
Abstract
The H1N1 pandemic in 2009 and the H5N1 outbreak in 2005 have shocked the world as millions of people were infected and hundreds of thousands died due to the infections by the influenza virus. Oseltamivir, the most common drug to block the viral life cycle by inhibiting neuraminidase (NA) enzyme, has been less effective in some resistant cases due to the virus mutation. Presently, the binding of 10 chalcone derivatives towards H5N1 and H1N1 NAs in the non-catalytic and catalytic sites was studied using molecular docking. The in silico study was also conducted for its drug-like likeness such as Lipinski Rule, mutagenicity, toxicity and pharmacokinetic profiles. The result demonstrates that two chalcones (1c and 2b) have the potential for future NA inhibitor development. Compound 1c inhibits H5N1 NA and H1N1 NA with IC50 of 27.63 µM and 28.11 µM, respectively, whereas compound 2b inhibits NAs with IC50 of 87.54 µM and 73.17 µM for H5N1 and H1N1, respectively. The in silico drug-like likeness prediction reveals that 1c is 62% better than 2b (58%) in meeting the criteria. The results suggested that 1c and 2b have potencies to be developed as non-competitive inhibitors of neuraminidase for the future development of anti-influenza drugs.
Similar content being viewed by others

Introduction
The novel Severe Acute Respiratory Syndrome (SARS) coronavirus, SARS-CoV-2, which was first discovered in December 2019 in Wuhan, China, infected approximately 64,000 people with about 1400 deaths announced at that time, mainly in China [1]. At almost the same time, the country was also reportedly dealing with an outbreak of the deadly H5N1 avian influenza in Hunan, an area that borders Hubei, the province where the new coronavirus emerged, according to the South China Morning Post [2]. As of February 1, 2020, local authorities had culled 17,828 poultries after the H5N1 outbreak, according to China’s Ministry of Agriculture and Rural Affairs statement [3]. Fortunately, there are no reported human cases of this H5N1 avian flu, although the transmission of the disease to humans had been reported since the first infection to humans recorded in 2003 [4].
Since the H5N1 outbreak in 2005, the world has been prepared for the impending influenza pandemic, and in 2009, H1N1 strains emerged and threatened humanity with its spread in more than 70 countries [5]. However, the severity of the 2009 H1N1 pandemic was modest compared to the H5N1 outbreak. H5N1 strain is considered particularly dangerous because of its human fatality rate of over 50% to date and because of the risk that the virus may develop the ability to pass efficiently between humans. As of October 2020, the World Health Organization (WHO) reported a total of 861 confirmed human cases which resulted in the deaths of 455 people since 2003 [6]. Thus, these instances in history warrant an enhanced pandemic preparedness, especially in this COVID-19 season. Although there have been no detected complications across the population in either seasonal or pandemic H1N1 influenza, the elderly patient has a higher risk for hospitalization and mortality during the seasonal H1N1 influenza [7]. In particular, patients having specific chronic conditions such as heart disorder, lung disease, hyperglycemia, renal failure, rheumatics, dementia, and thromboembolism are at high risk of influenza complications [8, 9].
The influenza virus is known to be rapidly mutated in which oral anti-influenza, oseltamivir (Tamiflu), has been less potent to the H1N1 infection [10]. Therefore, a study on finding a new anti-influenza agent should be continuously encouraged. Neuraminidase is the most common targeted protein in the therapy of influenza, although hemaglutinin and M2 ion channels were also studied [11]. NA has an important role in cleaving new virions from the infected cell [12]. Once it is blocked, the new virus could not spread over other organs [13]. Reverting to the problem of virus resistance, a new chemical that has not been recognized by the virus is highly desired [14].
A series of chalcone compounds have been reported to demonstrate anti-neuraminidase activity [15] together with their quantitative-structure activity relationships (QSARs) study. The QSAR model suggested that a functional group having electronic characters would increase the activity of the chalcone. In contrast, the steric effect would decrease its activity against H1N1 NA. Other chalcones having anti-NA activities were isolated from Glycyrrhiza uralensis [16], Erythrina addisoniae [17], Glycyrrhiza inflate [18], Polygala karensium [18], and Angelica keiskei [19] further supporting the claim of chalcones as potential NA inhibitors.
However, upon inspection of the chemical structure, chalcone might not be a favorable scaffold for NA inhibitors which usually belong to the shikimic acid structure. The shikimic acid inhibitors such as oseltamivir and zanamivir bind to the catalytic site of NA, which is a small pocket surrounded by basic sub-pocket, acidic sub-pocket as well as hydrophobic sub-pocket alongside its conserved amino acid residues [20]. Chalcone is a phenyl ring extended by an α,β-unsaturated carbonyl with various alkyl or aryl moieties next to the carbonyl group, making it too bulky to fit into the NA catalytic site [21]. A chalcone compound, namely 2-amino-5-[3-[4-[(E)-4-chloro-3-oxobut-1-enyl]anilino]propyl]-4-methyl-1H-pyrimidin-6-one (NSC89853) is reported to show H5N1 NA inhibition from the screening of 20 compounds from NCI database. This inhibition may be due to the binding to the protein with an alternative mode, that is different from the known drugs (oseltamivir or zanamivir). Although the potency of such a compound was not as strong as the known drugs, it may overcome the drug i.e. H274Y for the N1 protein [22].
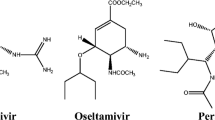
In one of the more recent research, safflomin A (SA) was reported to show inhibition against NAs from H1N1 and H3N2 influenza viruses. The antiviral assay using MDCK cells which was infected by H1N1 and H3N2 influenza virus exhibited the synergistic effect of SA with oseltamivir in viral cell proliferation. The kinetics study of SA demonstrated a non-competitive inhibition against N1 and N2 NA. Furthermore, a molecular docking study predicted that SA interacted with N1 and N2 NA at the non-catalytic site. These results suggested that SA, which has a chalcone backbone, may serve as a potential therapeutic option to the currently available anti-influenza agents to overcome the drug resistance [23]. The structure of NSC89853, SA, oseltamivir and zanamivir are presented in Fig. 1.

The structure of a NSC89853, and b SA, which are suggested to be non-catalytic site inhibitors of H5N1 and H1N1, respectively, whereas c oseltamivir, and d zanamivir are known NA inhibitors targetting the catalytic sites
In this present study, we examined 10 chalcone derivatives for their predicted binding affinities into the non-catalytic site of H1N1 and H5N1 NA using a molecular docking study. The non-catalytic location is at the back of the NA’s catalytic site within loop 150. Molecular docking of chalcones into the NA catalytic site was also conducted to compare their predicted binding affinities with the ones in the non-catalytic site. Further in silico studies to predict the drug-like likeness were also carried out employing Lipinski Rule, pharmacokinetic profile and toxicity prediction. The study was then followed by testing for their biological activities against H5N1 and H1N1 NA using in vitro MUNANA assay. In addition, the safety index was also calculated by studying the cytotoxicity effect of the most active compound toward a normal cell. Two chalcones demonstrated potential activity as neuraminidase of H5N1 and H1N1 inhibitors, respectively, with low toxicities to the normal cell lines and predicted to have considerably good drug-like structures.
Results
Molecular docking
The ten chalcone derivatives have been predicted for their molecular interactions using docking study as presented in Table 1. The ΔGbind ranged at − 6.12 to − 7.97 kcal/mol and − 6.34 to − 8.31 kcal/mol at H1N1 NA and H5N1 NA non-catalytic sites, respectively. These results describe that chalcone has a potency from a low to moderate micromolar activity (Ki 32.71 to 1.44 µM at H1N1 NA; and Ki 22.58 to 0.82 µM at H5N1 NA) to bind at this non-catalytic site and disrupt the enzyme activity. The superimposition of ten chalcone derivatives in the non-catalytic site of H1N1 and H5N1 NAs is illustrated in Fig. 2.

The superimposition of 10 chalcone derivatives (yellow sticks) in the non-catalytic site of a H1N1 and b H5N1 NA. The NA is presented in the surface model and the ligands are in the stick models, docked in the behind of the catalytic site (yellow circles)
To compare the binding mode of the ligands to the common site, a molecular docking study was also conducted into the H1N1 and H5N1 NA catalytic sites. The parameterization of molecular docking was controlled by re-docking oseltamivir-triazole into the H1N1 NA and oseltamivir into the H5N1 NA catalytic site. This resulted in RMSD 1.02 Å of the most populated cluster (85%) with ΔGbind − 9.35 kcal/mol at H1N1 NA, whereas the RMSD of oseltamivir at H5N1 NA active site was 1.9 Å of the most populated cluster (74%) with the lowest energy (ΔGbind − 7.52 kcal/mol). This defines that the parameter is accepted for further docking of chalcone compounds. The control docking pose of ligands to their individual NA which are overlapped with its initial pose is presented in Additional file 1: Figure S1.
The control docking pose closely interacts with the conserved amino acid residues such as ARG118, ASP151, TRP179, ARG293, ARG368 and TYR402 via hydrogen bondings. The parameter of that control docking is then used to dock 10 chalcone derivatives into both H1N1 and H5N1 NA active site binding pockets. The docking results of 10 chalcone derivatives are presented in Table 2 represented by the ΔGbind, interacting residues and the predicted Ki. The docking into H1N1 results in ΔGbind − 6.37 to − 8.34 kcal/mol, whereas it is ΔGbind − 5.05 to − 6.93 kcal/mol into H5N1 for the overlapped-docked pose which is presented in Fig. 3. The calculated energies of docked chalcones into the H1N1 catalytic site are likely lower than that of H1N1 at the non-catalytic one. In contrast, the chalcones docked into the H5N1 have lower free energy of binding in the non-catalytic than in the catalytic sites. However, except for compounds 1a, 2a and 3a, its long alkyl chain escaped from the catalytic site, indicating their non-fit binding into both H1N1 and H5N1 NA’s catalytic sites. Table 2 tabulated the docking results of the 10 compounds in the active site of H1N1, H5N1 NA, and their superposition as illustrated in Fig. 3.

The superimposition of 10 chalcone derivatives (yellow sticks) in the active site of a H1N1 NA (blue) and b H5N1 NA (red) in the ribbon model
Lipinski rule
According to the Lipinski Rule, an ideal drug should have a molecular weight (MW) which is less than 500 Da, < 5 log P, ≤ 5 Hydrogen Bond Donor (HBD), ≤ 10 Hydrogen Bond Acceptor (HBA), ≤ 10 rotatable bonds, and ≤ 140 Å surface area [24, 25]. The potency of the compound to be drug is commonly expressed by IC50, which is affected by its MW. The more potent drug should have a higher MW to minimize its dose in performing a pharmacological activity. Nevertheless, the MW should not be higher than 500 considering the drug absorption via the intestinal membrane [24].
The log P is the concentration of the compound in n-octanol divided by its concentration in water. Therefore, it reflects the balance of the compound’s solubility in water during oral dissolution steps with its oral bioavailability in the blood system [24, 25]. The ideal log P < 5 indicates that the compounds are well soluble in the body fluid as well as absorbed through the gastrointestinal cell membrane, which then be transferred into the blood vascular. On the other hand, the number of HBD or HBA is associated with polarity to interact with water during the dissolution process as well as their molecular interaction via hydrogen bond with its receptor [24]. The rotatable bonds may influence their stability during ADME (absorption, distribution, metabolism, and excretion) and the receptor binding. Thus, the less flexible the chain in the molecule, the more stable the drug is to perform its activity [25]. The Polar Surface Area (PSA) is related to the permeability of drugs across the cell membrane in which the higher the PSA, the poorer the cell permeability (oral bioavailability) is [25].
In general, the results show that all chalcones meet the MW, the number of HBD, HBA and the rotatable bonds requirements. However, compounds 2b and 3b slightly deviate on the log P limit, whereas compounds 2c, 3c and 4b do not fulfill the limit of the surface area requirement. Table 3 presents the chalcones with their Lipinski Rule profiles.
Mutagenicity and toxicity profiles
The in silico toxicity predictions have become a routine before processing compounds to be drug candidates. In this study, we also predicted the mutagenicity and toxicity profiles of the 10 chalcones. Generally, a compound’s mutagenic properties are usually confirmed using the AMES test [26, 27]. The human Maximum Tolerated Dose (hMTD) is acceptable when the predicted toxic dose threshold in humans is more than 0.477. The potassium channels that mediate the cardiac repolarization in humans are represented by the values of hERG I and II. Hence, inhibiting this kind of protein would cause a long QT syndrome development that might lead to a fatal arrhythmia. The in vivo toxicity is often expressed by LD50 value which can be defined as the required dose given to cause 50% death of a group of rats in evaluating a compound acute toxicity represented by ORAT (log LD50 value) prediction.
LOAEL (lowest-observed-adverse-effect level) associates with the compound’s lowest concentration which causes an adverse effect in human physiology. This is indicated by the alteration of morphology, function, growth, or development. The safety of a compound is higher as the LOAEL value increases. The liver injury commonly reflects the drug hepatotoxic properties, whereas the potential dermal adverse effects are determined by skin sensitization properties. The value of toxic endpoints, however, is measured by T. pyriformis and minnow toxicity [28].
Nowadays, the drugs’ safety to the environment is a concern. Low environmental damage is demonstrated by the values of T. pyriformis and minnow toxicity which are to be respectively higher than 0.5 and − 0.3. Based on the AMES test, two compounds i.e. 2a and 4b are predicted not to be toxic, while 4b might have the possibility to be hepatotoxic. No chalcones inhibit the hERG I protein, but 1c, 2b, 2c and 3c inhibit hERG II. Most of the chalcones have a low maximum dose which is weakly tolerated in humans, except for 2a, 2b and 4b. In addition, all chalcones exhibit no skin sensitization. The oral rat acute toxicity LD50 can be classified as very toxic (≤ 5 mg/kg), toxic or moderately toxic (> 5 to < 500 mg/kg), harmful or slightly toxic (> 500 to < 2000 mg/kg), and non-toxic (> 2000 mg/kg) [29]. All compounds are predicted to be potentially less toxic with LD50 ranging from 194.01 to 399 mg/kg. Oral rat LOAEL value shows that the ingestion chronic toxicity is relative to the limit of concentration and length of exposure, however, LOAEL does not solely represent drug safety accurately [30, 31]. This prediction showed that compounds 2a, 2c, 3a and 4b have lower values than the other compounds, which can be interpreted as they are able to cause any observable chronic toxicity at the lower amount and/or shorter time of exposure. Several compounds (2c, 3a, 3b and 3c) demonstrate T. Pyriformis toxicity potency, whereas, based on the minnow toxicity prediction, compounds 1a, 2a, 3a and 4b are deemed to be safe. Table 4 presents the AMES test results of the chalcones for mutagenicity prediction along with other toxicity profiles.
Pharmacokinetic profiles
Table 5 demonstrates that all chalcones are well absorbed through the human intestinal with near values to 100% into the blood system. In general, the water solubility of all chalcones are most likely poor as their log S value are lower than − 4, except for 3a which has a log S value of − 2.601; indicating that it may dissolve readily in the dissolution step. Furthermore, oral absorption prediction using Caco2 cell model [32] with the required value is higher than 0.90. Therefore, except for 2c and 4b, chalcones show good human gastrointestinal absorption. The skin permeability of chalcones is most likely suitable for the transdermal route because the values are approximately − 2.5. A protein transport namely P-glycoprotein (P-gp) is crucial during the pharmacokinetics steps, however, this could have either advantages or disadvantages in therapeutic effect [33]. A compound is supposed to not inhibit P-gp, either P-gp I or P-gp II. In the ideal situation, it should not be acting as P-gp substrate either. According to the prediction, compounds 1c and 2b are predicted to inhibit the P-gp I activity, whereas compounds 2c, 3c, and 4b inhibited both P-gp I and P-gp II. In contrast, 2a acts like the substrate for P-gp, accordingly.
One of the distribution parameters is defined by the number of VDss or volume of distribution at a steady state, which is directly proportional with the amount of drug distributed into tissue; a higher VD indicates a greater amount of tissue distribution. As a rule, VDss should be ≥ − 0.15. Among the 10 chalcones, three compounds (2c, 3a, and 3c) do not meet these criteria. The fraction unbound (fu) for all chalcones are predicted to be ≤ 0.15, indicating that more fractions of drug molecule are bound to the plasma protein. Besides VDss, the chalcones were also predicted for their ability to cross the brain membrane which is important as the compounds may affect the Central Nervous System (CNS) [34]. Values < − 1 indicate poor Blood–Brain Barrier (BBB) permeability and < − 3 for CNS permeability. In other words, the compound is poorly distributed to the brain and unable to penetrate CNS. From the results, all the 10 compounds should be carefully managed as there is potential for these chalcones to enter the CNS especially for 1a, 1b, 2b and 3b which can also penetrate BBB. The distribution profiles of the chalcones as predicted by software are presented in Table 6.
Metabolisms are also an important indicator of good drug-like properties. CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4 are among the cytochrome P450 subfamilies, which play significant roles in drug metabolism [35]. CYP2D6 and CYP3A4 are found in the brain and intestines, respectively, and are most likely responsible to metabolize the drug in their surrounding areas. Furthermore, CYP3A4 also affects oral bioavailability by first-pass metabolism. A compound that binds strongly to these CYPs could be acting as the substrate or inhibitor leading to the lower / higher bioavailability as well as activity of other drugs. This could be potential for the drug causing clinical drug-drug interactions, leading to adverse reactions or therapeutic failures. In this prediction, neither chalcones act as the substrate nor inhibitor for CYP2D6. However, except for 3a, all compounds act as CYP3A4 substrate, whereas 2c and 3c act as this enzyme’s inhibitor. Furthermore, chalcones are likely to inhibit the CYP1A2, CYP2C19, and CYP2C9 presented in Table 7, which should be of concern when the compound is consumed with other drugs.
Total clearance describes the compound’s rate while being removed from the body [36]. One of the main renal uptake transporters to remove the drug from the blood is the OCT2 transporter. It plays a pivotal role in the removal and renal clearance of mostly cationic drugs as well as endogenous compounds [37]. Inhibition of OCT2 (such as by cimetidine) elevates OCT2-dependent renal clearance drugs, hence altering pharmacokinetics and pharmacodynamics profiles. Compound 3a is predicted to be the fastest compound excreted from the body due to its highest total clearance. In contrast, compound 2b is the slowest compound to be removed from the body due to its lowest total clearance. All chalcones have low total clearance (log Cl < 0.763), yet, it is generally desirable to develop a drug for oral administration without a high dosage regimen [38]. One chalcone, 2b, is predicted to act as renal OCT2 substrate that might lead to undesirable side effects. Figure 4 illustrates the total clearance of all chalcones that reflects their rate to be eliminated from the body system.

The histogram of total renal clearance profiles of the chalcones with its OCT2-dependence clearance, green block = OCT2-independent and yellow block = OCT2-dependent
Neuraminidase assay
The chalcone derivatives have been examined for their inhibitions toward the activity of H1N1 and H5N1 influenza neuraminidase. The first screening using 100 µg/ml of the sample’s concentration showed that five compounds exhibited 0–10% inhibition toward H1N1 NA, while only one compound showed activity in the same range against H5N1. Furthermore, three compounds exhibited 10–50% inhibition toward H1N1, while five compounds demonstrated inhibition against H5N1 at the same inhibition percentage. In the H5N1 NA inhibition assay, two compounds (1b and 2c) were inactive. Interestingly, two compounds (1c and 2b) showed more than 50% inhibition to both H5N1 and H1N1. Therefore, they were selected to be further studied on their IC50 as well as CC50. Table 8 presents the assay results of ten chalcone compounds compared to vanillin as the positive control.

The chalcone backbone scaffold of 10 chalcone derivatives
The IC50 of 1c was then estimated as 28.11 µM and 27.63 µM for H1N1 and H5N1, respectively. Compound 2b also demonstrated similar % inhibition toward H1N1 and H5N1 NA with its IC50 87.54 µM and 73.17 µM, respectively. This is quite interesting because the compound showed similar IC50 when it was applied to both H5N1 and H1N1 NA. Figure 6 presents the drug-dose depending curve of 1c and 2b in inhibiting the H1N1 and H5N1 NA.

The drug–dose depending curve of 1c and 2b in inhibiting H1N1 and H5N1 NA
Cytotoxicity assay
In the cytotoxicity assay results (Fig. 7), 1c showed a high concentration to inhibit the Vero cell proliferation with CC50 968.16 µM. The safety index of this compound calculated with the safety index (SI) values was 34.44 and 35.69 for H1N1 and H5N1 NA, respectively. In addition, 2b showed CC50 757.18 µM with the SI values = 8.65 and 10.35 for H1N1 and H5N1, respectively. The graph plotting log concentration of 1c and 2b vs % cell viability against Vero cell in the cytotoxicity studies is presented in Fig. 7a. Furthermore, the cell imaging in Fig. 7b exhibited the cell proliferation survival, when there was no treatment to the cell, which was indicated by the formation of formazan crystals upon MTT reaction. Formazan crystal indicates the ability of the cell to express NADPH-dependent oxidoreductase to reduce the MTT, which leads to cell proliferation. In contrast, the formazan crystal formation will be reduced along with the treatment of 1c and 2b on the Vero cell leading to their cytotoxicity properties (Fig. 7a).

The cytotoxicity profiles of 1c and 2b plotting a log concentration of vs % Vero cell viability of 1c and 2b, whereas b is the Vero cell imaging without any compound exposure (negative control), and c with 1c treatment to the cells. The orange arrows indicate the formazan crystal formation which is higher in 1c absence than its presence, accordingly
Discussion
The 2009 H1N1 NA is one of several 2009 pandemic influenza virus mutants in which the I223 changed to R223 (I223R). This mutation results in shrinkage of the enzyme’s active site affecting the binding of NA inhibitor [39]. Another mutant is H274Y (H1N1), which was also reported to be resistant to oseltamivir as confirmed in about 20% isolates from humans in Europe [40, 41]. NA’s active site is composed of eight functional residues (ARG118, ASP151, ARG152, ARG224, GLU276, ARG292, ARG371 and TYR406) and surrounded by 11 framework residues (GLU119, ARG156, TRP178, SER179, ASP198, ILE222, GLU227, HIS274, GLU277, ASN294 and GLU425) [42].
Structurally, chalcone consists of one aromatic ring extended by a predominantly trans-configuration of α,β-unsaturated carbonyl. The relatively short carbon–carbon distance between α,β-unsaturated alongside the relatively long C–C distances is 1.326 Å and 1.46 Å, which is consistent with a localized double bond in the enone unit in this structure [43]. The chain next to the carbonyl group is usually prolonged by either alkyl or aryl moieties with various functional groups being attached. The conjugated double bond could make this type of compound less flexible. However, the C=O bond can present as either S-cis or S-trans conformation with respect to the vinylic double bond due to the free rotation along with the single bond between C-carbonylic and Cα. This, therefore, could increase the conformation stability of such compounds during a dynamic environment [44].
The combination of in silico and in vitro studies has suggested a new binding mode for chalcone compounds into N1 NA. The active site of NA is decorated by a relatively small pocket surrounded by some sub-pockets determining the binding region of such enzyme with a particularly striking feature in the catalytic domain referred to as the ‘150 loop’ [39, 45]. The shikimic acid scaffold has been classically patterned for NA inhibitors due to its capability to form a stable chair conformation transition state of the non-aromatic six-member ring with the rather flat oxonium cation, and thereby arrange the position of important residues to interact with the shikimic binding groups [45]. Taking this into consideration, some aromatic compounds have been devised to mimic this transition state leading to their inhibition against NA [46, 47].
In the present study, two chalcones (1c and 2b) have been observed to have the best in vitro results compared to the other 8 chalcones. Figures 8 and 9 illustrate the molecular interactions of 1c and 2b, respectively, while binding into the H1N1 (non- and catalytic sites) and H5N1 (non- and catalytic sites). During the binding into the non-catalytic site of H1N1 NA, 1c interacts via H-bond with only THR438. The other interactions are vdW interactions with ASN146 and HIS144; amide pi-stacked with HIS144 and ILE149; and pi-alkyl interactions with VAL116, ALA138 and SER145. In contrast, the binding to the catalytic site of H1N1 NA shows that 1c interacts with ASP151, ARG293, ASN344, ARG368, and TYR402 via H-bonds, whereas pi-alkyl and alkyl-alkyl interactions are also observed with LYS150 and LEU134, respectively. In addition, pi-anion interaction is observed with GLU119. Compound 1c interacts with the non-catalytic site of H5N1 NA via H-bonds with VAL116, ARG118, and ARG430; amide pi-stacked with ILE117; and pi-alkyl with VAL116, ARG118, ARG430 and PRO431. Furthermore, in its catalytic site, the interactions are observed via H-bonds with ARG152, ARG371, TYR347, and ARG430; alkyl interactions with TRP178, ARG224, and PRO431; pi-cation as well as pi-cation interactions are observed with ARG118 and ASP151, respectively.

The molecular interactions of 1c with a non-catalytic H1N1, b catalytic H1N1, c non-catalytic H5N1, and d catalytic H5N1 NA sites. The green, pale green, purple, magenta, pink, cyan, and orange represent H-bond, vdW, pi-sigma, amide pi-stacked, pi-alkyl, pi-halogen, and pi-cation/anion, respectively

The molecular interactions of 2b with a non-catalytic H1N1, b catalytic H1N1, c non-catalytic H5N1, and d catalytic H5N1 NA sites. The green, pale green, purple, magenta, pink, cyan, yellowish-green and orange represent H-bond, vdW, pi-sigma, amide pi-stacked, pi-alkyl, pi-halogen, pi-lone pair electrons, and pi-cation/anion, respectively
The second best compound (2b) also interacts via H-bond with the non-catalytic site of H1N1 NA with LYS150; pi-alkyl with PRO154, VAL177, PRO198 and VAL205. In contrast, the interactions with the catalytic site via H-bonds are absent. However, the remaining interactions of 2b are observed with ARG118 and ARG152 via pi-alkyl interactions; GLU277, ARG293 and ASN295 via pi-halogen interactions; with TRP179 via pi-lone pair electron interaction. The non-catalytic site of H5N1 NA shows interactions with chalcones via H-bonds with GLY147 and pi-alkyl with VAL116, ALA138, VAL149. In addition, amide pi-stacked with HIS155 is also observed. In the H5N1 NA catalytic site, chalcones make interactions via H-bonds with ARG118, ARG156 and ARG371. Furthermore, there are also amide pi-stacked interaction with TYR347, as well as pi-cation/anion with ASP151 and ARG292, respectively.
The possibility of chalcone to interact with the active site of NA is only in one aromatic ring augmented by the binding group such as OH or OCH3. The α,β-unsaturated chain most likely protruded outside of the active pocket. Although the ΔGbind of chalcone in the active site is considered acceptable, it is inconsistent with the in vitro results. Compound 1c has relatively higher ΔGbind in both NAs’ active sites than other compounds with poor inhibition against NAs. However, the in vitro results showed that 1c is the most active compared to others; indicating a poor correlation between in vitro and in silico studies. In contrast, although 2c is inactive against NAs in vitro, the ΔGbind in both NA’s active sites is considered fair.
Notably, compounds with –NO2 group (2a and 4b) show the lowest binding energy in either non-catalytic or catalytic sites for both H1N1 and H5N1 NAs. However, the in vitro results demonstrated a low percentage of inhibition toward both NAs. Nitro group is the strongest electron-withdrawing, which reactively interacts with atoms having an electron-donating group to form H-bond as well as electronic interactions [48]. This false-positive results could be due to the stability of the –NO2 group, which is easily reduced by the acidic pH turning into an amine group (nitro reduction) [49], which is measured in pH 6.5 in this MUNANA system. This conversion could make the 2b and 4b lose their interactions as suggested by the docking study, leading to their low inhibition against NA. The inconsistent results of 2b and 3b are not fully understood as well. Therefore, we suggest that a molecular dynamics simulation is performed to elucidate this phenomenon in future studies. On the other hand, there are inconsistent results between docking and in vitro results when the chalcones were docked into either non-catalytic and catalytic sites of H5N1. Therefore, it is recommended to study the kinetics of NA inhibition for the two most active compounds (1c and 2b) to confirm whether the mode of inhibition is competitive or non-competitive.
The kinetic assay can be carried out using methods such as Biacore or Isothermal Titration Calorimetry (ITC). Biacore measures the real-time binding association and dissociation rates using Surface Plasmon Resonance (SPR). In this method, the protein is immobilised onto a biosensor surface while the drug ligand is continuously flowing across the biosensor surface, where it binds to the immobilised receptor. The binding is measured by the kinetic association and dissociation rates (ka/kd) for several different ligand concentrations [50].
The second method is ITC, which measures the heat transfer during binding that enables accurate determination of binding constants (KD), reaction stoichiometry (n), enthalpy (∆H) and entropy (ΔS). This provides a complete thermodynamic profile of the molecular interaction. This deeper understanding of structure–function relationships enables more confident decision making in hit selection and lead optimization [51].
In this present study, an array of in silico predictions has been performed on the chalcones including Lipinski Rule, mutagenicity, toxicity and pharmacokinetic profiles. The chalcones have diverse functional groups conferring the distinction of their drug-like properties, mutagenicity, toxicity, and pharmacokinetics, which will contribute to their overall therapeutic effects. Given that the two compounds (1c and 2b) are active in the in vitro study, we are motivated to understand the possibility of these compounds to be considered as lead candidates for optimization. They have good Lipinski Rule profiles by meeting the requirements of MW, log P, number of HBD-HBA, number of rotatable bonds and the surface area. Both compounds are also not responsive against the AMES test describing their non-mutagenic potency. Unfortunately, 2b is predicted to have a low tolerance in the maximum dose. In addition, both compounds are responsive toward hERG II as inhibitors and having an acute toxic dose of around 200 mg/kg, but irresponsive to hERG I as an inhibitor, not toxic in chronic ingestion, and neither hepatotoxic nor skin sensitized. In environmental damage, both are non-toxic to T. pyriformis as well as minnow species.
These two compounds are insoluble in water. Thus, consideration should be given for a suitable delivery system. They have good in Caco2 permeability, intestinal human absorption and skin permeability; reflecting their good absorption profile either in oral or topical administration. Both compounds have potencies to inhibit the protein carrier during absorption but neither of them acts as the protein substrate. In the distribution profiles, both compounds bind tightly in the plasma protein, which could reduce the therapeutic dose. Furthermore, the BBB and CNS permeability should be taken into account. Interaction with other drugs should be evaluated as well, as these two compounds are predicted to act as the CYP3A4 substrate, CYP1A2, CYP2C19, and CYP2C9 inhibitors. In contrast, they are irresponsive toward CYP2D6 and CYP3A4. The excretion of 1c is faster than 2b since the total clearance of 1c is higher than 2b in addition to the potential of this compound as Renal OCT2’s substrate. Overall, 1c meets the requirements of drug-like properties at approximately 62%, whereas 2b is at 58%. These percentages are above 50%, therefore, these two compounds should be given the opportunity to be optimized and developed further as drug candidates. This conclusion is supported by the in vitro cytotoxicity study against normal cell lines confirming that both compounds have a good safety index augmenting their potency as the potential H5N1 and H1N1 NA inhibitors.
Materials and methods
Chemistry
The chalcone compounds (Fig. 10) were synthesized using the established method in our laboratory [21, 52, 53]. We scale up the production of the compounds and confirm their purity using a thin-layer chromatographic method and melting point test with the data of published compounds as the reference.

The structure of 10 chalcone derivatives (1a–4b) with different set of functional groups
Molecular docking
The proteins used are the resistant I223R NA mutant of H1N1 2009 pandemic influenza virus (PDB ID 4B7M) [39] and the wild type H5N1 NA (PDB ID 2HU0) [54]. The protein structure was processed using AutoDockTools 1.5.6 (www.autodock.scripps.edu) with the ligand separated from the protein. The grid for docking to the non-catalytic site of H1N1 NA and H5N1 NA was set to 94 × 72 × 96 and 80 × 80 × 80 with its spacing set to 0.375 Å. The center of mass of the ligand was set to x = 26.852, y = − 32.014, z = − 1.019 for H1N1 NA, and x = − 7.07, y = 28.242, z = 107.729 for H5N1 NA. Genetic Algorithm was chosen for docking calculation in AutoDock 4.2.3. The searching parameters were set to the default values (Population size = 150, maximum number of evals = 2,500,000, maximum of generations = 27,000, maximum number of top individuals that automatically survives = 1). The number of GA run was set to 100. Docking parameters such as random number generation, energy parameters, and step size were also set to the default values. The results were analyzed by checking the RMSD values, ligand–protein interactions, free energy of binding (FEB) as well as the number of conformations that exist in a population cluster [55]. For the subsequent molecular docking, the chalcone structure derivatives were sketched and energetically optimized using Hyperchem Professional version 8.0 (www.hyper.com) with MM + force field and Polak-Ribiere (Conjugate Gradient). The visualization of ligand–protein interaction was conducted using Biovia Discovery Studio 2016 (www.accelrys.com).
The docking of compounds to the catalytic site of NA was carried out by using PDB 2HU0 (H5N1 NA in complex with oseltamivir) [54] as well as PDB 6HP0 (H1N1 NA in complex with oseltamivir triazole) [56] with the same default protocol being used in PDB 4B7M, except for the number of points and its center of mass. For 6HP0, the number of points is 40 × 40 × 40 with the center of mass (x = 43.641; y = 0.53; z = 20.263). For 2HU0, the number of points is 40 × 40 × 40 with the center of mass (x = 1.763; y = 19.33; z = 108.34).
Lipinski rule of five
The Lipinski Rule profiles were individually predicted by inputting SMILES string, which is automatically done by the server. The value of molecular weight (MW), log P, the number of hydrogen bond donors (HBD), the number of hydrogen bond acceptors (HBA), the number of rotatable bonds and the surface area were observed and then tabulated.
Mutagenicity and toxicity studies
Using the same protocol as in “Lipinski rule of five” section, the mutagenic potency of the ligands was represented by the AMES test results. Other parameters such as maximum tolerated dose (human) (hMTD), hERG I inhibitor, hERG II inhibitor, oral rat acute toxicity (log LD50), oral rat chronic toxicity (log LOAEL), hepatotoxicity, skin sensitization, T. pyriformis toxicity, and minnow toxicity represented the toxicity properties of the ligands.
Pharmacokinetics study
Using the same protocol in 4.3, the ADME (absorption, distribution, metabolism, and excretion) profiles of the ligands were predicted. Subsequently, the absorption is influenced by water solubility, Caco2 permeability, skin permeability, P-glycoprotein substrate, P-glycoprotein I inhibitor, and P-glycoprotein II inhibitor instead of human gastrointestinal absorption. The distribution is represented by VDss (human), fraction unbound (human), blood–brain barrier (BBB) permeability, and central nervous system (CNS) permeability. The metabolism is represented by the CYP2D6 substrate, CYP3A4 substrate, CYP1A2 inhibitor, CYP2C19 inhibitor, CYP2C9 inhibitor, CYP2D6 inhibitor, and CYP3A4 inhibitor. Lastly, the excretion is represented by the total clearance and renal OCT2 substrate. The Lipinski Rule, mutagenicity, toxicity and pharmacokinetic prediction were carried out using pkCSM online tool (http://biosig.unimelb.edu.au/pkcsm/prediction) [28].
Neuraminidase assay
The H1N1 NA assay followed the general procedure of Fluorometric Neuraminidase Assay [57]. The H1N1 NA (A/California/04/2009) and H5N1 NA (A/Anhui/1/2005) enzymes were purchased from Sinobio. The fixed concentrations of H1N1 NA (0.3 u/mL) and MUNANA (100 mM) were optimized employing the previously described method. Vanillin was used as the positive control inhibitors [58], and the H1N1 neuraminidase assay was prepared by mixing an assay buffer, tested samples (at concentrations 100 μg/mL in 1% of DMSO-Buffer), and a constant 0.3 unit/mL of neuraminidase which were pre-incubated at 37 °C for 30 min with 200 rpm. After the addition of 100 μM substrates, the reaction assays were incubated at 37 °C for 60 min with 200 rpm. To stop the reaction, 100 μl of glycine stop solution was added. The assays were carried out in triplicate. The assay protocol for H5N1 inhibition assay is similar to that of H1N1 but the concentrations being used were 0.15 u/mL, and 50 µM for the enzyme and substrate, respectively. A series of concentrations were prepared for those demonstrating > 50% of enzyme inhibition to calculate the IC50. The fluorescence intensity of NANA was measured by Modulus Microplate Reader with a UV optical kit at λ 340/440 nm. The drug-dose dependent curve and its statistical analysis (95% confident interval) were generated using GraphPad Prism 5.0 (https://graphpad-prism.software.informer.com/5.0/).
Cytotoxicity assay
The cytotoxicity of each compound on Vero cells was determined using MTT assay. Vero cell is a non-tumorigenic cell from the kidney tissue of African green monkeys [59]. Cells will proliferate by expressing the NADPH-dependent oxidoreductase in mitochondria that reduces the MTT reagent into the reduction state of formazan crystal [60]. Cells (1 × 104/well) were seeded in 96-well flat-bottomed plates and incubated with each sample at various concentrations for 24 h. Compound solutions were prepared in the following concentrations: 10, 20, 40, 80, 160, 320, 640 and 1280 µg/mL. 30 μL of MTT solution (5 mg/mL in PBS) was added to each well and the plate was incubated at 37 °C for another 4 h. Then, the medium was discarded and 150 µl of DMSO was added to dissolve the formazan crystals. The absorbance of each sample was read at 595 nm using a microplate reader. Results were expressed as a percentage of cell viability with respect to untreated control cells (as 100%) [61]. The drug-dose dependent curve and its statistical analysis (95% confident interval) were generated using GraphPad Prism 5.0 (https://graphpad-prism.software.informer.com/5.0/).
Conclusions
Computational studies using molecular docking, ADME-Tox prediction and in vitro study, including their H5N1 and mutant H1N1 NA inhibitory activity and cytotoxicity towards Vero cells of the 10 synthesized chalcone derivatives have been employed to predict and evaluate the inhibitory mechanism and the safety of these chalcone derivatives. In conclusion, 1c ((E)-3-(4-butoxyphenyl)-1-(4-hydroxy-3-methoxyphenyl)prop-2-en-1-one) and 2b ((E)-1-(3-(cyclopentyloxy)phenyl)-3-(4-fluorophenyl)prop-2-en-1-one) have potencies to be developed as anti-influenza drugs by inhibiting H5N1 and H1N1 NA in the non-catalytic site.
Availability of data and materials
All data generated or analysed during this study are included in this published article [and its supplementary information files].
Change history
27 August 2023
A Correction to this paper has been published: https://doi.org/10.1186/s13765-023-00809-y
References
Chen N, Zhou M, Dong X et al (2020) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395:507–513. https://doi.org/10.1016/S0140-6736(20)30211-7
South China Morning Post (2020) China reports outbreak of deadly bird flu among chickens in Hunan province, close to coronavirus epicentre of Wuhan. https://amp.scmp.com/news/china/society/article/3048566/china-reports-outbreak-deadly-bird-flu-among-chickens-hunan
Reuters (2020) China reports H5N1 bird flu outbreak in Hunan province. https://www.reuters.com/article/health-birdflu-china-idUSL4N2A10GC
South China Morning Post (2020) H5N1 bird flu virus. https://www.scmp.com/topics/h5n1-virus
Coker RJ, Hunter BM, Rudge JW et al (2011) Emerging infectious diseases in southeast Asia: regional challenges to control. Lancet 377:599–609. https://doi.org/10.1016/S0140-6736(10)62004-1
WHO (2020) Cumulative number of confirmed human cases for avian influenza A (H5N1) reported to WHO, 2003–2020. Epidemic Pandemic Alert Response World Heal Organ 1–3
Rothberg MB, Haessler SD (2010) Complications of seasonal and pandemic influenza. Crit Care Med 38:e91–e97. https://doi.org/10.1097/CCM.0b013e3181c92eeb
Glezen WP, Decker M, Perrotta DM (1987) Survey of underlying conditions of persons hospitalized with acute respiratory disease during influenza epidemics in Houston, 1978–1981. Am Rev Respir Dis 136:550–555. https://doi.org/10.1164/ajrccm/136.3.550
Izurieta HS, Thompson WW, Kramarz P et al (2000) Influenza and the rates of hospitalization for respiratory disease among infants and young children. N Engl J Med 342:232–239. https://doi.org/10.1056/NEJM200001273420402
Collins PJ, Haire LF, Lin YP et al (2008) Crystal structures of oseltamivir-resistant influenza virus neuraminidase mutants. Nature 453:1258–1261. https://doi.org/10.1038/nature06956
von Itzstein M (2007) The war against influenza: discovery and development of sialidase inhibitors. Nat Rev Drug Discov 6:967–974. https://doi.org/10.1038/nrd2400
Bertram S, Glowacka I, Steffen I et al (2010) Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev Med Virol 20:298–310. https://doi.org/10.1002/rmv.657
Ikram NKK, Durrant JD, Muchtaridi M et al (2015) A virtual screening approach for identifying plants with anti H5N1 neuraminidase activity. J Chem Inf Model 55:308–316. https://doi.org/10.1021/ci500405g
Yusuf M, Mohamed N, Mohamad S et al (2016) H274Y’s effect on oseltamivir resistance: what happens before the drug enters the binding site. J Chem Inf Model 56:82–100. https://doi.org/10.1021/acs.jcim.5b00331
Yaeghoobi M, Frimayanti N, Chee CF et al (2016) QSAR, in silico docking and in vitro evaluation of chalcone derivatives as potential inhibitors for H1N1 virus neuraminidase. Med Chem Res 25:2133–2142. https://doi.org/10.1007/s00044-016-1636-5
Ryu YB, Kim JH, Park S-J et al (2010) Inhibition of neuraminidase activity by polyphenol compounds isolated from the roots of Glycyrrhiza uralensis. Bioorg Med Chem Lett 20:971–974. https://doi.org/10.1016/j.bmcl.2009.12.106
Nguyen PH, Na M, Dao TT et al (2010) New stilbenoid with inhibitory activity on viral neuraminidases from Erythrina addisoniae. Bioorg Med Chem Lett 20:6430–6434. https://doi.org/10.1016/j.bmcl.2010.09.077
Dao TT, Nguyen PH, Lee HS et al (2011) Chalcones as novel influenza A (H1N1) neuraminidase inhibitors from Glycyrrhiza inflata. Bioorg Med Chem Lett 21:294–298. https://doi.org/10.1016/j.bmcl.2010.11.016
Park J-Y, Jeong HJ, Kim YM et al (2011) Characteristic of alkylated chalcones from Angelica keiskei on influenza virus neuraminidase inhibition. Bioorg Med Chem Lett 21:5602–5604. https://doi.org/10.1016/j.bmcl.2011.06.130
Muchtaridi M, Lestari D, Khairul Ikram NK, Gazzali AM, Hariono M, Wahab HA (2021) Decaffeination and neuraminidase inhibitory activity of arabica green coffee (Coffea arabica) beans: chlorogenic acid as a potential bioactive compound. Molecules 26:3402. https://doi.org/10.3390/molecules26113402
Riswanto FDO, Rawa MSA, Murugaiyah V et al (2019) Anti-cholinesterase activity of chalcone derivatives: synthesis, in vitro assay and molecular docking study. Med Chem. https://doi.org/10.2174/1573406415666191206095032
Chintakrindi A, Martis E, Gohil D et al (2016) A computational model for docking of noncompetitive neuraminidase inhibitors and probing their binding interactions with neuraminidase of influenza virus H5N1. Curr Comput Aided-Drug Des 12:272–281. https://doi.org/10.2174/1573409912666160713111242
Yu M, Wang Y, Tian L et al (2015) Safflomin A inhibits neuraminidase activity and influenza virus replication. RSC Adv 5:94053–94066. https://doi.org/10.1039/C5RA17336A
Lipinski CA (2000) Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods 44:235–249. https://doi.org/10.1016/S1056-8719(00)00107-6
Armstrong JD, Hubbard RE, Farrell T, Maiguashca B (2006) Structure-based drug discovery: an overview
Huerta E, Grey N (2007) Cancer control opportunities in low- and middle-income countries
McCormick DL (2017) Preclinical evaluation of carcinogenicity using standard-bred and genetically engineered rodent models, 2nd edn. Elsevier Inc, Amsterdam
DE Pires V, Blundell TL, Ascher DB (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58:4066–4072. https://doi.org/10.1021/acs.jmedchem.5b00104
Gadaleta D, Vuković K, Toma C et al (2019) SAR and QSAR modeling of a large collection of LD50 rat acute oral toxicity data. J Cheminform 11:1–16. https://doi.org/10.1186/s13321-019-0383-2
Barlow S, Chesson A, Collins JD et al (2009) Use of the benchmark dose approach in risk assessment. EFSA J 1150:1–72
Sorell TL (2016) Approaches to the development of human health toxicity values for active pharmaceutical ingredients in the environment. AAPS J 18:92–101. https://doi.org/10.1208/s12248-015-9818-5
de Angelis I, Turco L (2011) Caco-2 cells as a model for intestinal absorption. Curr Protoc Toxicol. https://doi.org/10.1002/0471140856.tx2006s47
Lin JH, Yamazaki M (2003) Role of P-glycoprotein in pharmacokinetics. Clin Pharmacokinet 42:59–98. https://doi.org/10.2165/00003088-200342010-00003
Ballabh P, Braun A, Nedergaard M (2004) The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis 16:1–13. https://doi.org/10.1016/j.nbd.2003.12.016
Kinirons MT, O’Mahony MS (2004) Drug metabolism and ageing. Br J Clin Pharmacol 57:540–544. https://doi.org/10.1111/j.1365-2125.2004.02096.x
Rosenbaum SE (2016) Basic pharmacokinetics and pharmacodynamics, an integrated textbook and computer simulations, 2nd edn. https://doi.org/10.1111/j.1365-2125.2011.04077.x
Burt HJ, Neuhoff S, Almond L et al (2016) Metformin and cimetidine: physiologically based pharmacokinetic modelling to investigate transporter mediated drug-drug interactions. Eur J Pharm Sci 88:70–82. https://doi.org/10.1016/j.ejps.2016.03.020
Toutain PL, Bousquet-Mélou A (2004) Plasma clearance. J Vet Pharmacol Ther 27:415–425. https://doi.org/10.1111/j.1365-2885.2004.00605.x
van der Vries E, Collins PJ, Vachieri SG et al (2012) H1N1 2009 pandemic influenza virus: resistance of the I223R neuraminidase mutant explained by kinetic and structural analysis. PLoS Pathog 8:e1002914. https://doi.org/10.1371/journal.ppat.1002914
Lackenby A, Hungnes O, Dudman SG et al (2008) Emergence of resistance to oseltamivir among influenza A(H1N1) viruses in Europe. Eurosurveillance 13:3–4. https://doi.org/10.2807/ese.13.05.08026-en
Baranovich T, Saito R, Suzuki Y et al (2010) Emergence of H274Y oseltamivir-resistant A(H1N1) influenza viruses in Japan during the 2008–2009 season. J Clin Virol 47:23–28. https://doi.org/10.1016/j.jcv.2009.11.003
Samson M, Pizzorno A, Abed Y, Boivin G (2013) Influenza virus resistance to neuraminidase inhibitors. Antiviral Res 98:174–185. https://doi.org/10.1016/j.antiviral.2013.03.014
Essandoh E (2010) Structural studies of organic crystals of pharmaceutical relevance. Correlation of crystal structure analysis with recognised non-bonded structural motifs in the organic solid state
Evranos Aksöz B, Ertan R (2011) Chemical and structural properties of chalcones I. Fabad J Pharm Sci 36:223–242
Kim CU, Chen X, Mendel DB (1999) Neuraminidase inhibitors as anti-influenza virus agents. Antivir Chem Chemother 10:141–154. https://doi.org/10.1177/095632029901000401
Jedrzejas MJ, Singh S, Brouillette WJ et al (1995) Structures of aromatic inhibitors of influenza virus neuraminidase. Biochemistry 34:3144–3151. https://doi.org/10.1021/bi00010a003
Chand P, Babu YS, Bantia S et al (1997) Design and synthesis of benzoic acid derivatives as influenza neuraminidase inhibitors using structure-based drug design 1. J Med Chem 40:4030–4052. https://doi.org/10.1021/jm970479e
Poenitzsch VZ, Winters DC, Xie H et al (2007) Effect of electron-donating and electron-withdrawing groups on peptide/single-walled carbon nanotube interactions. J Am Chem Soc 129:14724–14732. https://doi.org/10.1021/ja0750827
Kumar JSD, Ho MM, Toyokuni T (2001) Simple and chemoselective reduction of aromatic nitro compounds to aromatic amines: reduction with hydriodic acid revisited. Tetrahedron Lett 42:5601–5603. https://doi.org/10.1016/S0040-4039(01)01083-8
Jason-Moller L, Murphy M, Bruno J (2006) Overview of Biacore systems and their applications. Curr Protoc Protein Sci 19:13. https://doi.org/10.1002/0471142301.ps1914s45
Lewis EA, Murphy KP (2005) Isothermal titration calorimetry. Methods Mol Biol 305:1–16. https://doi.org/10.1385/1-59259-912-5:001
Hariono M, Ngah N, Wahab HA, Abdul Rahim AS (2012) 2-Bromo-4-(3,4-dimethyl-5-phenyl-1,3-oxazolidin-2-yl)-6-methoxyphenol. Acta Crystallogr Sect E Struct Rep Online 68:o35–o36. https://doi.org/10.1107/S1600536811051269
Hariono M, Wahab HA, Tan ML et al (2014) 9-Benzyl-6-benzylsulfanyl-9 H-purin-2-amine. Acta Crystallogr Sect E Struct Rep Online 70:o288–o288. https://doi.org/10.1107/S1600536814001986
Russell RJ, Haire LF, Stevens DJ et al (2006) The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 443:45–49. https://doi.org/10.1038/nature05114
Morris GM, Lim-Wilby M (2008) Molecular docking. pp 365–382
Zima V, Albiñana CB, Rojíková K et al (2019) Investigation of flexibility of neuraminidase 150-loop using tamiflu derivatives in influenza A viruses H1N1 and H5N1. Bioorg Med Chem 27:2935–2947. https://doi.org/10.1016/j.bmc.2019.05.024
Potier M, Mameli L, Bélisle M et al (1979) Fluorometric assay of neuraminidase with a sodium (4-methylumbelliferyl-α-d-N-acetylneuraminate) substrate. Anal Biochem 94:287–296. https://doi.org/10.1016/0003-2697(79)90362-2
Hariono M, Abdullah N, Damodaran KV et al (2016) Potential new H1N1 neuraminidase inhibitors from ferulic acid and vanillin: molecular modelling, synthesis and in vitro assay. Sci Rep 6:38692. https://doi.org/10.1038/srep38692
Contreras G, Bather R, Furesz J, Becker BC (1985) Activation of metastatic potential in African green monkey kidney cell lines by prolonged in vitro culture. In Vitro Cell Dev Biol 21:649–652
Hariono M, Rollando R, Karamoy J et al (2020) Bioguided fractionation of local plants against matrix metalloproteinase9 and its cytotoxicity against breast cancer cell models: in silico and in vitro study. Molecules 25:1–17. https://doi.org/10.3390/molecules25204691
Borra RC, Lotufo MA, Gagioti SM et al (2009) A simple method to measure cell viability in proliferation and cytotoxicity assays. Braz Oral Res 23:255–262. https://doi.org/10.1590/S1806-83242009000300006
Acknowledgments
We thank to Scripps Institute, ACDLabs, and Biovia for providing free softwares.
Funding
This study was funded by Faculty of Pharmacy, Sanata Dharma University under Internship Grant 2019 for the financial support.
Author information
Authors and Affiliations
Contributions
HAW and MH organises the project and the article writing and review. Investigation, PH, JCK, CFA, EA and EJC are students under MH supervision that contribute by doing the experiments.
Corresponding author
Ethics declarations
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of the article was revised: The competing interest statement was updated.
Supplementary Information
Additional file 1: Figure S1.
The control docking results of a oseltamivir triazole into H1N1 catalytic site and b oseltamivir into H5N1 catalytic site.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hariyono, P., Kotta, J.C., Adhipandito, C.F. et al. A study on catalytic and non-catalytic sites of H5N1 and H1N1 neuraminidase as the target for chalcone inhibitors. Appl Biol Chem 64, 69 (2021). https://doi.org/10.1186/s13765-021-00639-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-021-00639-w