isolated from the Arctic fungus Eutypella sp., have high cytotoxicity and antibacterial activity [23]. New steroid compounds isolated from the Antarctic October coral Anthomastus bathyproctus exhibited anticancer effects on the human cancer cell lines MDA-MB-23, A-549, and HT-29 [24]. However, there is limited understanding of the biological activities of samples from the polar regions [25]. Therefore, natural compounds from polar regions are untapped resources for the development of natural compound-based medicines [26].
2,5,6-trimethoxy-p-terphenyl (TP1) is isolated from the Antarctic lichen S. alpinum, with a p-terphenyl backbone. p-Terphenyls are 1,4-diphenyl benzene derivatives with three phenyl rings connected by a single C–C bond [27]. p-Terphenyls possess several biological activities, including neuroprotective, antithrombotic, and anticoagulant activities [28]. However, studies have not yet investigated the anti-inflammatory effects and molecular mechanisms of several bioactive p-terphenyls. Therefore, this study aimed to investigate the anti-inflammatory effects and molecular mechanisms of TP1 in LPS-stimulated Raw264.7 macrophages.
Materials and methods
Reagents
TP1 was extracted by the Korea Polar Research Institute and dissolved in dimethyl sulfoxide (DMSO). Dulbecco’s modified Eagle’s medium (DMEM; high glucose) was obtained from Hyclone Laboratories Inc. (Marlborough, MA, USA), Fetal bovine serum (FBS) was purchased from Corning (Corning, NY, USA). Penicillin–streptomycin–glutamine (PSQ) was proucured from Gibco (Waltham, MA, USA). MTT and LPS from Escherichia coli O127:B8, N-(1-naphthyl) ethylenediamine dihydrochloride, phosphoric acid, sulfanilic acid, and nitrite ion standard solutions were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Dulbecco’s phosphate-buffered saline (DPBS) and Hank’s balanced salt solution (HBSS) were obtained from WELGENE (Gyeongsan, Korea). 2′7-Dicholrodihydrofluorecsein diacetate (DCF-DA) was procured from Cayman Chemical Company (Ann Arbor, MI, USA). Primers for quantitative reverse transcription-polymerase chain reaction (qRT-PCR) were purchased from Macrogen (Seoul, Korea). Primary antibodies against iNOS (#13120), COX-2 (#12282), phospho-p65 (#13346), phospho-JNK (#9255), phospho-ERK (#4370), and β-actin (#8457) were obtained from Cell Signaling Technology Inc. (Danvers, MA, USA). Goat anti-rabbit IgG (5220-0036) and goat anti-mouse IgG (5220-0341) antibodies were purchased from SeraCare Life Sciences, Inc. (Gaithersburg, MD, USA).
Cell culture
The mouse macrophage cell line RAW264.7 was provided by Dr. Sung Ho Ryu’s lab (POSTECH, Korea). The cells were cultured in DMEM supplemented with 10% FBS and 1% PSQ at 37 ℃ in a 5% CO2 incubator.
Cell viability
Raw264.7 macrophages were seeded in a 48-well plate (3 × 104 cells/well). After 24 h, cells were pretreated with TP1 for 30 min, treated with 0.1 µg/mL of LPS, and incubated for 24 h at 37 °C under 5% CO2. After removing the culture medium, the cells were treated with the MTT solution (1 mg/mL) for 2 h. Thereafter, the MTT solution was removed, followed by the addition of 200 µL of DMSO to dissolve the formed formazan crystals. Absorbance was measured at 570 nm using a microplate reader (Varioskan LUX Multimode Microplate Reader, Thermo Fisher Scientific Co.).
Measurement of nitric oxide production
Nitric oxide (NO) levels in the cell culture supernatants were measured using the Griess assay. Briefly, Raw264.7 macrophages were seeded in a 24-well plate (6 × 104 cells/well). After 24 h, the cells were pretreated with 25 μM of TP1 for 30 min, and then treated with LPS (0.1 μg/mL) and incubation at 37 °C in a 5% CO2 incubator for 24 h. The supernatants were collected from each well, transferred to a 96-well plate, and Griess reagent was added (reagents A and B at a 1:1 ratio). After 15 min, absorbance was measured at 550 nm using a microplate reader.
Measurement of reactive oxygen species production
The DCF-DA assay was used to measure reactive oxygen species (ROS) production. Briefly, Raw264.7 macrophages were seeded in a 24-well (6 × 104 cells/well) and incubated for 24 h. Thereafter, the cells were pretreated with TP1 (25 μM) for 30 min, and then treated with LPS (0.1 μg/mL) and incubated at 37 °C in a 5% CO2 incubator for 24 h. Cells were treated with DCF-DA for 30 min at 37 °C, followed by three washes with HBSS. ROS levels were detected by a fluorescence microscope (Zeiss, Jena, Germany) and analyzed using FACS (BD FACSVerse™, BD Biosciences).
qRT-PCR
Raw264.7macrophages were seeded in a 12-well plate (3 × 105 cells/well). After 24 h, cells were pretreated with TP1 (25 µM) for 30 min, and then treated with 0.1 µg/mL of LPS. After 24 h, the cells were washed thrice with DPBS and harvested. Total RNA was isolated from the cells using TRIzol reagent and reverse-transcribed to generate cDNA using a SimpliAmp Thermal Cycler (Applied Biosystems Co., Waltham, MA, USA). qRT-PCR was performed on a StepOnePlus Real-Time PCR System Cycler (Applied Biosystems Co.) using a Power SYBR Green PCR mix Cycler (Applied Biosystems Co.) and specific primers. The threshold cycle (Ct) values for the genes of interest were standardized with respect to the β-actin reference. The primer sequence for iNOS (Forward 5′-TGAAGAAAACCCCTTGTGCT-3′, Reverse 5′-TTCTGTGCTGTCCCAGTGAG-3′), COX-2 (Forward 5′-GAAGATTCCCT CCGGTGTTT-3′, Reverse 5′-CCCTTCTCACTGGCTTATGTAG-3′), Tnf-α (Forward 5′-ACGTGGAACTGGCAGAAGAG-3′, Reverse 5′-GGTCTGGGCCATAGAACTGA-3′), Il-1β (Forward 5′-AGGTCAAAGGTTTGGAAGCA-3′, Reverse 5′-TGAAGCAGCTATGGCA ACTG-3′), Il-6 (Forward 5’-TCTGAAGGACTCTGGCTTTG-3’, Reverse 5’-GATGGAT GCTACCAAACTGGA-3′), and β-actin (Forward 5′-ATGGAGGGGAATACAGCCC-3′, Reverse 5′-TTCTTT GCAGCTCCTTCGTT-3’).
Western blot analysis
Raw264.7 macrophages were seeded in a 60 mm plate (6 × 105 cells/well). Cells were pretreated with TP1 (25 µM) for 30 min, and then treatment with 0.1 µg/mL of LPS. Proteins were isolated from 100 µL of each sample at 30 min and 24 h intervals using RIPA buffer (10 mM Tris [pH 7.4], 150 mM of NaCl, 1 mM of EDTA [pH 8], 1% Triton X-100, 1% sodium deoxycholate, 30 mM of NaF, 1.5 mM of NaVO4, 1 mM of PMSF, and 1 mg/mL each of aprotinin, leupeptin, and pepstatin A), followed by sonication for 5 s thrice. The cell lysate was centrifugated at 130,000 rpm for 10 min at 4 ℃, and the protein content of the supernatant was collected and quantified using Bradford assay (Abcam, Cambridge, UK). Proteins were separated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto nitrocellulose (NC) membranes. Thereafter, the membranes were blocked with skim milk (5%) in 1X TBS-T buffer (Tris-buffered saline with 0.1% Tween-20) containing NaN3 for 30 min, followed by overnight incubation with primary antibodies at room temperature (RT). After washing with TBS-T for 30 min, the membranes were incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit/mouse IgG antibodies for 1 h at RT. Finally, the antibody reactions were detected using an ECL reagent (Cytiva, Malborough, MS, USA), and the bands were visualized using Chemidoc (iBright™ CL1500, Invitrogen). The intensities of the bands were analyzed using ImageJ software, with β-actin as the loading control.
Immunocytochemistry
Raw264.7 macrophages were seeded in a 6-well plate (5 × 105 cells/well). Cells were pretreated with TP1 (25 µM) for 30 min, and then treated with 0.1 µg/mL of LPS. After 30 min, the cells were washed with DPBS, followed by fixation with 4% paraformaldehyde (PFA, Sigma-Aldrich; Merck KGaA) for 20 min. Subsequently, they were incubated with 0.2% Triton X-100 and 0.1% citrate (Sigma-Aldrich, Merck KGaA) in PBS for 5 min. Thereafter, the cells were blocked with 2% bovine serum albumin (BSA; Sigma-Aldrich; Merck KGaA) for 30 min and probed with rabbit anti-p65 NF-κB antibody (1:500; cat. no. 8242; Cell Signaling Technology) for 2 h. Subsequently, washed the cells with 2% BSA, and then probed with fluorescein goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody Alexa Fluor 555 (4 µg/mL; cat. no. A-21428; Invitrogen Inc., Middlesex, MA, USA) for 1 h. After washing, the nuclei were stained with 1 mg/mL of DAPI solution for 5 min. Finally, fluorescence was visualized by a fluorescence microscope (Zeiss Microscope, Carl Zeiss AC).
Data analysis
The results are presented as mean ± standard deviation (SD). The student’s t-test for comparisons between two groups. Statistical significance was defined at p < 0.05 (*).
Results
TP1 inhibits the differentiation of Raw264.7 macrophage without inducing cytotoxicity
p-Terphenyls (Fig. 1A) are cytotoxic owing to their biological activities [29]. Therefore, we investigated the potential cytotoxic effects of TP1 in Raw264.7 macrophages using an MTT assay and found that TP1 treatment (5, 15, and 25 µM) for 24 h was not cytotoxic to Raw264.7 macrophages (Fig. 1B). Macrophage morphology has been linked to inflammation [30]. LPS treatment activates Raw264.7 macrophages, resulting in an increase in cell size and the formation of pseudopodia [31]. Macrophages, characterized by numerous fibrous cytoplasmic projections on their cell surface, are associated with the induction of inflammation [32, 33]. TP1 treatment significantly suppressed the LPS-induced transformation of Raw264.7 macrophages into the inflammatory phenotype compared with that in the LPS-only group (Fig. 1C).

Effects of TP1 on the viability and morphology of Raw264.7 macrophages. a Chemical structure of TP1. b The cells were pretreated with TP1 (0, 5, 15, and 25 µM) for 30 min, followed by treatment with LPS (0.1 µg/mL) for 24 h, after which cell viability was measured using MTT assay. The data are presented as mean ± standard deviation (SD). c To assess the anti-inflammatory effects of TP1, the cells were pretreated with TP1 (25 µM) for 30 min. Afterwards, morphological changes in Raw264.7 macrophages were examined after treatment with LPS (0.1 µg/mL) for 24 h. The data are presented as mean ± SD. Three independent experiments were performed, and representative data are presented
TP1 suppresses LPS-induced NO and ROS production in Raw264.7 macrophages
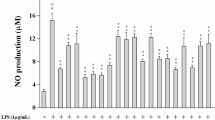
ROS and NO serve as central mediators of inflammatory response [34]. Excessive ROS production leads to endothelial dysfunction and tissue injury [35]. NO plays multiple roles in inflammatory responses, including the regulation of leukocyte activity and tissue cytotoxicity, which leads to vasodilation and edema formation [36]. To examine the anti-inflammatory effects of TP1, we measured the NO and ROS levels in Raw264.7 macrophages. TP1 treatment caused a significant dose-dependent decrease in LPS-induced NO production in Raw264.7 macrophages compared with that in the LPS-only group (Fig. 2A). Additionally, FACS (Fig. 2B and C) and fluorescence microscopy (Fig. 2D) confirmed that TP1 decreased ROS production in a dose-dependent manner. Collectively, these data demonstrate that TP1 can decrease NO and ROS production in LPS-induced Raw264.7 macrophages.

Effects of TP1 on nitric oxide (NO) and reactive oxygen species (ROS) production in LPS-induced Raw264.7 macrophages. The cells were pretreated with TP1 (0, 5, 15, and 25 µM), followed by treatment with LPS (0.1 µg/mL) for 24 h. a The NO level of the cell culture supernatant was measured. b, d ROS production was measured by FACS (b) and fluorescence microscopy d. c Quantitative analysis of panel b. *p < 0.05 compared with the LPS group. The data are presented as the means ± standard deviation (SD). Three independent experiments were performed, and representative data are presented
TP1 decreases the mRNA expression of pro-inflammatory enzymes and cytokines in LPS-induced Raw264.7 macrophages
LPS-induced activation of macrophages promotes the production of various inflammatory enzymes, such as iNOS and COX2, and inflammatory cytokines, including IL-1β, TNF-α, and IL-6 [37]. iNOS is an important marker of inflammation that leads to NO production [38]. COX-2 produces prostaglandin E2 (PGE2), which mediates the inflammatory cascade [39]. These components play pivotal roles in mediating inflammatory processes [40]. Therefore, we investigated the effects of TP1 on the mRNA expression of pro-inflammatory enzymes (iNOS and COX-2) and cytokines (IL-1β, TNF-α, and IL-6) using qRT-PCR. TP1 pretreatment significantly inhibited LPS-induced increase in the mRNA expression of pro-inflammatory enzymes (iNOS and COX-2) and cytokines (IL-1β, TNF-α, and IL-6) 24 h after LPS treatment (Fig. 3A–E). Overall, these data show that TP1 downregulates the gene expression of pro-inflammatory enzymes and cytokines in LPS-induced Raw264.7 macrophages.

Effects of TP1 on the mRNA expression of pro-inflammatory mediators. The cells were pretreated with TP1 (25 µM), followed by treatment with LPS (0.1 µg/mL) for 24 h. a iNOS, b COX-2, c TNF-α, d IL-1β, and e IL-6 mRNA expression levels were measured using qRT-PCR. *p < 0.05 compared with the LPS group. The data are presented as mean ± standard deviation (SD). Three independent experiments were performed, and representative data are presented
TP1 reduces the protein expression of pro-inflammatory enzymes in LPS-induced Raw264.7 macrophages
iNOS and COX-2 are crucial pro-inflammatory enzymes that play central roles in mediating inflammatory processes [41]. The final products of iNOS and COX2 (NO and PGE2, respectively) can accelerate inflammation and are involved in the pathogenesis of inflammatory diseases [42]. Given that the mRNA levels of iNOS and COX-2 were decreased by TP1, we examined the effect of TP1 on the protein levels of iNOS and COX-2 in LPS-induced Raw264.7 macrophages. Consistent with the mRNA expression levels, TP1 treatment significantly decreased iNOS and COX-2 protein expression levels compared with those in the LPS group (Fig. 4). Collectively, these data show that TP1 downregulates the protein expression of pro-inflammatory enzymes.

Effects of TP1 on the protein expression levels of pro-inflammatory enzymes. Raw264.7 macrophages were pretreated with TP1 (25 µM), followed by treatment with LPS (0.1 µg/mL) for 24 h. a Protein expression levels were measured using western blot analysis. b and c iNOS/β-actin and COX-2/β-actin expression levels were quantified by ImageJ software. *p < 0.05 compared with the LPS group. The data are presented as mean ± standard deviation (SD). Three independent experiments were performed. Among them, representative data are presented
TP1 modulates the phosphorylation of AKT/NF-κB pathway proteins in LPS-induced Raw264.7 macrophages
NF-κB is a key transcription factor that modulates immune cells, including inflammatory T cells, and triggers the expression of pro-inflammatory enzymes and cytokines after LPS stimulation [43, 44]. AKT activation can trigger the degradation of the inhibitor of nuclear factor B (IκB) protein and enhance the phosphorylation of the p65 NF-κB subunit, facilitating its translocation to the nucleus. Moreover, inhibiting AKT downregulates the nuclear translocation of NF-κB [45]. Therefore, we investigated the effects of TP1 on AKT and p65 phosphorylation in LPS-induced Raw264.7 macrophages. Additionally, immune-cytochemistry was performed to assess the nuclear translocation of p65. TP1 treatment significantly decreased AKT and p65 phosphorylation and suppressed p65 nuclear translocation (Fig. 5A–E). Overall, these results indicate that TP1 effectively downregulates the AKT and NF-κB signaling pathways.

Effects of TP1 on the phosphorylation of proteins of the AKT/ NF-κB pathway. Raw264.7 macrophages were pretreated with 25 µM of TP1, followed by treatment with LPS (0.1 µg/mL) for 2 h. a AKT and (c) p65 phosphorylation levels were measured by western blot analysis. b p-AKT/β-actin and d p-p65/β-actin expression levels were quantified using ImageJ software. e p65 translocation was visualized using an immunofluorescence assay. *p < 0.05 compared with the LPS group. The data are presented as mean ± standard deviation (SD). Three independent experiments were performed. Among them, representative data are presented
TP1 modulates the phosphorylation of MAPK pathway proteins in LPS-induced Raw264.7 macrophages
The MAPK signaling pathway plays an important role in initiating cellular inflammatory responses, including the production of pro-inflammatory cytokines, in response to LPS stimulation [46]. ERK1/2, JNK, and p38 phosphorylation regulates the generation of pro-inflammatory cytokines, such as TNF-α and IL-6 [47, 48]. Therefore, we examined the effects of TP1 on ERK, JNK, and p38 phosphorylation and found that TP1 treatment reduced LPS-induced phosphorylation of ERK in Raw264.7 macrophages (Fig. 6). Overall, these results suggest that TP1 exerts its anti-inflammatory effects by suppressing the ERK signaling pathway.

Effects of TP1 on the phosphorylation levels of MAPK pathway proteins. Raw264.7 macrophages were pretreated with TP1 (25 µM), followed by treatment with LPS (0.1 µg/mL) for 30 min. a ERK, c JNK, and e p38 phosphorylation levels were measured by western blot analysis. b p-ERK /β-actin, d p-JNK/β-actin, and f p-p38/β-actin expression levels were quantified by ImageJ software. *p < 0.05 compared with the LPS group. The data are presented as mean ± standard deviation (SD). Three independent experiments were performed. Among them, representative data are presented
Discussion
Strategies for ameliorating inflammation may be effective in protecting host tissues from the negative effects of long-term or excessive inflammation [49]. Currently available anti-inflammatory drugs show diverse side effects depending on the individual and treatment duration and concentration [50]. Nonsteroidal anti-inflammatory drugs (NSAIDs) used in the treatment of rheumatoid arthritis exert their therapeutic activity by inhibiting COX-2 [51]. For example, acetaminophen, a member of the NSAIDs family, selectively inhibits COX-1/2. However, it is associated with serious side effects, such as upper gastrointestinal complications [52].
Polar organisms produce various secondary metabolites for survival and adaptation to harsh environments, and these secondary metabolites possess several biological activities, such as antioxidant, anti-inflammatory, and antiparasitic activities [25]. Therefore, polar regions are considered large reservoirs of bioactive compounds [53]. However, studies on the anti-inflammatory effects of compounds derived from these organisms are limited. In the present study, TP1 treatment inhibited the expression of iNOS and COX2, crucial inflammatory enzymes, at both the mRNA and protein levels without causing any cytotoxic effects. The stimulation of PRRs on innate immune cells following pathogen infection or tissue damage triggers the activation of MAPKs, including the p38, ERK, and JNK subfamilies, resulting in the expression of multiple inflammatory genes [54]. To elucidate the potential anti-inflammatory mechanism of TP1, we examined the phosphorylation of MAPKs (p38, ERK, and JNK). TP1 treatment decreased LPS-induced phosphorylation of ERK but did not affect LPS-induced JNK and p38 phosphorylation. The MAPK signaling cascade comprises three main kinases: MAPK kinase kinase (MAP3K), which activates MAPK kinase (MAP2K), which in turn activates MAPK [55]. Each MAPK (ERK, JNK, and p38) is activated by distinct upstream MAP3K and MAP2K [56]. Therefore, the results of the present study suggest that TP1 can downregulate upstream molecules, including kinases that affect ERK but not p38 or JNK. Similar to TP1, tetrandrine derived from Stephania Tetrandra S. Moore inhibits inflammatory responses by modulating the ERK signaling pathway in primary rat mesangial cells [57].
Recently, several natural compounds have been studied for their ability to modulate inflammation and treat sepsis [58]. For example, 7-MCPA extracted from E. longifolia protected C57BL/6 mice against LPS-induced death [59]. Additionally, mangiferin from mangoes and papaya alleviates sepsis by reducing acute lung injury [60]. Moreover, silymarin isolated from milk thistle is effective in protecting mice against LPS-induced sepsis [61]. In line with these previous studies, it is anticipated that TP1 treatment could exert an anti-inflammatory effect in a murine model with LPS-induced sepsis.
Conclusively, the results suggest that TP1 exhibits anti-inflammatory effects in LPS-stimulated macrophages via MAPK pathway-mediated suppression of pro-inflammatory mediators. Overall, TP1 has potential applications in the development of anti-inflammatory drugs for treating inflammatory diseases such as sepsis.
Availability of data and materials
The datasets that support the finding of this study are available from the corresponding author on reasonable request.
References
Hannoodee S, Nasuruddin DN (2021) Acute inflammatory response. StatPearls Publishing, St. Petersburg
Gao H-M, Hong J-S (2008) Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol 29(8):357–365
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140(6):805–820
Chen L et al (2018) Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 9(6):7204
Kawasaki T, Kawai T (2014) Toll-like receptor signaling pathways. Front Immunol 5:461
Tucureanu MM et al (2018) Lipopolysaccharide-induced inflammation in monocytes/macrophages is blocked by liposomal delivery of Gi-protein inhibitor. Int J Nanomed 13:63
Hankittichai P et al (2020) Oxyresveratrol inhibits IL-1β-induced inflammation via suppressing AKT and ERK1/2 activation in human microglia, HMC3. Int J Mol Sci 21(17):6054
Hankittichai P et al (2020) Artocarpus lakoocha extract inhibits LPS-induced inflammatory response in RAW 264.7 macrophage cells. Int J Mol Sci 21(4):1355
Liu X et al (2021) Discovery of phenolic glycoside from Hyssopus cuspidatus attenuates LPS-induced inflammatory responses by inhibition of iNOS and COX-2 expression through suppression of NF-κB activation. Int J Mol Sci 22(22):12128
Hwang J-S et al (2019) Glucosamine improves survival in a mouse model of sepsis and attenuates sepsis-induced lung injury and inflammation. J Biol Chem 294(2):608–622
Gabarin RS et al (2021) Intracellular and extracellular lipopolysaccharide signaling in sepsis: avenues for novel therapeutic strategies. J Innate Immun 13(6):323–332
Rhee C et al (2015) Comparison of trends in sepsis incidence and coding using administrative claims versus objective clinical data. Clin Infect Dis 60(1):88–95
Fink MP, Warren HS (2014) Strategies to improve drug development for sepsis. Nat Rev Drug Discovery 13(10):741–758
Cohen J et al (2015) Sepsis: a roadmap for future research. Lancet Infect Dis 15(5):581–614
Garot D et al (2011) Population pharmacokinetics of ceftriaxone in critically ill septic patients: a reappraisal. Br J Clin Pharmacol 72(5):758–767
Hermans PE, Wilhelm MP (1987) Vancomycin. Mayo Clin Proc 66(11):1165–1170. https://doi.org/10.1016/s0025-6196(12)65799-1
Mohkam M et al (2007) Ceftriaxone associated nephrolithiasis: a prospective study in 284 children. Pediatr Nephrol 22:690–694
Shimamoto Y et al (2013) Systemic inflammatory response syndrome criteria and vancomycin dose requirement in patients with sepsis. Intensive Care Med 39:1247–1252
Koparde AA, Doijad RC, Magdum CS (2019) Natural products in drug discovery, in Pharmacognosy-medicinal plants. IntechOpen. https://doi.org/10.5772/intechopen.82860
Harvey AL (2008) Natural products in drug discovery. Drug Discovery Today 13(19–20):894–901
Bhattarai HD et al (2013) A new pseudodepsidone from the Antarctic lichen Stereocaulon alpinum and its antioxidant, antibacterial activity. J Antibiot 66(9):559–561
Seo C et al (2009) Protein tyrosine phosphatase 1B inhibitory effects of depsidone and pseudodepsidone metabolites from the Antarctic lichen Stereocaulon alpinum. Bioorg Med Chem Lett 19(10):2801–2803
Lu XL et al (2014) Pimarane diterpenes from the Arctic fungus Eutypella sp. D-1. J Antibiot 67(2):171–174
Mellado GG et al (2005) Steroids from the Antarctic octocoral Anthomastus bathyproctus. J Nat Prod 68(7):1111–1115
Tian Y, Li Y-L, Zhao F-C (2017) Secondary metabolites from polar organisms. Mar Drugs 15(3):28
Liu J-T et al (2013) Bioactive natural products from the antarctic and arctic organisms. Mini Rev Med Chem 13(4):617–626
Phi K-H et al (2022) Bioactive terphenyls isolated from the Antarctic lichen Stereocaulon alpinum. Molecules 27(7):2363
Wang L et al (2017) Novel p-terphenyl glycoside with a rare 2, 6-dideoxyhexopyranose moiety from Actinomycete strain SF2911 that inhibits cancer cell migration. J Antibiot 70(10):987–990
Wang D et al (2019) Cytotoxic p-terphenyls from a marine-derived Nocardiopsis species. J Nat Prod 82(12):3504–3508
Merecz-Sadowska A et al (2020) Anti-inflammatory activity of extracts and pure compounds derived from plants via modulation of signaling pathways, especially PI3K/AKT in macrophages. Int J Mol Sci 21(24):9605
Cao Y et al (2019) Punicalagin prevents inflammation in LPS-induced RAW264. 7 macrophages by inhibiting FoxO3a/autophagy signaling pathway. Nutrients 11(11):2794
Yao Y, Xu X-H, Jin L (2019) Macrophage polarization in physiological and pathological pregnancy. Front Immunol 10:792
Heinrich F et al (2017) Morphologic, phenotypic, and transcriptomic characterization of classically and alternatively activated canine blood-derived macrophages in vitro. PLoS ONE 12(8):e0183572
Agita A, Alsagaff MT (2017) Inflammation, immunity, and hypertension. Acta Med Indones 49(2):158
Rizwan S, ReddySekhar P, MalikAsrar B (2014) Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 20:1126–1167. https://doi.org/10.1089/ars.2012.5149
Tripathi P et al (2007) The role of nitric oxide in inflammatory reactions. FEMS Immunol Med Microbiol 51(3):443–452
Lee S et al (2011) Anti-inflammatory function of arctiin by inhibiting COX-2 expression via NF-κB pathways. J Inflamm 8:1–9
Zhou J-P et al (2021) Rosiglitazone alleviates lipopolysaccharide-induced inflammation in RAW264, 7 cells via inhibition of NF-κB and in a PPARγ-dependent manner. Exp Ther Med 22(1):1–8
Simon LS (1999) Role and regulation of cyclooxygenase-2 during inflammation. Am J Med 106(5):37S-42S
Han S et al (2019) Procyanidin A1 Alleviates Inflammatory Response induced by LPS through NF-κB, MAPK, and Nrf2/HO-1 Pathways in RAW264. 7 cells. Sci Rep 9(1):15087
Murakami A, Ohigashi H (2007) Targeting NOX, INOS and COX-2 in inflammatory cells: chemoprevention using food phytochemicals. Int J Cancer 121(11):2357–2363
Dilshara MG et al (2014) Downregulation of NO and PGE2 in LPS-stimulated BV2 microglial cells by trans-isoferulic acid via suppression of PI3K/Akt-dependent NF-κB and activation of Nrf2-mediated HO-1. Int Immunopharmacol 18(1):203–211
Liu T et al (2017) NF-κB signaling in inflammation. Signal Transduct Target Ther 2(1):1–9
Xu X et al (2014) Punicalagin inhibits inflammation in LPS-induced RAW264. 7 macrophages via the suppression of TLR4-mediated MAPKs and NF-κB activation. Inflammation 37:956–965
Bai D, Ueno L, Vogt PK (2009) Akt-mediated regulation of NFκB and the essentialness of NFκB for the oncogenicity of PI3K and Akt. Int J Cancer 125(12):2863–2870
Thalhamer T, McGrath M, Harnett M (2008) MAPKs and their relevance to arthritis and inflammation. Rheumatology 47(4):409–414
Dong N et al (2020) Astragalus polysaccharides alleviates LPS-induced inflammation via the NF-κB/MAPK signaling pathway. J Cell Physiol 235(7–8):5525–5540
Roy PK et al (2008) Role of the JNK signal transduction pathway in inflammatory bowel disease. World J Gastroenterol WJG 14(2):200
Panigrahy D et al (2021) Resolution of inflammation: An organizing principle in biology and medicine. Pharmacol Ther 227:107879
Pirlamarla P, Bond RM (2016) FDA labeling of NSAIDs: Review of nonsteroidal anti-inflammatory drugs in cardiovascular disease. Trends Cardiovasc Med 26(8):675–680
Vane JR, Botting RM (1998) Anti-inflammatory drugs and their mechanism of action. Inflamm Res 47:78–87
Bruno A, Tacconelli S, Patrignani P (2014) Variability in the response to non-steroidal anti-inflammatory drugs: mechanisms and perspectives. Basic Clin Pharmacol Toxicol 114(1):56–63
Bratchkova A, Ivanova V (2011) Bioactive metabolites produced by microorganisms collected in Antarctica and the Arctic. Biotechnol Biotechnol Equip 25(sup1):1–7
Arthur JSC, Ley SC (2013) Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol 13(9):679–692
Zhang W, Liu HT (2002) MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 12(1):9–18
Hommes D, Peppelenbosch M, Van Deventer S (2003) Mitogen activated protein (MAP) kinase signal transduction pathways and novel anti-inflammatory targets. Gut 52(1):144–151
Wu C-J et al (2011) Tetrandrine down-regulates ERK/NF-κB signaling and inhibits activation of mesangial cells. Toxicol In Vitro 25(8):1834–1840
Alikiaii B et al (2021) The role of phytochemicals in sepsis: a mechanistic and therapeutic perspective. BioFactors 47(1):19–40
Hai Dang N et al (2016) 7-Methoxy-(9H-β-Carbolin-1-il)-(E)-1-propenoic acid, a β-Carboline alkaloid from Eurycoma longifolia, exhibits anti-inflammatory effects by activating the Nrf2/Heme oxygenase-1 Pathway. J Cell Biochem 117(3):659–670
Gong X et al (2013) Anti-inflammatory effects of mangiferin on sepsis-induced lung injury in mice via up-regulation of heme oxygenase-1. J Nutr Biochem 24(6):1173–1181
Kang JS et al (2004) Protection against lipopolysaccharide-induced sepsis and inhibition of interleukin-1β and prostaglandin E2 synthesis by silymarin. Biochem Pharmacol 67(1):175–181
Acknowledgements
Not applicable.
Funding
This research was supported by the Korea Polar Research Institute (KOPRI) grant funded by the Ministry of Oceans and Fisheries (KOPRI project No. * PE23900).
Author information
Authors and Affiliations
Contributions
MK, JA, SAS, MK, SC, HK, and KHP performed the experiments. MK wrote the manuscript under the guidance of CSL. SYM, JHL, UJY, and HHP contributed intellectually to the present study. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, M., An, J., Shin, SA. et al. Anti-inflammatory effects of TP1 in LPS-induced Raw264.7 macrophages. Appl Biol Chem 67, 16 (2024). https://doi.org/10.1186/s13765-024-00873-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-024-00873-y