
Abstract
Nine morpholine-derived halogenated chalcone derivatives (MHC1-MHC9) were synthesized, and their inhibitory activity against monoamine oxidase (MAO) was evaluated. MHC5 showed the highest inhibitory activity against MAO-B with an IC50 value of 0.065 μM, followed by MHC7 (IC50 = 0.078 μM) and MHC6 (IC50 = 0.082 μM). The para-F substituent MHC4 was also potent (IC50 = 0.095 μM). The selectivity index values of all the compounds were high for MAO-B over MAO-A, and the values for MHC5 and MHC4 were 66.15 and 80.11, respectively. MHC5 and MHC4 were competitive MAO-B inhibitors with Ki values of 0.024 ± 0.00062 and 0.041 ± 0.0028 μM, respectively. In reversibility tests, the changes in residual activity before and after the dialysis of MHC5 and MHC4 were similar to those of safinamide, a reversible MAO-B reference inhibitor. Additionally, molecular docking and dynamic simulations predicted that the lead molecules MHC5 and MHC4 could strongly bind to the MAO-B active site with docking scores of –10.92 ± 0.08 and –10.64 ± 0.14 kcal/mol, respectively. Additionally, MHC4 and MHC5 exhibited favorable ADME features, including blood–brain barrier permeability. The experiments confirmed that MHC5 and MHC4 are reversible and potent selective inhibitors of MAO-B and are promising candidates for the treatment of neurodegenerative diseases (human health).
Similar content being viewed by others
Introduction
Monoamine oxidases (MAOs) substantially inactivate various biogenic amines in central and peripheral tissues [1]. Several neuropsychiatric and neurodegenerative disorders, such as Alzheimer's disease (AD) and Parkinson’s disease (PD) are primary treatment targets of these enzymes [2]. MAO-A and MAO-B are two isoenzymes found on the mitochondrial outer membrane that produce hydrogen peroxide [3]. The quest for selective MAO inhibitors is currently a crucial focus in the field of drug discovery. Understanding the catalytic mechanism, substrate selectivity, and inhibitor-binding mode can accelerate the development of selective MAO inhibitors [4]. Early-stage PD was successfully and safely treated with monotherapy using MAO-B inhibitors. In contrast, adjuvant MAO-B inhibitors are routinely used to treat severe diseases [5]. Significant progress has been made in the development of selective MAO-B inhibitors during the last five years, including outcomes, structures, structure–activity correlations (SARs), and medicinal chemistry approaches [6].
The MAO-B inhibitor selegiline, which is used either alone in monotherapy or in combination with levodopa, is effective in treating the symptoms of PD. First-pass metabolism converts selegiline into R(–)amphetamine and R(–)methamphetamine, which may have detrimental effects on the cardiovascular and neurological systems [7]. The third-generation MAO-B inhibitor safinamide, a reversible and selective MAO-B inhibitor, has favorable pharmacokinetic and pharmacodynamic features for the treatment of PD [8]. Recently, several studies have suggested that many structural scaffolds exhibit remarkable MAO-B inhibition with a reversible and competitive mode of inhibition. These include chalcones, pyrazolines, isatins, coumarins, chromones, and benzyloxy-derived compounds [9,10,11,12,13,14,15,16,17,18].
Chalcones are bioactive compounds containing carbonyl-conjugated systems and two electrophilic sites. These traditional substances are the biogenetic antecedents of various therapeutically useful substances, including flavonoids and isoflavonoids [19]. Chalcones have cis and trans isomers owing to the presence of olefinic bonds, although the thermodynamically more stable trans form is more prevalent. Additionally, this unusual conjugated form of ketones serves as a Michael acceptor in several cellular biochemical signaling pathways [20]. The physicochemical properties of chalcones, which are provided by the components that make up the complete molecule, are directly related to how well they block selective MAO-B enzymes [11]. Many chalcones linked to FDA-approved drugs have recently shown remarkable MAO-B inhibition [21,22,23,24]. The electrophilic nature of the enone unit of chalcones is influenced by the differential electron density of the aromatic and heterocyclic nuclei caused by the presence of different substituents [11]. The ongoing efforts over the past several years to develop potent MAO-B inhibitors have led to the identification of novel compounds. However, based on the aforementioned data, the parent chalcone scaffold continued to play a significant role in inhibiting MAO-B.
Morpholine is a tetrahydro-1,4-oxazine containing a saturated heterocyclic ring. It is a nonplanar heteronuclear structure comprising two ethylene bridges connecting electronegative oxygen and nitrogen atoms. The morpholine ring is an integral part of central nervous system (CNS)-acting FDA-approved drugs such as phendimetrazine-anorectic, moclobemide-reversible MAO-A inhibitory, reboxetine-antidepressant, and rocuronium-neuromuscular agents (Fig. 1) [25]. Recently, morpholine-containing chalcones were shown to exhibit potent and selective MAO-B inhibitory activity [26, 27]. The current study focuses on the MAO inhibitory effect of the morpholine heterocyclic system on the para position of ring B of the phenyl system and the fine-tuning of various halogens on the phenyl ring A in the chalcones.

Morpholine structure-based drugs approved by FDA
Materials and methods
Synthesis
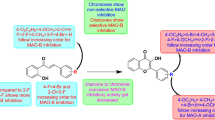
Equimolar amounts of 4(4-formylphenyl) morpholine (0.01 M) and the corresponding acetophenones (0.01 M) were combined in a beaker and dissolved in 7.5 mL of 40% NaOH and 15–20 mL of methanol. The reaction mixture was stirred for 14–24 h using a magnetic stirrer. After completion of the reaction, ice cubes were added to the mixture and the precipitate was collected. The product was filtered and rinsed with ice-cold water to remove excess base. Methanol was used to recrystallize the product. The synthesis route is shown in Scheme 1.

Synthetic route of halogenated chalcones bearing morpholine ring
(2E)-3-[4-(morpholin-4-yl)phenyl]-1-phenylprop-2-en-1-one (MHC1)
1H NMR (500 MHz, CDCl3): δ 8.00–7.99 (d, 2H), 7.78–7.75 (d, 1H), 7.57–7.53 (m, 3H), 7.49–7.46 (d, 2H), 7.39–7.36 (d, 1H), 6.89–6.87 (d, 2H), 3.86–3.84 (m, 4H), 3.26–3.24(m, 4H); 13C NMR (125 MHz, CDCl3) δ 190.63, 152.77, 145.03, 144.02, 138.76, 132.41, 130.15, 128.54, 128.39, 125.82, 118.73, 114.64, 66.64, 48.00. Molecular formula C19H19NO2 (HRMS) Calculated = 293.3596, Observed = 293.3595. Elemental analysis calculated: C, 77.79; H, 6.53; N, 4.77; O, 10.91, Observed: C, 77.81; H, 6.51; N, 4.74; O, 10.94.
(2E)-1-(4-chlorophenyl)-3-[4-(morpholin-4-yl)phenyl]prop-2-en-1-one (MHC2)
1H NMR (500 MHz, CDCl3): δ 7.95–7.93 (d, 2H), 7.78–7.75 (d, 1H), 7.57–7.54 (d, 2H), 7.46–7.43 (m, 2H), 7.34–7.31 (d, 1H), 6.89–6.87 (d, 2H), 3.86–3.84 (m, 4H), 3.27–3.25 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 189.18, 152.88, 145.25, 138.74, 137.06, 130.26, 129.80, 128.82, 125.56, 118.03, 114.58, 66.63, 47.92. Molecular formula C19H18ClNO2 (HRMS) Calculated = 327.8047, Observed = 327.8046. Elemental analysis calculated: C, 69.62; H, 5.53; Cl, 10.81; N, 4.27; O, 9.76, Observed: C, 69.61; H, 5.554; Cl, 10.80; N, 4.30; O, 9.74.
(2E)-1-(4-bromophenyl)-3-[4-(morpholin-4-yl)phenyl]prop-2-en-1-one (MHC3)
1H NMR (500 MHz, CDCl3): δ 7.88–7.85 (m, 2H), 7.78–7.75 (d, 1H), 7.63–7.60 (m, 2H), 7.57–7.54(d, 2H), 7.33–7.30 (d, 1H), 6.90–6.87 (m, 2H), 3.86–3.84 (m, 4H), 3.28–3.26 9 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 189.40, 152.89, 145.60, 137.49, 131.80, 130.27, 129.93, 127.40, 125.55, 118.01, 114.58, 66.63, 47.92. Molecular formula C19H18BrNO2 (HRMS) Calculated = 372.2557, Observed = 372.2556. Elemental analysis calculated: C, 61.30; H, 4.87; Br, 21.46; N, 3.76; O, 8.60, Observed: C, 61.30; H, 4.87; Br, 21.46; N, 3.76; O, 8.60.
(2E)-1-(4-fluorophenyl)-3-[4-(morpholin-4-yl)phenyl]prop-2-en-1-one (MHC4)
1H NMR (500 MHz, CDCl3) δ: 8.05–8.02 (m, 2H), 7.78–7.75 (d, 1H), 7.57–7.55 (d, 2H), 7.36–7.33 (d, 1H), 7.17–7.13 (m, 2H), 6.90–6.87 (d, 2H), 3.86–3.84 (m, 4H), 3.27–3.25 (m, 4H); 13C NMR (125 MHz, CDCl3) δ: 199.87, 166.40, 164.39, 152.79, 145.20, 135.06, 135.04, 130.95, 130.87, 130.19, 125.69, 116.16, 115.68, 115.51, 114.63, 47.98. Molecular Formula C19H18FNO2 (HRMS) Calculated = 311.35012, Observed = 311.3501. Elemental analysis calculated: C, 73.30; H, 5.83; F, 6.10; N, 4.50; O, 10.28, Observed: C, 73.31; H, 5.82; F, 6.09; N, 4.51; O, 10.29.
2E)-1-(3-chlorophenyl)-3-[4-(morpholin-4-yl)phenyl]prop-2-en-1-one (MHC5)
1H NMR (500 MHz, CDCl3) δ: 7.96–7.95 (m, 1H), 7.87–7.86 (m, 1H), 7.79–7.76 (d, 1H), 7.57–7.52 (d, 2H), 7.52–7.51 (m, 1H), 7.43–7.47 (m, 1H), 7.32–7.26 (m, 1H), 6.89–6.87 (d, 2H) 3.86–3.84(m, 4H) 3.28–3.26 (m, 4H); 13C NMR (125 MHz, CDCl3) δ: 189.10, 152.94, 145.86, 140.40, 134.70, 132.29, 130.35, 129.86, 128.86, 126.44, 125.46, 117.95, 115.75, 77.32, 77.07, 47.89. Molecular Formula C19H18ClNO2 (HRMS) Calculated = 327.8047, Observed = 327.8046. Elemental analysis calculated: C, 69.62; H, 5.53; Cl, 10.81; N, 4.27; O, 9.76, Observed: C, 69.60; H, 5.54; Cl, 10.82; N, 4.28; O, 9.75.
(2E)-1-(3-bromophenyl)-3-(4-morpholinophenyl)prop-2-en-1-one (MHC6)
1H NMR (500 MHz, CDCl3) δ: 8.12–8.11 (d, 1H), 7.92–7.90 (d, 1H), 7.79–7.76 (d, 1H), 7.68–7.66 (m, 1H), 7.58–7.56 (m, 2H), 7.37–7.34 (d, 1H), 7.31–7.28 (d, 1H), 6.90–6.88 (d, 2H), 3.86–3.85 (m, 4H), 3.28–3.26 (m, 4H); 13C NMR (125 MHz, CDCl3) δ: 189.00, 152.93 145.89, 140.60, 135.18, 131.36, 130.34, 126.86, 125.46, 122.84, 117.91, 114.54, 66.60, 47.88. Molecular Formula C19H18BrNO2 (HRMS) Calculated = 372.2557, Observed = 372.2557. Elemental analysis calculated: C, 61.30; H, 4.87; Br, 21.46; N, 3.76; O, 8.60, Observed: C, 61.28; H, 4.89; Br, 21.44; N, 3.77; O, 8.61.
(2E)-1-(3-fluorophenyl)-3-[4-(morpholin-4-yl)phenyl]prop-2-en-1-one (MHC7)
1H NMR (500 MHz, CDCl3) δ: 7.80–7.77 (m, 2H), 7.69–7.68 (m, 1H), 7.57–7.56 (d, 2H), 7.48–7.44 (m, 1H), 7.33–7.30 (d, 1H), 7.27–7.25 (m, 1H), 6.90–6.88 (d, 2H), 3.86–3.84 (m, 4H), 3.28–3.26 (m, 4H); 13C NMR (125 MHz, CDCl3) δ: 189.16, 163.85, 161.88, 152.92, 140.91, 130.12, 125.52, 119.42, 115.27, 115.57, 77.03, 77.07, 76.79, 47.91. Molecular Formula C19H18FNO2 (HRMS) Calculated = 311.3501, Observed = 311.3500. Elemental analysis calculated: C, 73.30; H, 5.83; F, 6.10; N, 4.50; O, 10.28, Observed: C, 73.28; H, 5.84; F, 6.11; N, 4.52; O, 10.26.
(2E)-1-(2-chlorophenyl)-3-[4-(morpholin-4-yl)phenyl]prop-2-en-1-one (MHC8)
1H NMR (500 MHz, CDCl3) δ: 7.49–7.48 (m, 2H), 7.45–7.43 (m, 2H), 7.40–7.35 (m, 3H), 6.97–6.94 (d, 2H), 6.87–6.84 (m, 2H), 3.85–3.83 (m, 4H), 3.27–3.25(m, 4H); 13C NMR (125 MHz, CDCl3) δ: 193.99, 152.98, 146.80, 139.61, 131.20, 130.99, 130.34, 130.20, 129.23, 126.70, 125.19, 123.10, 114.53, 77.30, 77.05, 76.79, 47.86. Molecular Formula C19H18ClNO2 (HRMS) Calculated = 327.8080, Observed = 327.8046. Elemental analysis calculated: C, 69.62; H, 5.53; Cl, 10.81; N, 4.27; O, 9.76, Observed: C, 69.60; H, 5.52; Cl, 10.80; N, 4.28; O, 9.77.
(2E)-1-(2-fluorophenyl)-3-[4-(morpholin-4-yl)phenyl]prop-2-en-1-one (MHC9)
1H NMR (500 MHz, CDCl3): δ 7.80–7.76 (m, 1H), 7.70–7.66 (m, 1H), 7.55–7.52 (m, 2H), 7.51–7.46 (m, 1H), 7.26–7.21 (m, 2H), 7.16–7.12 (m, 1H), 6.89–6.86 (m, 2H), 3.86–3.84 (m, 4H), 3.27–3.25(m, 4H); 13C NMR (125 MHz, CDCl3) δ 189.18, 162.01, 160.00, 152.87, 145.24, 133.43, 130.88, 127.74, 125.56, 124.42, 122.43, 116.53, 116.34, 114.57, 66.63, 47.93. Molecular formula C19H18FNO2 (HRMS) Calculated = 311.3501, Observed = 311.3501. Elemental analysis calculated: C, 73.30; H, 5.83; F, 6.10; N, 4.50; O, 10.28, Observed: C, 73.32; H, 5.84; F, 6.09; N, 4.48; O, 10.27.
Biochemistry
Inhibition studies of MAO-A and MAO-B
The activities of MAO-A and MAO-B were assayed using kynuramine (0.06 mM) and benzylamine (0.30 mM), respectively, by continuously measuring the changes in absorbance at 316 and 250 nm, respectively [28]. Toloxatone, clorgyline, safinamide, and pargyline were used as reference compounds. Recombinant human MAO-A, MAO-B, benzylamine, kynuramine, safinamide mesylate salt, pargyline, clorgyline, and toloxatone were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Enzyme kinetics
The enzyme activity of MAO-B was assayed at five substrate concentrations around the Km value (0.0375 – 0.6 μM) without inhibitor for enzyme kinetics. Inhibition kinetics were determined prior to the inhibition study. As an initial screening step, residual activity was analyzed by measuring the change in absorbance in the presence of the inhibitor at 10 μM. IC50 values were determined for potential compounds with residual activity of less than 80% from residual activity curves that were plotted using GraphPad Prism software 5 (San Diego, CA, USA) [29]. The limit of IC50 was set to 40 μM. If necessary, a concentration of 1 μM was used for potent inhibitors. The selectivity index (SI) of MAO-B was calculated by dividing IC50 of MAO-A / IC50 of MAO-B [30]. The type of inhibition of the leading compound for MAO-B was determined at three inhibitor concentrations, ~ 1/2 × , 1 × , and 2 × IC50, as well as at five different substrate concentrations [31]. Enzyme kinetic patterns and Ki values were determined by comparing Lineweaver–Burk (LB) plots and their secondary plots [32].
Reversibility studies
The reversibilities of the leading compounds for MAO-A and MAO-B were evaluated by comparing the undialyzed (AU) and dialyzed (AD) residual activities at a concentration approximately twice the IC50 after preincubation for 30 min before measurement. The time-dependency curve was constructed by measuring the residual activity after preincubation for 0, 5, 10, 20, 30, and 60 min at the concentration, and the preincubation time was selected. Two types of reference inhibitors were used for MAO-A and MAO-B: reversible inhibitors toloxatone and safinamide mesylate (for MAO-A and MAO-B, respectively) and irreversible inhibitors clorgyline and pargyline (for MAO-A and MAO-B, respectively) [33]. A dialysis kit (6–8 kDa, DiaEasy™) was purchased from BioVision (St. Louis, MA, USA).
Computational studies
Molecular docking
A molecular docking study of the lead compounds, MHC5 and MHC4 from the enzyme inhibition analysis was conducted using the Schrödinger suite [34]. The X-ray structure of human MAO-B (hMAO-B, PDB ID:2V5Z) was obtained from the Protein Data Bank [35]. The protein preparation wizard from the Schrödinger suite, which also performed energy minimization, hydrogen atom addition, protonation state correction, and protonation state addition, was used to enhance and optimize the crystal structures. The ligand structure was constructed using the LigPrep software. The automated center of the grid box comprised the co-crystallized ligands. The Extra Precision (XP) docking protocol default parameters and Force Field OPLS4 default settings were applied to the docking simulations [36, 37].
Molecular dynamic simulation
The molecular dynamics (MD) simulations were conducted using Schrodinger LLC's Desmond simulation program (Shaw, 2021). For the Desmond system builder panel, a protein–ligand combination was initially developed utilizing the compounds MHC5 and MHC4 against MAO-B using an aqueous solvent system. The simulation parameters were 100 ns at 300 K, 1.01325 bar pressure, and 1000 frames for full protein–ligand simulations and stability trajectory analysis such as root-mean-square deviation (RMSD), root mean square fluctuation (RMSF), and protein–ligand interactions [36, 37].
MM-GBSA
The Molecular Mechanics Generalized Born and Surface Area (MM-GBSA) solvation method was employed to calculate the binding energies of ligands to proteins. Multiple poses from molecular dynamics (MD) simulations of the docked complex were utilized to assess both macromolecular stability and the affinity between proteins and ligands. The free energy was determined using the specified formula during the post-processing stage following the MD studies.
The molecular mechanics framework designates the internal, electrostatic, and van der Waals energies as Eint, Eele, and Evdw, respectively. The equation includes representations for the free energy contributions of polar and non-polar solvation systems, indicated by Gpol and Gnp, respectively. S stands for an entropy estimate, and T represents the absolute temperature. The formula employed to calculate the binding free energy, ΔG Bind, between the ligand and the protein is as follows:
where PL is the complex, p is the protein, and L is the ligand.
ADME prediction
The ADME of a potential therapeutic agent is considered throughout the drug discovery process to ensure appropriate pharmacokinetic parameters. Using web-based software such as SwissADME (http://www.swissadme.ch/) and pkCSM (http://biosig.unimelb.edu.au/pkcsm/), pharmacokinetic parameters were determined in silico [38, 39].
Results and discussion
Chemistry
To synthesize morpholine-containing unsaturated ketones, benzaldehyde-based morpholines, and appropriate halogen-derived methyl ketones were combined using the Claisen-Schmidt condensation method in the presence of an alcoholic basic medium (Scheme 1). The H1 and H2 protons of the morpholine ring resonated at 3.28–3.23 and 3.83–3.86 ppm as triplets, respectively, according to the 1H NMR spectra of MHC1-MHC9. Chalcones containing morpholine had sharp doublets of Hα and Hβ protons at 7.33–7.58 and 7.80–7.70 ppm, respectively. The large coupling constant of 15 Hz indicated a double bond in the trans configuration of the chalcones. The existence of a sharp deshielded sp2 carbonyl carbon at 193–189 ppm and the morpholine ring containing (CH2)-N, and (CH2)-O carbon of shielded area range between 40 and 70 ppm in the 13C-NMR data supported the hypothesis that the compound was an α, β-unsaturated ketone system. Owing to the nature of the targeted compounds, all chalcones containing morpholine exhibited intense molecular ions in their mass spectra (Additional file 1).
Enzyme inhibition studies
Inhibition studies of MAO-A and MAO-B
All compounds tested showed low residual activity of < 50% for MAO-B at a concentration of 1 μM, while six showed low residual activity of < 50% for MAO-A at 10 μM (Table 1). MHC5 had an IC50 value of 0.065 μM, showing the greatest inhibitory ability against MAO-B, followed by compounds MHC7 (IC50 = 0.078 μM) and MHC6 (IC50 = 0.082 μM). The compound with the highest MAO-A inhibitory activity was MHC2, with an IC50 of 0.82 μM. All the compounds showed higher inhibitory activity against MAO-B compared to MAO-A, with high SI values for MAO-B. The SI value of MHC8 was 102.47, followed by MHC4 (80.11) and MHC5 (66.15). Comparing the MHC series of compounds with reference compounds for MAO-B inhibition, MHC5 had lower potency than safinamide (a reversible MAO-B inhibitor); however, it had similar or better potency than pargyline (an irreversible MAO-B inhibitor).
All the compounds in the present study were chalcone-based derivatives with a morpholine ring bonded to the B-ring. Previously, we synthesized 1-(4-morpholinophenyl) prop-2-en-1-one (chalcones). In this design strategy, a morpholine ring was placed on the A ring of chalcones, and various electron-donating and electron-withdrawing groups were added to the para position of the phenyl B ring of the basic scaffold.
The MHC series contained different substituents based on the type and location of the halogen atom in the A-ring of chalcone. Considering the IC50 values, we found that the MAO-B inhibitory effect was the best when a halogen was bonded to the meta-site of ring A, compared to the para- or ortho-sites, which had similar IC50 values. When comparing the meta-derivative sites, the inhibitory ability was in the order of–Cl > –F > –Br, although the difference was insignificant. The structure–activity relationships (SARs) are shown in Fig. 2.

SARs of morpholine-based compounds
Enzyme kinetics
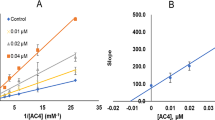
Enzyme kinetics and inhibition studies were performed at five substrate concentrations and three inhibitor concentrations for para-Cl substituted MHC5 and meta-F substituted MHC4. In the Lineweaver-Bulk plot, MHC5 and MHC4 appeared to be competitive MAO-B inhibitors (Fig. 3A, C). From the secondary plots, the Ki values of both compounds were found to be 0.024 ± 0.00062 and 0.041 ± 0.0028 μM, respectively (Fig. 3B, D). These results suggested that MHC5 and MHC4 are acting as competitive inhibitors of MAO-B.

Lineweaver–Burk plots for MAO-B inhibitions by MHC5 (A) and MHC4 (C), and their respective secondary plots (B and D) of the slopes vs. inhibitor concentrations
Reversibility studies
The reversibility of MAO-B inhibition by MHC5 and MHC4 was analyzed using the dialysis method after 30 min of pre-incubation. The preincubation time was selected as 30 min, based on their time-dependency studies (Additional file 1: Figure S10). Concentrations of MHC5, MHC4, safinamide, and pargyline used were twice their IC50 values (0.13, 0.19, 0.038, and 0.22 μM, respectively). Recovery patterns were compared using undialyzed (AU) and dialyzed (AD) relative activities. The inhibition of MAO-B by compounds MHC5 and MHC4 recovered from 17.35% to 72.39% and from 9.02% to 74.70%, respectively (Fig. 4). The recovery of the compound was similar to that of safinamide (reversible type, 24.98–70.03%) and could be distinguished from that of pargyline (irreversible type, 20.76–10.35%). These results suggested that MHC5 and MHC4 are reversible inhibitors of MAO-B.

Recovery of MAO-B inhibition by MHC5 and MHC4 in residual activities measured using dialysis experiments
Computational studies
Molecular docking
Using molecular docking, we further analyzed the hypothetical binding modes of the lead compounds (MHC4, MHC5, MHC6, and MHC7) as effective selective MAO-B inhibitors. Using the Glide module, we docked successful compounds from virtual screening into the binding cavity of 2V5Z. Using increased precision (XP), we examined the hits which showed docking scores ranging from -10.915 to -10.078 kcal/mol (Table 2). To validate the docking approach, we co-crystallized the ligand in the binding site of 2V5Z with 0.90 Å RMSD. The binding affinities of these substances were comparable to that of the co-crystallized ligand (safinamide, XP = -11.313 kcal/mol). As shown in Fig. 6, all lead molecules were bound to the same binding site of the co-crystallized ligand.
In the binding site of 2V5Z, MHC5 and MHC4 interacted with important amino acids, such as Tyr60, Phe99, Pro102, Phe103, Phe104, Trp119, Leu164, Leu167, Phe168, Leu171, Cys172, Tyr188, Ile198, Ile199, Gln206, Tyr398, Tyr435, Ile316, Tyr326, Leu328, and Phe343 (Fig. 5). In particular, Tyr326 interacted through pi-pi stacking with the phenyl ring attached to morpholine in both ligands (MHC5 and MHC4). Analysis of the orientation of all lead molecules revealed that their A rings were in close proximity to the FAD cofactor (Fig. 6).

2D interaction of MHC5 (A) and MHC4 (B) with MAO-B binding pocket

3-D Superimposition of all ligands that bind close to the FAD cofactor (yellow). MHC4, MHC5, MHC6, and MHC7 are represented by the colors, violet, blue, green, and red, respectively
MD simulation
MD simulation is a widely used and well-known method that aids in our understanding of the stability and interaction of enzyme-ligand complexes in the process of computer-aided drug development. We used 100 ns MD simulations on the best lead molecules, MHC5 and MHC4 to determine the stability of the receptor-ligand complex interacting inside the 2V5Z binding pocket after performing molecular docking against MAO-B.
Root mean square deviation (RMSD)
The root-mean-square deviation (RMSD) value derived from the MD simulation trajectory is one of the most crucial markers for identifying multiple structural conformations of the protein backbone over time during system equilibration. A stable protein structure is represented by low and stable RMSD values. The protein should be close to equilibrium, which means that the RMSD value should remain constant to evaluate the ligand–protein interaction. A comparison of the RMSD of lead and the reference molecule with that of MAO-B is shown in Fig. 7. RMSD was used to measure the alpha carbon atoms in the protein backbone and evaluate the stability of the protein throughout the simulation. The average RMSD values for MHC5 and MHC4 were 2.79 and 2.37, respectively. Both lead compounds exhibited similar behavior throughout the 100 ns. At 20–35 ns after stabilization, the MHC5 cells showed a visual change. In the case of MHC4, modest variations were observed between 10 and 30 ns and between 40 and 60 ns. Therefore, it can be concluded that MHC5 differs slightly from MHC4 in its interactions; however, overall, both molecules exhibited stability in protein–ligand complexes.

RMSD plots of MHC5 and MHC4 to MAO-B
Root mean square fluctuation (RMSF)
The flexibility and mobility of particular amino acids are described by the root mean square fluctuation (RMSF). Given the reduced RMSF, the residue might have been less active and mobile. It may be argued that the bond between the protein and ligand is stronger when the RMSF value is low at residues near the active site or at residues where the ligand interacts with the receptor protein. Higher values (peaks) indicate the existence of twists, loose bonding loops, terminal ends, and loose bonding, demonstrating the flexibility of the structure. Low values indicated secondary structures such as β-sheets and α-helices, which suggested structural stability. The RMSF variation for MHC5 and MHC4 with the 2V5Z protein was revealed to be 0.50–3.0 Å (Fig. 8). By comparing the RMSF values of both lead compounds, MHC5 exhibited higher fluctuation than MHC4, i.e., overall MHC5 and MHC4 for 0.97 Å and 0.92 Å, respectively. When MHC5 was simulated, important contacts were observed in 24 amino acids such as Leu88 (0.67 Å), His90 (0.76 Å), Gly101 (0.98 Å), Phe99 (0.87 Å), Pro102 (0.91 Å), Pro104 (0.83 Å), Trp119 (0.79 Å), Leu164 (0.81 Å), Leu167 (0.70 Å), Phe168 (0.66 Å), Ile171 (0.66 Å), Cys172 (0.65 Å), Tyr188 (0.58 Å), Cys192 (0.57 Å), Ile198 (0.58 Å), Ile199 (0.68 Å), Gly205 (0. 69 Å), Gln206 (0.64 Å), Ile316 (0.69 Å), and Tyr326 (0.60 Å), Phe343 (0.59 Å), Leu345 (0.66 Å), Tyr398 (0.50 Å), and Tyr435 (0.45 Å). In case of MHC4, important contacts were in 21 amino acid such as Tyr60 (0.54 Å), His90 (0.81 Å), Pro102 (0.90 Å), Pro104 (0.85 Å), Trp119 (0.73 Å), Leu164 (0.89 Å), Leu167 (0.72 Å), Phe168 (0.66 Å), Ile171 (0.66 Å), Cys172 (0.66 Å), Ile198 (0.71 Å), Ile199 (0.86 Å), Gln206 (0.87 Å), Ile316 (0.70 Å), Tyr326 (0.66 Å), Leu328 (0.58 Å), Met341 (0.54 Å) Phe343 (0.70 Å), Leu345 (0.65 Å), Tyr398 (0.50 Å), and Tyr435 (0.44 Å). Both lead molecules with almost identical amino acids demonstrated interactions during the simulation with very minor RMSF variations.

RMSF plots of MHC5 and MHC4 to MAO-B
Protein–ligand contact
Hydrogen bonds are considered a crucial type of binding interaction in drug design that aids in understanding the metabolism, adsorption, and specificity of drug candidates. The pattern by which docked protein–ligand poses changed was clearly demonstrated throughout the MD simulation period, owing to the important contributions of non-covalent interactions such as hydrophobic contacts, π-cation; π-π; polar or ionic interactions, and the creation of a water-bridge hydrogen bond. In the presence of active-site amino acid residues in the binding pocket of the 2V5Z protein, the lead molecules, MHC5 and MHC4 showed similar binding patterns (Fig. 9). MD simulations showed that the major interactions between chemicals and proteins were hydrophobic contacts, hydrogen bonds, and polar (water-mediated) hydrogen bonding interactions. Tyr435 and Tyr398 were crucial amino acids for the binding site, together with Phe343, Tyr326, and Leu171. These interlocking residues were discernible in the matched docked complexes, indicating that the docked ligands remained in the active pocket throughout the simulation. The catalytic pocket of 2V5Z was significantly occupied by bioactive chemicals based on protein–ligand contact mapping. Based on the number of molecular interactions generated throughout the 100 ns MD simulation time, MHC5 and MHC4 were determined to be potent MAO-B inhibitors. This enables the identification of the bioactive features of these molecules.

Protein − ligand contact histogram analysis for MHC5 (A) and MHC4 (B). Green, hydrogen bond; blue, water bridge; grey, hydrophobic interaction
MM-GBSA
The estimation of free binding energy for the optimal molecule MHC5, possessing the highest docking energy and activity value prediction, was conducted based on its MD simulation frames. The total average energies for ΔG Bind, ΔG Bind H-bond, ΔG Bind Lipo, and ΔG Bind vdW were found to be –143.08, –11.90, −32.41, and −116.13, respectively, over the 10 to 100 ns MD snapshot. The analysis of these energies revealed that ΔG Bind vdW and ΔG Bind Lipo had the most significant impact on the average binding energy across all interactions, as outlined in Table 3.
Specifically, the ΔG Bind vdW values for the interactions of MHC5 with protein complexes indicated stable van der Waals interactions with amino acid residues. Consequently, the MM-GBSA calculations, derived from MD simulation trajectories, exhibited consistency with the binding energies obtained from the docking results. Notably, the molecule demonstrated very low free binding energy, suggesting a strong binding affinity toward the receptor. As a result, it can be inferred that the MHC5 compound exhibits robust affinity for the MAO-B protein.
ADME/T properties
A biologically effective molecule needs to be present at the target site in the body in a bioactive form for a sufficiently long period for the anticipated biological activities to occur for it to be effective as a medication. To produce new drugs, it is necessary to evaluate absorption, distribution, metabolism, and excretion (ADME) at progressively earlier stages of the discovery process when the number of potential compounds is high, but physical sample access is constrained. In this context, the solubility of the preferred compounds varies from −3.58 to −4.87, according to the logarithm of the molar concentration. Log S values of the lead compounds MHC5 and MHC4 were −4.56 and −4.12 log mol/L, respectively. The intake of oral medicines can be predicted based on Caco-2 cell permeation. The molecule should have a Papp value greater than 8 × 10–6 cm/s for remarkable permeation. Interestingly, all the compounds exhibited significant permeability. If log VDss is greater than 0.45, the medicine may be distributed particularly in tissues as compared to plasma; therefore, most of the molecules are distributed throughout the tissues. The drug is minimally attached to blood proteins according to the fraction-bound method of assessing drug efficiency and is therefore more freely distributed. The GI permeability was evaluated, and the preferred compounds exhibited high GI permeability. BBB permeation is crucial for treating neurodegenerative diseases. In this study, SwissADME and pkCSM were used to evaluate the BBB permeability (Table 4). While substances with log BB values of -1 are assumed to be poorly distributed in the brain, substances with log BB values of > 0.3 are thought to easily cross the BBB. In this study, all compounds had a value greater than 0.3. The log BB values of the lead compounds MHC5 and MHC4 were 0.364 and 0.422, respectively, indicating that they could easily permeate the BBB. While substances with log PS value < −3 are assumed to poorly penetrate the CNS, substances with log PS values of > −2 are thought to easily cross the CNS. All compounds investigated showed CNS permeability. Every substance uniquely interacts with cytochromes, regardless of whether it serves as a substrate or an inhibitor. The total clearances of the lead compounds MHC5 and MHC4 were 0.243 and 0.175 log ml/min/kg, respectively. Specifically, MHC5 was predicted to have no hepatotoxicity. Most compounds exhibited favorable ADME characteristics, making them promising candidates (Fig. 10).

Distribution prediction of the lead compounds MHC5 and MHC4
Collectively, nine halogenated chalcones bearing a morpholine ring were synthesized, and their inhibitory effects on MAOs were assessed. MHC5 and MHC4 are selective MAO-B inhibitors with reversible and competitive modes of inhibition. The ring B of MHC5 and MHC4 connected to the amino acid Tyr326 of MAO-B through a pi-pi stacking with docking scores of -10.64 and -10.95 kcal/mol, respectively, in molecular docking analysis. This could be the primary cause of the efficient inhibition of MAO-B by compounds MHC5 and MHC4. These findings suggested that MHC5 and MHC4 have the potential to be used in the management of neurological disorders.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
References
Guglielmi P, Carradori S, Ammazzalorso A, Secci D (2019) Novel approaches to the discovery of selective human monoamine oxidase-B inhibitors: is there room for improvement? Expert Opin Drug Discov 14:995–1035
Manzoor S, Hoda N (2020) A comprehensive review of monoamine oxidase inhibitors as anti-Alzheimer’s disease agents: a review. Eur J Med Chem 206:112787
Ramsay RR (2013) Inhibitor design for monoamine oxidases. Curr Pharm Des 19:2529–2539
Bhawna Kumar A, Bhatia M, Kapoor A, Kumar P, Kumar S (2022) Monoamine oxidase inhibitors: a concise review with special emphasis on structure activity relationship studies. Eur J Med Chem 242:114655
Tan YY, Jenner P, Chen SD (2022) Monoamine oxidase-B inhibitors for the treatment of Parkinson’s disease: past, present, and future. J Parkinson’s Dis 12:477–493
Guglielmi P, Carradori S, D’Agostino I, Campestre C, Petzer JP (2022) An updated patent review on monoamine oxidase (MAO) inhibitors. Expert Opin Ther Pat 32:849–883
Finberg JP, Rabey JM (2016) Inhibitors of MAO-A and MAO-B in psychiatry and neurology. Front Pharmacol 7:340
Sanchez Alonso P, De La Casa-Fages B, Alonso-Cánovas A, Martínez-Castrillo JC (2023) Switching from rasagiline to safinamide as an add-on therapy regimen in patients with levodopa: a literature review. Brain Sci 13:276
Mateev E, Georgieva M, Mateeva A, Zlatkov A, Ahmad S, Raza K, Azevedo V, Barh D (2023) Structure-based design of novel MAO-B inhibitors: a review. Molecules 28:4814
Pérez-González A, Castañeda-Arriaga R, Guzmán-López EG, Hernández-Ayala LF, Galano A (2022) Chalcone derivatives with a high potential as multifunctional antioxidant neuroprotectors. ACS Omega 7:38254–38268
Guglielmi P, Mathew B, Secci D, Carradori S (2020) Chalcones: unearthing their therapeutic possibility as monoamine oxidase B inhibitors. Eur J Med Chem 205:112650
Mathew B, Suresh J, Anbazhagan S, Mathew GE (2013) Pyrazoline: a promising scaffold for the inhibition of monoamine oxidase. Cent Nerv Syst Agents Med Chem 13:195–206
Mathew B, Parambi DGT, Sivasankarapillai VS, Uddin MS, Suresh J, Mathew GE, Joy M, Marathakam A, Gupta SV (2019) Perspective design of chalcones for the management of CNS disorders: a mini-review. CNS Neurol Disord Drug Targets 18:432–445
Rangarajan TM, Mathew B (2021) Recent updates on pyrazoline derivatives as promising candidates for neuropsychiatric and neurodegenerative disorders. Curr Top Med Chem 21:2695–2714
Kumar S, Nair AS, Abdelgawad MA, Mathew B (2022) Exploration of the detailed structure-activity relationships of isatin and their isomers as monoamine oxidase inhibitors. ACS Omega 7:16244–16259
Koyiparambath VP, Prayaga Rajappan K, Rangarajan TM, Al-Sehemi AG, Pannipara M, Bhaskar V, Nair AS, Sudevan ST, Kumar S, Mathew B (2021) Deciphering the detailed structure-activity relationship of coumarins as Monoamine oxidase enzyme inhibitors-an updated review. Chem Biol Drug Des 98:655–673
Mathew B, Mathew GE, Petzer JP, Petzer A (2017) Structural exploration of synthetic chromones as selective MAO-B inhibitors: a mini review. Comb Chem High Throughput Screen 20:522–532
Sudevan ST, Rangarajan TM, Al-Sehemi AG, Nair AS, Koyiparambath VP, Mathew B (2022) Revealing the role of the benzyloxy pharmacophore in the design of a new class of monoamine oxidase-B inhibitors. Arch Pharm (Weinheim) 355:e2200084
Zhuang C, Zhang W, Sheng C, Zhang W, Xing C, Miao Z (2017) Chalcone: a privileged structure in medicinal chemistry. Chem Rev 117:7762–7810
Kar Mahapatra D, Asati V, Bharti SK (2019) An updated patent review of therapeutic applications of chalcone derivatives (2014-present). Expert Opin Ther Pat 29:385–406
Cao Z, Yang J, Xu R, Song Q, Zhang X, Liu H, Qiang X, Li Y, Tan Z, Deng Y (2018) Design, synthesis and evaluation of 4’-OH-flurbiprofen-chalcone hybrids as potential multifunctional agents for Alzheimer’s disease treatment. Bioorg Med Chem 26:1102–1115
Tian C, Qiang X, Song Q, Cao Z, Ye C, He Y, Deng Y, Zhang L (2020) Flurbiprofen-chalcone hybrid Mannich base derivatives as balanced multifunctional agents against Alzheimer’s disease: design, synthesis and biological evaluation. Bioorg Chem 94:103477
Xiao G, Li Y, Qiang X, Xu R, Zheng Y, Cao Z, Luo L, Yang X, Sang Z, Su F, Deng Y (2017) Design, synthesis and biological evaluation of 4’-aminochalcone-rivastigmine hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg Med Chem 25:1030–1041
Wang L, Wang Y, Tian Y, Shang J, Sun X, Chen H, Wang H, Tan W (2017) Design, synthesis, biological evaluation, and molecular modeling studies of chalcone-rivastigmine hybrids as cholinesterase inhibitors. Bioorg Med Chem 25:360–371
Kourounakis AP, Xanthopoulos D, Tzara A (2020) Morpholine as a privileged structure: a review on the medicinal chemistry and pharmacological activity of morpholine containing bioactive molecules. Med Res Rev 40:709–752
Mathew B, Baek SC, Thomas Parambi DG, Lee JP, Mathew GE, Jayanthi S, Vinod D, Rapheal C, Devikrishna V, Kondarath SS, Uddin MS, Kim H (2019) Potent and highly selective dual-targeting monoamine oxidase-B inhibitors: fluorinated chalcones of morpholine versus imidazole. Arch Pharm (Weinheim) 352:e1800309
Sasidharan R, Eom BH, Heo JH, Park JE, Abdelgawad MA, Musa A, Gambacorta N, Nicolotti O, Manju SL, Mathew B, Kim H (2021) Morpholine-based chalcones as dual-acting monoamine oxidase-B and acetylcholinesterase inhibitors: synthesis and biochemical investigations. J Enzyme Inhib Med Chem 36:188–197
Lee HW, Ryu HW, Kang MG, Park D, Lee H, Shin HM, Oh SR, Kim H (2017) Potent inhibition of monoamine oxidase A by decursin from Angelica gigas Nakai and by wogonin from Scutellaria baicalensis Georgi. Int J Biol Macromol 97:598–605
Oh JM, Kang Y, Hwang JH, Park JH, Shin WH, Mun SK, Lee JU, Yee ST, Kim H (2022) Synthesis of 4-substituted benzyl-2-triazole-linked-tryptamine-paeonol derivatives and evaluation of their selective inhibitions against butyrylcholinesterase and monoamine oxidase-B. Int J Biol Macromol 217:910–921
Baek SC, Park MH, Ryu HW, Lee JP, Kang MG, Park D, Park CM, Oh SR, Kim H (2019) Rhamnocitrin isolated from Prunus padus var. seoulensis: a potent and selective reversible inhibitor of human monoamine oxidase A. Bioorg Chem. 83:317–325
Baek SC, Lee HW, Ryu HW, Kang MG, Park D, Kim SH, Cho ML, Oh SR, Kim H (2018) Selective inhibition of monoamine oxidase A by hispidol. Bioorg Med Chem Lett 28:584–588
Oh JM, Jang HJ, Kim WJ, Kang MG, Baek SC, Lee JP, Park D, Oh SR, Kim H (2020) Calycosin and 8-O-methylretusin isolated from Maackia amurensis as potent and selective reversible inhibitors of human monoamine oxidase-B. Int J Biol Macromol 151:441–448
Lee HW, Ryu HW, Kang MG, Park D, Oh SR, Kim H (2016) Potent selective monoamine oxidase B inhibition by maackiain, a pterocarpan from the roots of Sophora flavescens. Bioorg Med Chem Lett 26:4714–4719
Shaw DE (2021) Desmond molecular dynamics system. Maestro Desmond interoperability tools. Research, Schrodinger release. Springer, New York
Binda C, Wang J, Pisani L, Caccia C, Carotti A, Salvati P, Edmondson DE, Mattevi A (2007) Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: safinamide and coumarin analogs. J Med Chem 50:5848–5852
Mathew B, Haridas A, Uçar G, Baysal I, Joy M, Mathew GE, Lakshmanan B, Jayaprakash V (2016) Synthesis, biochemistry, and computational studies of brominated thienyl chalcones: a new class of reversible MAO-B inhibitors. ChemMedChem 11:1161–1171
Parambi DGT, Oh JM, Baek SC, Lee JP, Tondo AR, Nicolotti O, Kim H, Mathew B (2019) Design, synthesis and biological evaluation of oxygenated chalcones as potent and selective MAO-B inhibitors. Bioorg Chem 93:103335
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717
Pires DE, Blundell TL, Ascher DB (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58:4066–4072
Acknowledgements
The authors extend their appreciation to the Researchers Supporting Project number (RSP2024R56), King Saud University, Riyadh, Saudi Arabia.
Funding
This study was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean Government (NRF-2022R1A2B5B01002536), Researchers Supporting Project (RSP2024R56), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
Conceptualization, BM and HK; synthesis, SKP, NKP, and SK; biological tests, JL; docking simulation and ADMET study, SK, AS, AME, LSW, and R.; writing—original draft preparation, JL, SK, and BM; writing—review and editing, BM and HK; supervision, BM and HK; funding acquisition, HK All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Structure of compound MHC 1. Figure S1.1 | 1H-NMR spectrum of compound MHC 1. Figure S1.2 | 13C-NMR spectrum of compound MHC 1. Figure S1.3 | HRMS of compound MHC 1. Figure S2. Structure of compound MHC 2. Figure S2.1 | 1H-NMR spectrum of compound MHC 2. Figure S2.2 | 13C-NMR spectrum of compound MHC 2. Figure S2.3 | HRMS of compound MHC 2. Figure S3. Structure of compound MHC 3. Figure S3.1 | 1H-NMR spectrum of compound MHC 3. Figure S3.2 | 13C-NMR spectrum of compound MHC 3. Figure S3.3 | HRMS of compound MHC 3. Figure S4. Structure of compound MHC 4. Figure S4.1 | 1H-NMR spectrum of compound MHC 4. Figure S4.2 | 13C-NMR spectrum of compound MHC 4. Figure S4.3 | HRMS of compound MHC 4. Figure S5. Structure of compound MHC 5. Figure S5.1 | 1H-NMR spectrum of compound MHC 5. Figure S5.2 | 13C-NMR spectrum of compound MHC 5. Figure S5.3 | HRMS of compound MHC 5. Figure S6. Structure of compound MHC 6. Figure S6.1 | 1H-NMR spectrum of compound MHC 6. Figure S6.2 | 13C-NMR spectrum of compound MHC 6. Figure S6.3 | HRMS of compound MHC 6. Figure S7. Structure of compound MHC 7. Figure S7.1 | 1H-NMR spectrum of compound MHC 7. Figure S7.2 | 13C-NMR spectrum of compound MHC 7. Figure S7.3 | HRMS of compound MHC 7. Figure S8. Structure of compound MHC 8. Figure S8.1 | 1H-NMR spectrum of compound MHC 8. Figure S8.2 | 13C-NMR spectrum of compound MHC 8. Figure S8.3 | HRMS of compound MHC 8. Figure S9. Structure of compound MHC 9. Figure S9.1 | 1H-NMR spectrum of compound MHC 9. Figure S9.2 | 13C-NMR spectrum of compound MHC 9. Figure S9.3 | HRMS of compound MHC 9. Figure S10. Time-dependency curves of MHC4 and MHC5. The residual activities were measured at the designated times.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, J., Parmbil, S.K., Pandit, N.K. et al. Development of morpholine ring-bearing halogenated α,β-unsaturated ketones as selective monoamine oxidase-B inhibitors. Appl Biol Chem 67, 6 (2024). https://doi.org/10.1186/s13765-024-00857-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-024-00857-y