
Abstract
All chronic renal disorders eventually lead to renal interstitial fibrosis (RIF). Chronic inflammation and pro-fibrotic substances are familiar companions of the fibrotic process. The Sulforaphane (Sul) molecule is particularly useful in protecting the liver from oxidative damage. To investigate the Sul effects on fibrosis markers and inflammatory proteins in the kidney of NRK52E cell line and rats and clarify the mechanism of TGF-β/Smad signaling pathway in a rat model of RIF were developed in the present study. Sul (50, 100, and 200 ng/ml) remarkably reduced the gene expressions of tumor necrosis factor (TNF-α), interleukin-6 (IL-6), interleukin (IL)-1β, collagen 3 (COL3A1), collagen 1 (COL1A1), and α-smooth muscle actin (α-SMA) in fibrotic NRK52E cells compared with those in cells inspired by transforming growth factor-α (TGF-α). Histopathological investigations showed that Sul administration retained renal tissue structure and decreased kidney tissue fibrosis in rats subjected to unilateral ureteral blockage (UUO). The expression level of TNF-α, IL-6, IL-1β, COL3A1, COL1A1, and α-SMA in the rats’ kidneys exposed to UUO was also suppressed by the treatment of Sul. In the present study, western blot analysis showed that Sul upregulated the expressions of fibrotic NRK52E cells Smad7 and rat model UUO groups while simultaneously decreasing the stimulation of Smad2/3 and the expressions of cyclooxygenase-2, NF-κB, Smad4, activator protein-1, and high-mobility group protein B1. Ultimately, Sul’s ability to inhibit the TGF-β/Smad pathway and the development of inflammation factors may mitigate RIF.
Similar content being viewed by others


Introduction
Fibrosis of the kidneys occurs when there is an inappropriate extracellular matrix (ECM) buildup in solid organs in response to damage [1]. This is considered permanent and may lead to the breakdown of normal tissue architecture and compromised organ function. Kidneys are quite susceptible to damage. Most individuals with CKD will develop renal fibrosis [2]. This pathological trait can lead to the development of end-stage renal disease (ESRD) and the subsequent need for a kidney transplant or dialysis to sustain life [3]. The global incidence of CKD ranges between 3 and 18%, significantly burdening public health systems. Therefore, there is an immediate need for a medication that effectively addresses renal fibrosis [4].
Several antifibrotic medicines have been tried out in human trials [5]. However, the lack of renal selectivity and low bioavailability are significant obstacles. Studies we’ve conducted show that the multikinase inhibitor sorafenib, which is used in cancer treatment, has antifibrotic properties [6]. The United States Food and Drug Administration (USFDA) has authorized sorafenib as an oral medication for hepatocellular carcinoma and renal cell carcinoma. The vascular endothelial growth factor receptor 2 (VEGF-R2) and the platelet-derived growth factor (PDGF) receptor are two of the tyrosine kinases that it blocks (PDGFR) [7, 8]. Gavage administration of sorafenib, a cancer drug, may reduce fibrosis caused by the UUO in rats and mice. However, sorafenib’s promise as a therapeutic agent for renal fibrosis therapy may be constrained by its low absorption and systemic adverse effects after long-term dosing [9]. Therefore, it is necessary to develop a method to enhance sorafenib absorption in fibrotic renal tissues while minimizing its adverse effects [10].
Inflammation, apoptosis, and oxidative stress are only a few of the signaling cascades and biological processes involved in RIF [11]. Renal fibrosis is a disease for which the specific molecular process is poorly understood [12]. Numerous signaling pathways, such as apoptosis signal-regulating kinase and transforming growth factor beta (TGF-β)/Smad, have been involved in renal fibrosis [4]. High TGF-β activation is commonly linked to fibrosis and disease progression, regardless of the underlying cause [4]. Previous investigation has shown that the NF-kB transcription factors may increase TGF-β expression, activating the TGF-β/Smad pathway and mediating its downstream biological impacts [13]. The Smad functions as TGF-β intracellular effectors. Located upstream of the kidney fibrotic and inflammatory methods, Smad4 is a critical factor in the Smad unit that may interact with Smad3 and Smad7 to influence the transcriptional activity [14]. The TGF-β/Smad pathway’s deregulation has also been shown as a significant contributor to tissue fibrosis. In fibrotic tissue, TGF-1β can upregulate the expression of Smad3 and Smad2; Smad7 negatively controls this process as part of a feedback loop [15]. However, prior research has linked RIF, which makes kidney cells more susceptible to TGF-β and further exacerbates fibrosis, to the long-term persistence of inflammatory molecules like interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) in renal tissues [16]. Macrophages, invading immune cells, have been identified as the primary mediators of this inflammatory response [17]. Numerous monocytes/macrophages generated by the kidney continually infiltrate the organ and induce inflammation by secreting inflammatory cytokines like TNF-α and IL-1β [18]. As previously described, reducing RIF’s severity may be possible by preventing inflammatory factor release. Based on these findings, we hypothesize that inflammatory proteins and the TGF-β/Smad pathway play critical roles in RIF progression [19].
The isothiocyanate sulforaphane (Sul), found in green cruciferous vegetables, has been demonstrated to have antioxidant and anti-inflammatory effects [20,21,22]. In addition to protecting the kidneys against cisplatin and maleate-stimulated type 2 diabetes and acute kidney injury (AKI), Sul enhances mitochondrial dynamics, mitophagy, and autophagy [23]. Yoon et al. demonstrated that Sul reduces fibrosis and inflammation in the UUO model by stimulating the activation of nuclear factor erythroid 2-related factor 2 (Nrf2), reducing oxidative stress of mitochondrial dysfunction [24]. This finding suggests that Sul may play a considerable role in reestablishing mitochondrial homeostasis. Sul enhances lipid metabolism and reduces lipid buildup in animal models of chronic kidney disease (CKD) and diabetic nephropathy (DN) [25]. Sul also controls proteins involved in lipid production in NAFLD [26]. However, it has been hypothesized that mitochondria function for Sul in the UUO models; whether this antioxidant reduces kidney damage by modifying mitochondrial homeostasis via the activation of mitochondrial bioenergetics and mitochondrial biogenesis improvement is not reported. Also, Sul function in lipid metabolism during UUO has not been investigated. We reasoned that Sul’s ability to promote mitochondrial biogenesis, enhance the electron transport system, and regulate lipid metabolism, autophagy, and mitophagy [27] in the UUO model would mitigate renal injury. We investigated that Sul decreased levels of the kidney damage indicators kidney injury molecule 1 (KIM-1), interleukin-1 beta (IL-1β), and alpha-smooth muscle actin α-SMA). Mitochondrial biogenesis was restored in the obstructed kidney, as demonstrated by the activation of peroxisome proliferator-stimulated receptor gamma co-activator 1 (PGC-1) and nuclear respiratory factor 1 (NRF1) [28]. This resulted in a decrease in indicators of renal injury. Therefore, Sul led to an increase in the mitochondrial marker voltage-dependent anion channel (VDAC). Additionally, Sul therapy led to a more robust structure in the mitochondria [29]. The activities of citrate synthase, aconitase 2 (ACO2), and complex III (CIII) were all raised when mitochondrial biogenesis was stimulated. Also, Sul was able to restore lipid metabolism by lowering the expression of CD36, sterol regulatory-element binding proteins 1 (SREBP1), diacylglycerol O-acyltransferase 1 (DGAT1), and fatty acid synthase (FASN), all of which play roles in the manufacture of triglycerides (TGs). By enhancing mitochondrial structure, Sul was able to restore autophagy flow by lowering autophagy and fission markers sinusotomies (p62) and beclin and increasing the microtubule-associated proteins 1 A/1B light chain 3 II and I (LC3II/LC3I) ratio and B-cell lymphoma (Bcl2).
Materials and methods
Reagents and materials
Sulforaphane (Sul) was purchased from Lumifore Biotech. (Hefei, China). FBS and TGF-β were acquired from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Cell proliferation Kit I MTT was obtained from Aladdin Chemical Co., Ltd. (Shanghai, China). 5% skimmed milk powder, Tween-20, Roswell Park Memorial Institute (RPMI) 1640 medium, and DAPI were purchased from Beyotime Institute of Biotechnology (Shanghai, China). Blood urea nitrogen and creatinine kits were purchased from Dojindo Molecular Technologies (Kumamoto Techno, Japan). Tissue protein extraction kits and bicinchoninic acid (BCA) protein concentration purpose were obtained from Seebio Biotech (Shanghai) Co., Ltd. Anti-α-smooth muscle actin (α-SMA) was acquired from Santa Cruz Biotechnology, Inc. NF-κB, α-SMA, Smad7, and Anti-phosphorylated (p)-Smad2/3, was obtained from BioLegend, Inc., U.S.A. HMGB1, activator protein-1, cyclooxygenase (COX)-2, Smad4, Smad3, Smad2, TGF-β, anti-collagen1 (COL1A1), GAPDH, secondary antibody-HRP-conjugated Affinipure Goat Anti-Rabbit IgG (H+L) were obtained from ZSGB-BIO Biotechnology Co., Ltd., Beijing, China. Deionized water (18.2 MΩ/cm) from a Milli-Q ultrapure system was utilized in this investigation.
MTT assay
Rat kidney epithelial NRK52E cells were acquired from the Shanghai Academy of Life Sciences and the Chinese Academy of Sciences. The cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum, 100 µg/ml streptomycin, and 100 U of penicillin in a humidified incubator at 37 °C and 5% CO2.
NRK-52E is a cell line exhibiting epithelial morphology isolated from a rat’s kidney. NRK52E cells were chosen as the NRK-52E cell model. First, the NRK52E cells were planted at a density of 5 × 103 cells/well in 96-well plates. After culturing for 24-h, the culture medium was replaced by a medium (100 µl) containing various concentrations of Sul (0, 50, 100, 200, 400, and 800 ng/ml). After incubation for another 24-h, the viability of the NRK52E cells was assessed using the MTT assay at the absorbance of 590 nm [30,31,32,33].
In vivo animal model
36 male Sprague-Dawley (SD) rats, 7 weeks old, weighing between 200 and 220 g, were acquired from the SPF Experimental Animal Center at Shanghai Donghai Senior Nursing Hospital Shanghai, China. All experimental procedures were carried out according to the protocol and international standards of care for animals to adhere to the requirements established by the ethical committee of the Department of Integrated Internal Medicine, Shanghai Donghai Senior Nursing Hospital Shanghai China, Shanghai-201304, China (W-22-10254A1). The rats were kept in an animal room with a 12-h light/dark cycle, a constant temperature of 20 °C, and a relative humidity of 60%. Before each test, rats were given a week to acclimate and fasted for 12-h. All procedures involving rats complied with the NIH’s recommendations for the experimental animals [34,35,36]. There was a concerted attempt to lessen the number of animals utilized and the pain they suffered.
Thirty-six total SD rats were randomly separated into six groups (n = 7): Sham group (group I); UUO (group II), high dose Sul (80 mg/kg) (group III); medium dose Sul (40 mg/kg) (group IV); low dose Sul (20 mg/kg) (group V).
In rats, unilateral ureteral obstruction (UUO) causes recurrent inflammatory cystitis (RIF). Rats had unrestricted access to water but were fasted for 12 h before the procedure. Following anesthesia with an intraperitoneal injection of pentobarbital (60 mg/kg), rats were secured on the operation table. The animal was first sterilized and shaved before a scratch was done on the left side, exposing the ureter and kidney on the left side. Starting postoperative day 2, rats in the low, medium, and single administration groups and high dosage administration groups received intragastrically administered Sul for 28 days. The control and experimental groups were given the same total volume of carboxymethyl cellulose (CMC-Na, 0.5% concentration). After anesthesia with an intraperitoneal injection of pentobarbital (60 mg/kg), blood samples were taken from the abdominal aorta of rats on day 28. After collecting blood samples in heparin tubes, they were centrifuged for 10 min at 4000×g and 4 °C. We used the manufacturer’s instructions to get the BUN and Scr values. The rats were sacrificed, and their right and left kidneys were removed, decapsulated, and stored in the oxygenated buffer at 4 °C before being divided in half. The kidney tissue was divided in half, frozen at − 20 °C until the testing, and preserved in 10% formaldehyde solution at room temperature for a week to facilitate histological analysis.
Histopathological examination
Standard histopathology procedure calls for fixing tissue samples (kidney organs) in 40% formaldehyde solution and then embedding them in paraffin into the wax block and a rotatory tissue cutter kidney tissue into slices 4 μm thick for histological analysis. Hematoxylin and eosin (H&E) were used for the histopathological investigation under an optical microscope. Following is a quick summary of the H&E staining procedure: After dehydrating the hematoxylin-stained slices for 4 min in 85 and 95% ethanol, the eosin-stained slices were left at 25 °C for another 4 to 5 min before being dried in three cylinders of 100% anhydrous ethanol. Neutral gum was used after n-butanol and xylene were used to clean the slices.
Immunofluorescence analysis
NRK52E cells at a density of 5 × 104 cells were planted on coverslips in a 24-well plate per well for 24-h. NRK52E cells after the treatments of TGF-β (10 ng/ml) and Sul (50, 100, and 200 ng/ml). After 24 h, cells were fixed in 4% paraformaldehyde for 10 min and permeabilized with 0.1% (wt/vol) Triton X-100 for 10 min. The cells were then prevented by 1% BSA for antibody anti-α-SMA (1:50) at 4 °C and stained with antibody Rhodamine-conjugated IgG h (1) + L (70)-conjugated secondary antibody to rabbit, respectively. Cell nuclei were stained with DAPI. Fluorescence microscopy was carried out with LSM 880 with Airyscan (Carl Zeiss, Jena, Germany).
Reverse transcription-quantitative PCR
Total RNA was obtained from NRK52E cells and dissected using an RNAiso Plus® Reagent Kit according to the manufacturer’s protocol (Takara Biotechnology Co., Ltd.). cDNA was generated using the QuantiNova Rev Transcription Kit according to the manufacturer’s instructions. QuantiNova SYBR Green PCR Kit and qPCR (ViiA™ 7 Real-time PCR system) were used to evaluate the mRNA levels by using specific primer for sGC made at the Personalbio, Shanghai, China. The results were normalized to the housekeeping gene β-actin and analyzed by the 2−ΔΔCt method.
Western blotting assay
The NRK52E cell extracts were prepared by lysing cells in RIPA buffer [150 mM NaCl, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50 mM Tris-HCl pH 7.4, 1 mM Ethylene Diamine Tetraacetic Acid (EDTA), 1 mM Phenylmethanesulfonyl fluoride (PMSF), and 1% Triton X-100]. Total protein was collected by centrifugation and quantified with a BCA Protein Assay Kit. Then, 50 µg of targeting protein was separated on an SDS-polyacrylamide gel, transferred to PVDF membranes (Millipore, Billerica, MA), and probed with specific antibodies. The target proteins were detected using an Odyssey Infrared Imaging System [37].
Statistical analysis
Data were verified as mean ± standard deviation (SD). A Student’s t-test was utilized to assess the difference between the two groups. At the same time, a one-way analysis of variance (ANOVA) was employed to determine the significance among the groups. The statistical analyses were utilized with GraphPad Prism version 8.00 software. Changes were regarded as significant if P < 0.05.
Results
Sul inhibits cell viability in the NRK52E line in a TGF-β-dependent manner. Sul toxicity was tested by incubating NRK52E cells with Sul at progressively higher concentrations throughout 24-h (0, 50, 100, 200, 400, and 800 ng/ml). The toxic effects of Sul on the NRK52E cell line were negligible below 200 ng/ml (Fig. 1A). The viability of TGF-β-induced NRK52E cells was similarly observed to be significantly suppressed by treatment with Sul (50, 100, and 200 ng/ml) (Fig. 1B). Based on these findings, it appears that Sul can counteract the enhancing outcomes of TGF-β on NRK52E cells’ survival. Sul at 50, 100, and 200 ng/ml concentrations was therefore desired for the following experiments.

Effects of Sulforaphane (Sul) on the fibrosis of NRK52E cells stimulated by TGF-β. A Cytotoxicity of Sulforaphane (Sul) on NRK52E cells. B The effects of Sulforaphane (Sul) on NRK52E cell proliferation with TGF-β. C Immunofluorescence examined the effect of Sulforaphane (Sul) on α-SMA expressions in NRK52E cells with TGF-β. Scale bar 50 μm
Sul inhibits TGF-β-induced fibrosis in cultured cells. The immunofluorescence staining identified the fibrotic markers expressions of α-SMA to examine the impact of Sul on cell fibrosis produced by TGF-β. NRK52E cell line treated with Sul (50, 100, or 200 ng/ml) had significantly lower amounts of α-SMA protein expression than cells treated with TGF-β alone (Fig. 1C). These findings suggested that Sul might effectively block TGF-β-induced cell fibrosis.
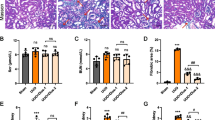
The role of Sul in rat models of renal impairment due to unintended overdose on urea (RIF) To examine Sul-affected renal function in rats after UUO. We collected blood for BUN and Scr analyses. BUN and Scr were considerably higher in the UUO group contrasted to the control (untreated) group (Fig. 2A). There was a statistically significant reduction in BUN levels across all three dosing groups compared to the UUO group (49.1%, 35.4%, and 33.9%, respectively) (Fig. 2B). Comparing the UUO groups to the other groups, Scr levels were reduced by 40.1, 27.2, and 18.5%, respectively (Fig. 2B). According to these results, Sul therapy in UUO rats can considerably lower BUN and Scr levels. As there was no statistically significant difference between the BUN and Scr levels of the Sul and sham groups, Sul at 80 mg/kg had a less negative impact on the rats’ renal function.

Effect of Sulforaphane (Sul) on RIF stimulated by rat model UUO. Effect of Sulforaphane (Sul) treatment on A BUN and B Scr ratio. C Effect of 20, 40 and 80 mg/kg Sulforaphane (Sul) on renal sections as assessed using H&E staining. Scale bar 100 μm
Sul influence on kidney disease in UUO-induced RIF. Kidney sections from rats in all the groups were histologically examined, and the damage to their tissues was assessed using H&E staining. In UUO groups, renal tubulars were enlarged, necrotized, and attenuated compared to their sham counterparts; this was accompanied by a significant increase in the infiltration of monocytes and lymphocytes (Fig. 2C). Renal tubules expanded or shrank after treatment with Sul (20, 40, or 80 mg/kg). The level of vacuole degeneration decreased compared to tissues in the UUO group (Fig. 2C). Based on these findings, Sul appears to be an effective treatment for reducing UUO-related kidney damage. Collagen fibrotic connection tissue deposition and proliferation were significantly higher in the kidney tissues of UUO-treated rats than in the sham group. On the other hand, Sul-treated rats showed substantially less collagen fibrous connection tissue deposition and proliferation in their renal than in the UUO group (20, 40, and 80 mg/kg, respectively) (Fig. 3). Treatment with Sul was associated with decreased blue and red fiber content, with the most significant drop occurring in the high-dosage group. Immunofluorescence investigation further revealed that UUO rat kidney tissue α-SMA expression was decreased after Sul treatment (Fig. 3). These data demonstrated that Sul significantly mitigated the RIF triggered by the UUO.

Effect of Sulforaphane (Sul) on α-SMA expressions in renal tissues following UUO in rats as examined using immunofluorescence. Magnification, ×400. Sulforaphane (Sul); UUO, unilateral ureteral obstruction
Effects of the expression sulforaphane
The expressions of COL1A1, α-SMA, and TGF-β were then assessed in the NRK52E cell line and rat kidneys to evaluate the influence of Sul on the expression of the fibrosis markers. Protein expression levels of COL1A1, α-SMA, and TGF-β were considerably greater in rats’ renal tissues after UUO than in the untreated group (NRK52E) and the sham (rat model). Moreover, Sul treatment of the NRK52E cell line and UUO rat group indicated dramatically decreased mRNA expression levels of COL1A1, α-SMA, and TGF-β (Fig. 4A–D). Treatment with Sul substantially decreased COL1A1, α-SMA, and TGF-β expression in TGF-β-treatment renal tissues and NRK52E cell line model of UUO models, compared with the expression levels in the UUO rat groups and controlled NRK52E cell (Fig. 5A, B). These findings imply that Sul reduced fibrosis severity in the UUO rat models and fibrotic cells.

Effect of Sulforaphane (Sul) on the mRNA expressions of inflammatory and factors fibrotic markers. A Effect of Sulforaphane (Sul) on the gene expressions of COL3A1, COL1A1 and α-SMA in NRK52E cells. B Effects of Sulforaphane (Sul) on the gene expressions of IL-6, IL-1 and TNF-α in NRK52E cells. C Effects of Sulforaphane (Sul) on gene expressions of COL3A1, COL1A1 and α-SMA in rat kidney tissues. D Effect of Sulforaphane (Sul) on the gene expressions of IL-6, IL-1 and TNF-α in rat kidney tissues

Effect of Sulforaphane (Sul) on the protein expressions of fibrotic markers in rat kidneys and NRK52E cells. A Effects of Sulforaphane (Sul) on COL1A1, α-SMA and TGF-β, protein expressions in NRK52E cells. B Effects of Sulforaphane (Sul) in rat kidneys on COL1A1, α-SMA and TGF-β
Effects of sulforaphane on the expressions of TGF-β/Smad
The impact of Sul on expressions of Smad, the regulator of TGF-β, was studied to understand better the mechanism by which Sul inhibits TGF-β-induced RIF (Fig. 6A–D). Smad4 expression and Smad2/3 phosphorylation were considerably higher in the rat renal tissues in the UUO and the TGF-β-treatment group than in the sham and control cell groups (Fig. 6A–D). TGF-β-treatment NRK52E cell line and rats’ model UUO showed considerably lower phosphorylation of Smad4 and Smad2/3 expressions after treatment with Sul (Fig. 6A–D). Smad7 expression went the opposite in both the in-vivo and in-vitro models. Based on these findings, therapy with Sul may reduce fibrosis by controlling the expression of Smad proteins.

Effects of Sulforaphane (Sul) on the expressions of important parts of the TGF-β/Smad pathway. A, B Effects of Sulforaphane (Sul) on the protein levels of Smad4, Smad7, p-Smad2/3, Smad3, and Smad2 in NRK52E cells. C, D Effect of Sulforaphane (Sul) on the protein levels of Smad2, Smad3, p-Smad2/3, Smad7 and Smad4 in rat kidneys
The expression of inflammatory proteins
Regulation of inflammatory protein expression by Sul. Renal fibrosis is often accompanied by inflammation. Thus, an examination of Sul affects the expression of inflammatory proteins. The inflammatory cytokines expression of the IL-6, IL-1β, and TNF-α was considerably decreased in the renal tissues of TGF-β and UUO treatment with NRK52E cell line after treatment with Sul (Fig. 7A, B). Additionally, the TGF-β-treatment NRK52E cells and rat renal tissues model UUO exhibited considerably increased COX-2, NF-κB, AP-1, and HMGB1 proteins compared to the sham and untreated groups (Fig. 7A, B). When administered to the NRK52E cells model and the rat renal tissues model UUO, Sul dramatically reduced the upregulated expression of COX-2, NF-κB, AP-1, and HMGB1 induced by TGF-β (Fig. 7A, B). In conclusion, our findings indicated that Sul could lessen fibrosis by preventing the production of inflammation proteins.

Effects of Sulforaphane (Sul) on the expressions of COX-2, NF-κB, AP-1, and HMGB1 in NRK52E cells and rat kidneys. A Effects of Sulforaphane (Sul) on COX-2, NF-κB, AP-1, and HMGB1 in NRK52E cells. B Effects of Sulforaphane (Sul) on COX-2, NF-κB, AP-1, and HMGB1 in rat kidneys
Discussion
As chronic kidney disease progresses, renal fibrosis often develops as a clinical consequence. Regularly recognized and strongly linked to declines in kidney function in individuals with chronic kidney disease (CKD), renal interstitial fibrosis (RIF) is a clinical symptom of kidney fibrosis [38, 39]. Patients with chronic kidney disease would greatly benefit from RIF preventive and intervention methods. Although much data has been collected over the past decade on the mechanism behind renal fibrosis, viable therapeutic techniques for the treatment and prevention of RIF continue to remain mysterious [39,40,41]. Sul has been shown to have various beneficial effects on the body, including an antifibrotic nature, anti-inflammatory, antibacterial, hypolipidemic, and antioxidant in the liver. In addition to its anti-inflammatory properties, this study determined that Sul has antifibrotic properties in the rat renal tissues, likely via modulating the TGF-β/Smad signaling route to reduce the production of inflammation markers [40].
The current analysis employed rat RIF models to examine Sul’s possible effects. Three basic techniques are being used to induce renal fibrosis in animals: I Surgical procedures, such as unilateral nephrectomy (UUO), bilateral nephrectomy (5/6), and the ischemia–reperfusion injury model (IRIM); ii) long-term drugs or toxic induction, such as with adenine, aristolochic acid, or cyclosporine; iii) complex models, such as folate-Phd14 gene knockout-induced transgenic and fibrosis toxic induction techniques were not employed in the current investigation due to the possibility of drug interactions [42,43,44]. Ischemia–reperfusion, 5/6 nephrectomy, and UUO models are frequently used in surgical procedures for renal fibrosis [45,46,47]. Two protocols are required to create a 5/6 nephrectomy model, and then patients must be monitored regularly for the next 5 weeks [48]. Compared to UUO, the time required to construct a model during this technique is much higher due to its complexity. Renal transplantation can reflect glomerulosclerosis, and renal fibrosis after ischemia–reperfusion (RIF) is the primary model used to mimic these changes in renal function. The UUO model simulates RIF because it produces similar results in pathological alterations in a short time (around a week or two), lower rates of animal death, simpler surgical procedures, and high repeatability [49,50,51]. Collagen accumulation in the kidney infiltration, interstitium, dilatation, and inflammatory cells of kidney tubules are all clinical symptoms that follow successful model establishment [52]. In addition, the serological markers BUN and Scr rose by 1.5 [53]. Therefore, the UUO rat animal model was selected for this investigation. H&E staining revealed that rats subjected to UUO had significantly more penetrating renal tubule dilatation or atrophy, nuclear deformation, and inflammatory cells in their kidney tissues than in the sham group. Serum BUN levels were three times higher in the UUO group compared to the sham group, while serum levels of Scr were 1.6 times higher in the UUO group compared to the sham group.
Subsequently, the consequences of Sul therapy on RIF were studied. The in vivo outcomes demonstrated that Sul treatment at 20 mg/kg, 40 mg/kg, and 80 mg/kg improved renal tubule dilation or atrophy and decreased inflammatory cell infiltration and vacuolar degeneration of UUO rats in the kidneys. Our findings imply that Sul can enhance renal dysfunctions and pathological alterations in rat model UUO. More specifically, Sul inhibited the growth of collagen fibers and collagen fibrous connective tissue deposition in the kidneys of UUO rats. The expressions of fibrosis markers α-SMA, COL1A1, and COL3A1 were further evaluated to examine Sul’s impact on RIF in rats. Connective tissue in the skin relies heavily on COL3A1 and COL1A1, whereas α-SMA expressions are a significant fibroblast marker that plays a crucial role in fibroblast migration [48]. Aberrant expressions of COL3A1 and COL1A1 mostly cause pathological alterations in dermal connective tissue and fibroplasia. We found that Sul significantly suppressed the expressions of the kidneys’ fibrotic markers of UUO rats, indicating that Sul has the potential to protect kidneys and prevent and function RIF in rat models.
TGF-β, a potent anti-inflammatory cytokine, and well-studied fibroblast-promoting factor, is controlled by Renal inflammation [49]. Growth, differentiation, and proliferation of cells are all reportedly affected by it. Renal fibrosis can be brought on by overexpression of TGF-β. The current investigation showed that inducing UUO improved TGF-β expression in the kidneys of rats, an effect that was suppressed by Sul administration. This finding suggests that Sul inhibits RIF via decreasing TGF-β expression in renal tissues. The major intracellular signal transducers of transforming growth factor beta (TGF-β) are Smad superfamily members, including 9 distinct protein isoforms. TGF-β may activate Smads Smad2 and Smad3 via their respective receptors. Smad7, on the other hand, is an inhibitory Smad that inhibits Smad phosphorylation in response to receptor activation [54]. Fibrosis can be prevented or slowed by Smad7’s ability to reduce the expression of Smad4, Smad3, and Smad2. To this end, the present study evaluated Sul effect on the TGF-β/Smad signaling pathway. TGF-β is commonly used to stimulate cell growth in vitro to induce fibrosis. This investigation aimed to determine if Sul could prevent TGF-β-induced cell fibrosis. In the NRK52E cell line and UUO kidney treated with TGF-β, Sul decreased the phosphorylation of Smad4 and Smad2/3 expressions while enhancing the expressions of Smad7. Evidence suggests Sul can suppress RIF by controlling its target protein’s Smad expression.
The TGF-β/Smad signaling system partially mediates renal fibrosis and inflammation, but other routes, such as the connective tissue growth factor (CTGF), MAPK, and Wnt signaling pathways, have been implicated. It has been discovered that the TGF-β pathway crosstalks with the Wnt signaling system, a significantly protected signaling pathway in the cell line. After MAPK protein activation, a vast group of protein kinases, with ERK5/MAPK, p38, JNK/ SAPK, ERK, and NF-κB, is the final product of this pathway [55]. By increasing the tubular synthesis of thrombospondin-1, JNK signaling can awaken the dormant form of the transforming growth factor. It has also been found that TGF-β may prevent enzymatic deconstruction and enhance the development of protease inhibitors of extracellular matrix (ECM) by stimulating the expressions of ECM in MSCs [53]. To promote the invention of extracellular matrix (ECM) proteins, TGF-β induces CTGF. Smad has been shown to have a critical regulatory function in various biological processes at the hub of the crosstalk network and intracellular signaling involving the CTGF, MAPK, and Wnt signaling pathways. Thus, these three routes may account for the antifibrotic properties of Sul.
Renal fibrosis is a progressive disease that begins with inflammation and cannot be separated from it. The expression of inflammatory cytokines can be suppressed once AP-1 and NF-κB are inhibited [56]. The buildup of extracellular matrix proteins like COL3A1 and COL1A1 is facilitated by the proliferation of stromal cells and intercellular stroma, which inflammatory stimuli can trigger. Histological results corroborated the current study’s finding that COL3A1 and COL1A1 expression are elevated in the fibrotic model cell line and the renal system of UUO rats [57]. Based on these results, Sul counteracts fibrosis, reducing inflammatory markers.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author upon request.
References
Gewin LS (2018) Renal fibrosis: primacy of the proximal tubule. Matrix Biol 68:248–262
Lawson J, Elliott J, Wheeler-Jones C, Syme H, Jepson R (2015) Renal fibrosis in feline chronic kidney disease: known mediators and mechanisms of injury. Vet J 203:18–26
Chen P-S, Li Y-P, Ni H-F (2019) Morphology and evaluation of renal fibrosis, renal fibrosis: mechanisms and therapies. Adv Exp Med Biol 1165:17–36
Meng X, Nikolic-Paterson DJ, Lan HY (2016) TGF-β: the master regulator of fibrosis. Nat Rev Nephrol 12:325–338
Sharma PS, Sharma R, Tyagi T (2011) VEGF/VEGFR pathway inhibitors as anti-angiogenic agents: present and future. Curr Cancer Drug Targets 11:624–653
Perona R (2006) Cell signaling: growth factors and tyrosine kinase receptors. Clin Transl Oncol 8:77–82
Sjöblom T, Shimizu A, O’Brien KP, Pietras K, Dal Cin P, Buchdunger E, Dumanski JP, Ostman A, Heldin C-H (2001) Growth inhibition of dermatofibrosarcoma protuberans tumors by the platelet-derived growth factor receptor antagonist STI571 through induction of apoptosis. Cancer Res 61:5778–5783
Pietras K, Ostman A, Sjöquist M, Buchdunger E, Reed RK, Heldin C-H, Rubin K (2001) Inhibition of platelet-derived growth factor receptors reduces interstitial hypertension and increases transcapillary transport in tumors. Cancer Res 61:2929–2934
Modi SJ, Kulkarni VM (2019) Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: medicinal chemistry perspective. Med Drug Discov 2:100009
Wood JM, Bold G, Buchdunger E, Cozens R, Ferrari S, Frei J, Hofmann F, Mestan J, Mett H, O’Reilly T (2000) PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res 60:2178–2189
Kanasaki K, Taduri G, Koya D (2013) Diabetic nephropathy: the role of inflammation in fibroblast activation and kidney fibrosis. Front Endocrinol 4:7
Li W, Cheng F, Songyang Y, Yang S, Wei J, Ruan Y (2020) CTRP1 attenuates UUO-induced renal fibrosis via AMPK/NOX4 pathway in mice. Curr Med Sci 40:48–54
Tieri P, Termanini A, Bellavista E, Salvioli S, Capri M, Franceschi C (2012) Charting the NF-κB pathway interactome map. PLoS ONE 7:e32678
Derynck R, Zhang YE (2003) Smad-dependent and smad-independent pathways in TGF-β family signalling. Nature 425:577–584
Meng X-M, Huang XR, Xiao J, Chung ACK, Qin W, Chen H, Lan HY (2012) Disruption of Smad4 impairs TGF-β/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int 81:266–279
Hu H-H, Chen D-Q, Wang Y-N, Feng Y-L, Cao G, Vaziri ND, Zhao Y-Y (2018) New insights into TGF-β/Smad signaling in tissue fibrosis. Chemico-Biol Interact 292:76–83
Jang HR, Rabb H (2015) Immune cells in experimental acute kidney injury. Nat Rev Nephrol 11:88–101
Campbell MT, Hile KL, Zhang H, Asanuma H, Vanderbrink BA, Rink RR (2011) Meldrum, toll-like receptor 4: a novel signaling pathway during renal fibrogenesis. J Surg Res 168:e61–e69
Pulskens WP, Rampanelli E, Teske GJ, Butter LM, Claessen N, Luirink IK, van der Poll T, Florquin S, Leemans JC (2010) TLR4 promotes fibrosis but attenuates tubular damage in progressive renal injury. J Am Soc Nephrol 21:1299–1308
Cardozo LFMF, Alvarenga LA, Ribeiro M, Dai L, Shiels PG, Stenvinkel P, Lindholm B, Mafra D (2021) Cruciferous vegetables: rationale for exploring potential salutary effects of sulforaphane-rich foods in patients with chronic kidney disease. Nutr Rev 79:1204–1224
Vanduchova A, Anzenbacher P, Anzenbacherova E (2019) Isothiocyanate from broccoli, sulforaphane, and its properties. J Med Food 22:121–126
Guerrero-Beltrán CE, Calderón-Oliver M, Pedraza-Chaverri J, Chirino YI (2012) Protective effect of sulforaphane against oxidative stress: recent advances. Exp Toxicol Pathol 64:503–508
Çakır I, Lining Pan P, Hadley CK, El-Gamal A, Fadel A, Elsayegh D, Mohamed O, Rizk NM (2022) Ghamari-Langroudi, sulforaphane reduces obesity by reversing leptin resistance. Elife 11:e67368
Yoon H-Y, Kang N-I, Lee H-K, Jang KY, Park J-W, Park B-H (2008) Sulforaphane protects kidneys against ischemia–reperfusion injury through induction of the Nrf2-dependent phase 2 enzyme. Biochem Pharmacol 75:2214–2223
Chang R (2022) Research advances in the protective effect of sulforaphane against kidney injury and related mechanisms. In: BIO web of conferences, EDP sciences, p 1006
Fawzy E, Nehad E (2011) Potential health benefits of sulforaphane: a review of the experimental, clinical and epidemiological evidences and underlying mechanisms. J Med Plants Res 5:473–484
Zeng X, Su W, Zheng Y, He Y, He Y, Rao H, Peng W, Yao H (2019) Pharmacokinetics, tissue distribution, metabolism, and excretion of naringin in aged rats. Front Pharmacol 10:34
Adil M, Kandhare AD, Ghosh P, Venkata S, Raygude KS, Bodhankar SL (2016) Ameliorative effect of naringin in acetaminophen-induced hepatic and renal toxicity in laboratory rats: role of FXR and KIM-1. Ren Fail. 38:1007–1020
El-Desoky AH, Abdel-Rahman RF, Ahmed OK, El-Beltagi HS, Hattori M (2018) Anti-inflammatory and antioxidant activities of naringin isolated from Carissa carandas L.: in vitro and in vivo evidence. Phytomedicine 42:126–134
Wang L, Mao N, Tan R, Wang H, Wen J, Liu Y, Furhad M, Fan J (2015) Ginsenoside Rg1 reduces aldosterone-induced autophagy via the AMPK/mTOR pathway in NRK-52E cells. Int J Mol Med 36:518–526
Münz S, Wolf L, Hoelzle LE, Chernyakov D, Edemir B, Föller M (2022) Impact of cytotoxic agents or apoptosis stimulants on αklotho in MDCK, NRK-52E and HK2 kidney cells. Aging 14:7282
Rovetta F, Stacchiotti A, Consiglio A, Cadei M, Grigolato PG, Lavazza A, Rezzani R, Aleo MF (2012) ER signaling regulation drives the switch between autophagy and apoptosis in NRK-52E cells exposed to cisplatin. Exp Cell Res 318:238–250
Davis MA, Smith MW, Chang SH, Trump BF (1994) Characterization of a renal epithelial cell model of apoptosis using okadaic acid and the NRK-52E cell line. Toxicol Pathol 22:595–605
Chen X, Zhouhua W, Jie Z, Xinlu F, Jinqiang L, Yuwen Q, Zhiying H (2015) Renal interstitial fibrosis induced by high-dose mesoporous silica nanoparticles via the NF-κB signaling pathway. Int J Nanomed. https://doi.org/10.2147/IJN.S73538
Wei S, Xu C, Zhang Y, Shi Z, Wu M, Yang B (2020) Ultrasound assisted a peroxisome proliferator-activated receptor (PPAR) γ agonist-loaded nanoparticle-microbubble complex to attenuate renal interstitial fibrosis. Int J Nanomed. 15:7315–7327
Zhang X, Xia C, Li S (2019) Efficacy of Cortex Mori-Liuwei Dihuang nanoparticle pills on renal interstitial fibrosis via TGF-β/Smad signaling. Nanosci Nanotechnol Lett 11:1661–1665
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2–∆∆CT method. Methods 25:402–408
Lan HY (2011) Diverse roles of TGF-β/Smads in renal fibrosis and inflammation. Int J Biol Sci 7:1056
Zhang M, Guo Y, Fu H, Hu S, Pan J, Wang Y, Cheng J, Song J, Yu Q, Zhang S (2015) Chop deficiency prevents UUO-induced renal fibrosis by attenuating fibrotic signals originated from Hmgb1/TLR4/NFκB/IL-1β signaling. Cell Death Dis 6:e1847
Nogueira A, Pires MJ, Oliveira PA (2017) Pathophysiological mechanisms of renal fibrosis: a review of animal models and therapeutic strategies. In Vivo 31:1–22
Gong W, Mao S, Yu J, Song J, Jia Z, Huang S, Zhang A (2016) NLRP3 deletion protects against renal fibrosis and attenuates mitochondrial abnormality in mouse with 5/6 nephrectomy. Am J Physiol-Ren Physiol 310:F1081–F1088
Lucarelli G, Mancini V, Galleggiante V, Rutigliano M, Vavallo A, Battaglia M, Ditonno P (2014) Emerging urinary markers of renal injury in obstructive nephropathy. BioMed Res Int 2014:303298
Xin B, Wang X, Jin W, Yan H, Cui B, Zhang X, Hua F, Yang H, Hu Z (2010) Activation of toll-like receptor 9 attenuates unilateral ureteral obstruction-induced renal fibrosis. Acta Pharmacol Sin 31:1583–1592
Lan HY, Mu W, Tomita N, Huang XR, Li JH, Zhu H-J, Morishita R, Johnson RJ (2003) Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J Am Soc Nephrol 14:1535–1548
Teng S, Liu G, Li L, Ou J, Yu Y (2022) CUX1 promotes epithelial–mesenchymal transition (EMT) in renal fibrosis of UUO model by targeting MMP7. Biochem Biophys Res Commun 608:128–134
Ai K, Li X, Zhang P, Pan J, Li H, He Z, Zhang H, Yi L, Kang Y, Wang Y (2022) Genetic or siRNA inhibition of MBD2 attenuates the UUO-and I/R-induced renal fibrosis via downregulation of EGR1, molecular therapy-nucleic acids. Mol Ther Nucleic Acids. 28:77–86
Lee JA, Shin M-R, Roh S-S (2022) Corni fructus alleviates UUO-induced renal fibrosis via TGF-β/Smad signaling. BioMed Res Int. 2022:5780964
Wang Q, Peng Z, Xiao S, Geng S, Yuan J, Li Z (2007) RNAi-mediated inhibition of COL1A1 and COL3A1 in human skin fibroblasts. Exp Dermatol 16:611–617
Biernacka A, Dobaczewski M, Frangogiannis NG (2011) TGF-β signaling in fibrosis. Growth Factors 29:196–202
Zhang X, Huang H, Zhang G, Li D, Wang H, Jiang W (2019) Raltegravir attenuates experimental pulmonary fibrosis in vitro and in vivo. Front Pharmacol 10:903
Zeglinski MR, Hnatowich M, Jassal DS, Dixon IMC (2015) SnoN as a novel negative regulator of TGF-β/Smad signaling: a target for tailoring organ fibrosis. Am J Physiol Heart Circ Physiol 308:H75–H82
Baek S, Jang MG, Kim J-W, Ko HC, Nam MH, Hur S-P, Park SA, Kim S-J (2022) Polymethoxyflavone-rich fraction from Citrus Sunki leaves alleviates renal dysfunction in mice with unilateral ureteral obstruction. Nat Prod Commun 17:1934578X221109412
Masola V, Carraro A, Granata S, Signorini L, Bellin G, Violi P, Lupo A, Tedeschi U, Onisto M, Gambaro G (2019) In vitro effects of interleukin (IL)-1 beta inhibition on the epithelial-to-mesenchymal transition (EMT) of renal tubular and hepatic stellate cells. J Transl Med 17:1–11
Hammad FT (2022) The long-term renal effects of short periods of unilateral ureteral obstruction. Int J Physiol Pathophysiol Pharmacol 14:60
Caraci F, Gili E, Calafiore M, Failla M, La Rosa C, Crimi N, Sortino MA, Nicoletti F, Copani A, Vancheri C (2008) TGF-β1 targets the GSK-3β/β-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol Res 57:274–282
Liu J-H, He L, Zou Z-M, Ding Z-C, Zhang X, Wang H, Zhou P, Xie L, Xing S, Yi C-Z (2018) A novel inhibitor of homodimerization targeting MyD88 ameliorates renal interstitial fibrosis by counteracting TGF-β1-induced EMT in vivo and in vitro. Kidney Blood Pressure Res 43:1677–1687
Liu B, Sun T, Li H, Qiu S, Li Y, Zhang D (2022) Proximal tubular RAGE mediated the renal fibrosis in UUO model mice via upregulation of autophagy. Cell Death Dis 13:1–12
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, Z., He, W. & Shi, W. Sulforaphane (Sul) reduces renal interstitial fibrosis (RIF) by controlling the inflammation and TGF-β/Smad signaling pathway. Appl Biol Chem 67, 8 (2024). https://doi.org/10.1186/s13765-024-00858-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-024-00858-x