
Abstract
There is accumulating evidence that compounds containing N-acylhydrazone or 4-chromenone moieties can be active against multiple cancer cell types, yet the combined effect of these chemical groups is unclear. This study aimed to develop more effective anti-cancer compounds by combining 4-chromenone and N-acylhydrazone. Thirteen derivatives were designed, synthesized, and characterized, and their structures were identified using nuclear magnetic resonance spectroscopy and high-resolution mass spectrometry. Most of the derivatives exhibited moderate to high efficacy in inhibiting the clonogenicity of HCT116 colon cancer cells. In particular, derivative 12, (E)-N'-((6-methoxy-4-oxo-4H-chromen-3-yl)methylene)isonicotinohydrazide, strongly inhibited clonogenicity (GI50 = 34.8 μM) of HCT116 cells and aurora kinase A (aurA) activity in vitro (IC50 = 1.4 μM). In silico docking experiment predicted that derivative 12 interacts with aurA based on computational docking and calculations of binding free energy. When derivative 12 was exposed to HCT116 cells, the phosphorylation of aurA at Thr288 was dose-dependently decreased within 60 min. Further analysis showed that derivative 12 destroyed the mitotic spindle in HCT116 cells. These results suggest that derivatives of 4-chromenone combined with N-acylhydrazone are feasible in the development of selective aurA inhibitor and could be considered potential chemotherapeutic agents.
Similar content being viewed by others

Introduction
Matrix N-acylhydrazone, which can be synthesized by the condensation of hydrazide and ketone, is a motif commonly used in medicinal chemistry. Compounds containing N-acylhydrazone moieties (Additional file 1: Fig. S1A) have been developed as drugs for diverse conditions due to their easy synthesis and diverse interactions with biological targets. For example, dantrolene and azumolene (Additional file 1: Fig. S1B) are effective against malignant hyperthermia, presenting with a fast heart rate and muscle rigidity [1, 2]. In addition, nifroxiazide (Additional file 1: Fig. S1C) and carbazochrome (Additional file 1: Fig. S1D) are used to treat diarrhea and hereditary hemorrhagic telangiectasia, respectively [3, 4]. Nitrofurantoin (Additional file 1: Fig. S1E) and nitrofurazone (Additional file 1: Fig. S1F) display anti-bacterial activities [5]. N-(4-tert-butylbenzoyl)-2-hydroxy-1-naphthaldehyde hydrazone, naphthyl salicylic acyl hydrazone, and 2-(3,4,5-trihydroxybenzylidene)hydrazinecarboxamide (Additional file 1: Fig. S1G and H) inhibit HIV-1 RNase H, ribonucleotide reductase, and Influenza virus PA endonuclease, respectively [6,7,8]. Combretastatin A4 (Additional file 1: Fig. S2A), isolated from Combretum caffrum, is an active microtubule-destabilizing agent consisting of two benzene rings connected via ethylene [9]. Its derivative, LASSBio (Additional file 1: Fig. S2B), which contains N-acylhydrazone in place of the ethylene group of Combretastatin A4, also displays microtubule-destabilizing activity [10]. Many other compounds containing N-acylhydrazone exhibit potentially beneficial biological activities, including anti-cancer properties. For example, diphenyl-N-acylhydrazone derivatives (Additional file 1: Fig. S2C) inhibit histone deacetylases [11], ortho-hydroxy-N-acylhydrazone derivatives (Additional file 1: Fig. S2D) activate procaspase-3 kinase [12], and N-Acylhydrazones with heterobivalent β-carbolines (Additional file 1: Fig. S2E) act as anti-cancer agents [13].
Like N-acylhydrazone-containing compounds, compounds containing 4-chromenone moieties have been shown to display various biological activities. Flavone is a 4-chromenone derivative with a phenyl group substituted at the 2-position. Luteolin (3′,4′,5,7-tetrahydroxyflavone) (Additional file 1: Fig. S3A) inhibits the proliferation of tumor cells, including MCF7/HER18 breast cancer cells, BxPC-3 human pancreatic cancer cells, and T98G/U87MG human glioblastoma cells [14]. Vitexin (apigenin-8-C-glucoside) (Additional file 1: Fig. S3B) inhibits hypoxia-inducible factor-1α in human hepatoma HepG2 cells [15]. In addition, nobiletin (4′,5,5′,6,7,8-hexamethoxyflavone) (Additional file 1: Fig. S3C) displays anti-cancer effects via modulating Nrf2/AKT/ERK pathways in an MDR xenograft model [16].
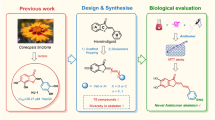
The aim of this study was to identify compounds displaying anti-cancer activities by designing derivatives of 4-chromenone combined with N-acylhydrazone. Unlike the phenyl group substitution in flavones, the 3-position of 4-chromenone was substituted with N-acylhydrazone in this study (Additional file 1: Fig. S4). Thirteen derivatives were synthesized, and their structures identified using nuclear magnetic resonance (NMR) spectroscopy and high-resolution mass spectrometry (HR/MS). The 13 derivatives synthesized here displayed similar structures, with halogen, methyl, or methoxy substituents at the 6-position of the 4-chromenone moiety, and a phenyl or pyrimidine group at the N-acylhydrazone moiety. The anti-cancer activities of the derivatives were measured using a long-term survival clonogenic assay for 7 days, which can distinguish differences in the survival of cancer cells caused by compounds with similar structures.
Aurora kinases are members of the mitotic serine/threonine kinase family and include aurora kinase A (aurA), aurora kinase B (aurB), and aurora kinase C (aurC). Among them, aurA is frequently overexpressed in various human cancer and has been shown to act as an oncogene, thereby developing aggressive tumors [17, 18]. Inhibition of aurA arrests cells at the G2/M phase and suppresses tumor growth in vivo [19,20,21].
In our previous study, compounds containing an N-acylhydrazone moiety showed inhibitory effects on aurA [22]. Therefore, in vitro aurA kinase assays were performed for all 13 derivatives synthesized here. In order to elucidate the binding mode between aurA and the most promising 4-chromenone derivative at the molecular level, in silico docking was performed. aurA inhibitory effect of the lead compound was analyzed by biochemical experiments, including western blot analysis and immunofluorescence microscopy.
Materials and methods
Chemical synthesis
The synthetic procedure for the preparation of derivatives of 4-chromenone combined with N-acylhydrazone 1–13 is shown in Scheme 1. First of all, the Vilsmeier–Haack reaction was performed between substituted 2-hydroxyacetophenone and N,N-dimethylformamide (DMF) in the presence of phosphorous oxychloride (POCl3)-furnished chromenone aldehyde (II). Excess quantities of DMF reacted with POCl3 to form a reaction intermediate active chloroiminium ion of DMF, which rapidly reacted with 2-hydroxyacetophenone to form chromenone. The chromenone intermediate reacted with additional chloroiminium ions to produce substituted 3-chromenone aldehyde (II). The substituted 3-chromenone aldehyde (II) was condensed with various benzohydrazides (III) in the presence of catalytic quantities of glacial acetic acid to produce 4-chromenone-N-acylhydazone hybrid compounds 1–13.

Synthetic procedures of thirteen N-acylhydrazone derivatives combined with 4-chromenone
Spectroscopic analysis
The structures of the thirteen 4-chromenone/N-acylhydrazone derivatives synthesized here were determined using NMR spectroscopy and HR/MS. All compounds were dissolved in deuterated dimethyl sulfoxide (DMSO-d6), diluted to approximately 50 mM, and transferred into 2.5 mm NMR tubes. All NMR experiments were performed on a Bruker AVANCE 400 spectrometer system (9.4 T; Bruker, Karlsruhe, Germany) at room temperature, and the chemical shifts were referenced to tetramethylsilane (0 ppm). The detailed experimental procedure followed has been reported previously [23]. Ultra-performance liquid chromatography-hybrid quadrupole-time-of-flight mass spectrometry (UPLC-TOFMS) was carried out on a Waters ACQUITY UPLC system (Waters, Milford, MA) [24]. All HR/MS data were collected as positive modes. The FT-IR spectra of the 13 derivatives are provided as Additional file 2.
Cell culture
Human HCT116 colon cancer cells were procured from the American Type Culture Collection (Manassas, VA). Cells were maintained in Dulbecco’s modified Eagle’s medium (ThermoFisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT) and penicillin–streptomycin (Sigma-Aldrich).
Clonogenic assay
HCT116 human colon cancer cell lines were counted and plated onto 24-well tissue culture plates (BD Falcon™; Becton Dickson Immunocytometry System, San Jose, CA) at a density of 4 × 103 cells/well in Dulbecco’s modified eagle’s medium supplemented with 10% fetal bovine serum. The cells were treated with various concentrations of derivative compounds (0, 5, 10, and 20 μM) for 7 days, fixed with 6% glutaraldehyde, and stained with 0.1% crystal violet, as described previously [25]. The cell growth inhibitory concentration values were measured using densitometry, and the half-maximal cell growth inhibitory concentration (GI50) values were calculated using the SigmaPlot program.
In vitro kinase assay
In vitro kinase assays were performed using the EMD Milliporesigma kinaseprofiler service assay protocol (MilliporeSigma Corp., St. Louis, MO). Aurora A kinase (aurA) was supplied by EMD Millipore Corp. The substrate for phosphorylation was 30 μM AKRRRLSSLRA and the concentration of ATP was 10 μM [26]. All experiments were repeated thrice at five different concentrations (0, 5, 10, 20, and 80 μM). The half-maximal inhibitory concentrations (IC50) were obtained on SigmaPlot software (SYSTAT, Chicago, IL, USA) using the sigmoid curve fit [27].
In silico docking
The 3D structure of aurA was available in the protein data bank (3uod.pdb) [28]. The 3D structure of derivative 12 was determined using the X-ray crystallographic 3D structure of (E)-4-hydroxy-N’-(3-methoxybenzylidene)benzohydrazide [29]. In silico docking experiments were performed as described previously [25] on an Intel Core 2 Quad Q6600 (2.4 GHz) Linux PC with SYBYL 7.3 (Tripos, St. Louis, MO) [30]. The binding site was determined using the LigPlot program [31], and 3D images were generated using the PyMOL program (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. Portland, OR, USA). The logP values were obtained using the SYBYL 7.3 program.
Western blot analysis
HCT116 cells were treated with derivative 12 either at two different concentrations (50 and 100 μM) for 1 h or at 50 μM for various periods (0, 1, and 3 h). The cells were lysed in a cell lysis buffer (20 mM HEPES (pH 7.2), 1% (v/v) Triton X-100, 10% (v/v) glycerol, 150 mM NaCl, 10 μg/mL leupeptin, 1 mM phenylmethylsulfonyl fluoride). Protein extracts (20—30 μg per sample) were electrophoresed on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes (Bio-Rad). After blocking in a solution of 5% nonfat dry milk in TTBS buffer (0.05% Tween-20 in 50 mM Tris-buffered saline, pH 7.6) for 30 min, the blots were incubated with primary antibodies against phospho (p)-aurA (Thr288)/aurB (Thr232)/aurC (Thr198) (Cell Signaling Technology, Beverly, MA, USA), or glyceraldehyde phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnology, Santa Cruz, CA) diluted to 1:1000 in the same nonfat dry milk solution for 4 h at 25 °C. After washing three times with TTBS, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies for 2 h at 25 °C. After washing with TTBS, blots were developed with an enhanced chemiluminescence detection system (GE Healthcare, Piscataway, NJ).
Immunofluorescence staining
HCT116 cells were seeded onto coverslips. After attachment, the cells were treated with 50 μM derivative 12 or the vehicle control (0.001% DMSO) for 24 h, followed by fixation with 4% (w/v) paraformaldehyde and permeabilization with 0.1% (v/v) Triton X-100, as described previously [32]. The slides were incubated with primary antibodies against phospho-histone H3 (Ser10; 1:500) and α/β-tubulin (1:500). After 2 h, the slides were incubated with secondary antibodies conjugated with Alexa Fluor 555 (for phospho-histone H3 (Ser10); red signal, Invitrogen) and Alexa Fluor 488 (for α/β-tubulin, green signal, Invitrogen) for 30 min. Nuclear DNA was counterstained with 1 μg/mL Hoechst 33,258 (Sigma-Aldrich) for an additional 10 min (blue signal). Stained cells were observed under an EVOS FL fluorescence microscope (Advanced Microscopy Group, Bothell, WA).
Statistical analysis
The data are plotted as means with S.D. Statistical comparisons were performed using a one-way ANOVA followed by Dunnett’s multiple comparisons test with the GraphPad Prism V8.3.1 software (GraphPad Software, San Diego, CA). A P-value < 0.05 was considered statistically significant.
Results and discussion
The structures and names of the 4-chromenone/N-acylhydrazone derivatives
The structures and names of the thirteen 4-chromenone/N-acylhydrazone derivatives synthesized here are listed in Table 1.

Spectroscopic data of the 13 derivatives, including NMR and HR/MS, are as follows:
(E)-N'-((6-chloro-4-oxo-4H-chromen-3-yl)methylene)-4-methoxybenzohydrazide (1)
1H NMR (400 MHz, DMSO-d6) δ 11.82 (s, 1H, NH), 8.81 (s, 1H, H-2), 8.58 (s, 1H, N=H), 8.03 (d, 1H, H-5, J = 2.6 Hz), 7.92 (d, 2H, H-2′, H-6′, J = 9.0 Hz), 7.87 (dd, 1H, H-7, J = 9.0, 2.6 Hz), 7.77 (d, 1H, H-8, J = 9.0 Hz), 7.04 (d, 2H, H-3′, H-5′, J = 9.0 Hz), 3.83 (s, 1H, 4′-OCH3); 13C NMR (400 MHz, DMSO-d6) δ 174.0 (C-4), 162.3 (NHC=O), 162.0 (C-4′), 154.5 (C-2), 154.3 (C-9), 136.2 (N=CH), 134.4 (C-7), 130.4 (C-6), 129.5 (C-2′, C-6′), 125.1 (C-1′), 124.4 (C-10), 124.1 (C-5), 121.2 (C-8), 118.5 (C-3), 113.7 (C-3′, C-5′), 55.4 (4′-OCH3); HR/MS (m/z): Calcd. for (M+H)+: 357.0642; Found: 357.0630; IR(cm−1): 3267(N–H), 1667(C=N), 1503(C–N), 1247, 1028(C–O).
(E)-N'-((6-bromo-4-oxo-4H-chromen-3-yl)methylene)-4-methoxybenzohydrazide (2)
1H NMR (400 MHz, DMSO-d6) δ 11.82 (s, 1H, NH), 8.81 (s, 1H, H-2), 8.58 (s, 1H, N=CH), 8.16 (d, 1H, H-5, J = 2.5 Hz), 7.98 (dd, 1H, H-7, J = 8.9, 2.5 Hz), 7.92 (d, 2H, H-2′, H-6′, J = 9.0 Hz), 7.71 (d, 1H, H-8, J = 8.9 Hz), 7.04 (d, 2H, H-3′, H-5′, J = 8.8 Hz), 3.83 (s, 1H, 4′-OCH3); 13C NMR (400 MHz, DMSO-d6) δ 174.0 (C-4), 162.3 (NHC=O), 162.0 (C-4′), 154.5 (C-2), 154.3 (C-9), 136.2 (N=CH), 134.4 (C-7), 130.4 (C-6), 129.5 (C-2′, C-6′), 125.1 (C-1′), 124.4 (C-10), 124.1 (C-5), 121.2 (C-8), 118.5 (C-3), 113.7 (C-3′,C-5′), 55.4 (4′-OCH3); HR/MS (m/z): Calcd. for (M+H)+: 401.0137; Found: 401.0134; IR(cm−1): 3214(N–H), 1640(C=N), 1503(C–N), 1249, 1028(C–O).
(E)-N'-((6-methoxy-4-oxo-4H-chromen-3-yl)methylene)benzohydrazide (3)
1H NMR (400 MHz, DMSO-d6) δ 11.92 (s, 1H, NH), 8.80 (s, 1H, H-2), 8.65 (s, 1H, N=CH), 7.93 (d, 2H, H-2′, H-6′, J = 7.6 Hz), 7.68 (d, 1H, H-8, J = 9.1 Hz), 7.59 (dd, 1H, H-4′, J = 7.3, 7.3 Hz), 7.52 (dd, 2H, H-3′, H-5′, J = 7.6, 7.3 Hz), 7.48 (d, 1H, H-5, J = 3.1 Hz), 7.43 (dd, 1H, H-7, J = 9.1, 3.1 Hz), 3.88 (s, 1H, 6-OCH3); 13C NMR (400 MHz, DMSO-d6) δ 174.7 (C-4), 162.9 (NHC=O), 156.9 (C-6), 154.2 (C-2), 150.5 (C-9), 140.5 (N=CH), 133.2 (C-1′), 131.8 (C-4′), 128.4 (C-3′, C-5′), 127.6 (C-2′, C-6′), 124.0 (C-10), 123.7 (C-7), 120.3 (C-8), 117.5 (C-3), 104.9 (C-5), 55.8 (6-OCH3); HR/MS (m/z): Calcd. for (M+H)+: 323.1032; Found: 323.0672; IR(cm−1): 3209(N–H), 1739(C=O), 1642(C=N), 1543(C–N), 1209, 1020(C–O).
(E)-N'-((6-methoxy-4-oxo-4H-chromen-3-yl)methylene)-3-bromobenzohydrazide (4)
1H NMR (400 MHz, DMSO-d6) δ 11.98 (s, 1H, NH), 8.80 (s, 1H, H-2), 8.63 (s, 1H, N=CH), 8.11 (s, 1H, H-2′), 7.93 (d, 1H, H-6′, J = 7.8 Hz), 7.79 (d, 1H, H-4′, J = 7.7 Hz), 7.67 (d, 1H, H-8, J = 9.1 Hz), 7.50 (dd, 1H, H-5′, J = 7.8, 7.7 Hz), 7.47 (d, 1H, H-5, J = 3.0 Hz), 7.43 (dd, 1H, H-7, J = 9.1, 3.0 Hz), 3.87 (s, 1H, 6-OCH3); 13C NMR (400 MHz, DMSO-d6) δ 174.7 (C-4), 161.3 (NHC=O), 156.9 (C-6), 154.3 (C-2), 150.5 (C-9), 141.1 (N=CH), 135.3 (C-1′), 134.5 (C-4′), 130.7 (C-5′), 130.1 (C-2′), 126.8 (C-6′), 124.0 (C-10), 123.7 (C-7), 121.7 (C-3′), 120.3 (C-8), 117.4 (C-3), 104.9 (C-5), 55.8 (6-OCH3); HR/MS (m/z): Calcd. for (M+H)+: 401.0137; Found: 401.0107; IR(cm−1): 3212(N–H), 1672(C=N), 1483(C–N), 1220, 1023(C–O).
(E)-N'-((6-methoxy-4-oxo-4H-chromen-3-yl)methylene)-3-fluorobenzohydrazide (5)
1H NMR (400 MHz, DMSO-d6) δ 11.95 (s, 1H, NH), 8.77 (s, 1H, H-2), 8.62 (s, 1H, N=CH), 7.78 (d, 1H, H-6′, J = 7.6 Hz), 7.72 (d, 1H, H-2′, J = 9.9 Hz), 7.63 (d, 1H, H-8, J = 9.1 Hz), 7.56 (dd, 1H, H-5′, J = 7.6, 6.2 Hz), 7.43 (d, 1H, H-5, J = 2.8 Hz), 7.42 (m, 1H, H-4′), 7.39 (dd, 1H, H-7, J = 9.1, 2.8 Hz), 3.85 (s, 1H, 6-OCH3); 13C NMR (400 MHz, DMSO-d6) δ 174.7 (C-4), 161.9 (C-3′), 161.5 (NHC=O), 156.9 (C-6), 154.3 (C-2), 150.5 (C-9), 141.1 (N=CH), 135.4 (C-1′), 130.7 (C-5′), 124.0 (C-10), 123.8 (C-6′), 123.6 (C-7), 120.3 (C-8), 118.7 (C-4′), 117.4 (C-3), 114.4 (C-2′), 104.9 (C-5), 55.8 (6-OCH3); HR/MS (m/z): Calcd. for (M+H)+: 341.0938; Found: 341.0691; IR(cm−1): 3179(N–H), 1739(C=O), 1632(C=N), 1540(C–N), 1207, 1028(C–O).
(E)-N'-((6-methoxy-4-oxo-4H-chromen-3-yl)methylene)-3-hydroxybenzohydrazide (6)
1H NMR (400 MHz, DMSO-d6) δ 11.85 (s, 1H, NH), 9.73(s, 1H, 3′-OH), 8.75 (s, 1H, H-2), 8.63 (s, 1H, N=CH), 7.63 (d, 1H, H-8, J = 9.0 Hz), 7.44 (d, 1H, H-5, J = 2.9 Hz), 7.39 (dd, 1H, H-7, J = 9.0, 2.9 Hz), 7.34 (d, 1H, H-6′, J = 8.0 Hz), 7.33 (s, 1H, H-2′), 7.30 (dd, 1H, H-5′, J = 8.0, 7.8 Hz), 6.98 (d, 1H, H-4′, J = 7.8 Hz), 3.85 (s, 1H, 6-OCH3); 13C NMR (400 MHz, DMSO-d6) δ 174.7 (C-4), 163.0 (NHC=O), 157.4 (C-3′), 156.9 (C-6), 154.1 (C-2), 150.5 (C-9), 140.4 (N=CH), 134.6 (C-1′), 129.5 (C-5′), 124.0 (C-10), 123.6 (C-7), 120.2 (C-8), 118.8 (C-4′), 118.1 (C-6′), 117.6 (C-3), 114.6 (C-2′), 104.9 (C-5), 55.8 (6-OCH3); HR/MS (m/z): Calcd. for (M+H)+: 339.0981; Found: 339.0959; IR(cm−1): 3284(N–H), 1652(C=N), 1540(C–N), 1204, 1001(C–O).
(E)-N'-((6-methoxy-4-oxo-4H-chromen-3-yl)methylene)-3-methoxybenzohydrazide (7)
1H NMR (400 MHz, DMSO-d6) δ 11.87 (s, 1H, NH), 8.80 (s, 1H, H-2), 8.66 (s, 1H, N=CH), 7.67 (d, 1H, H-8, J = 9.1 Hz), 7.51 (d, 1H, H-6′, J = 7.7 Hz), 7.49 (d, 1H, H-5, J = 3.1 Hz), 7.47 (s, 1H, H-2′), 7.43 (dd, 1H, H-7, J = 9.1, 3.1 Hz), 7.42 (dd, 1H, H-5′, J = 8.1, 7.7 Hz), 7.15 (d, 1H, H-4′, J = 8.1, 2.0 Hz), 3.88 (s, 1H, 6-OCH3), 3.84 (s, 1H, 3′-OCH3); 13C NMR (400 MHz, DMSO-d6) δ 174.7 (C-4), 162.6 (NHC=O), 159.2 (C-3′), 156.9 (C-6), 154.1 (C-2), 150.5 (C-9), 140.5 (N=CH), 134.5 (C-1′), 129.5 (C-5′), 124.0 (C-10), 123.6 (C-7), 120.2 (C-8), 119.8 (C-6′), 117.6 (C-4′), 117.5 (C-3), 112.7 (C-2′), 104.9 (C-5), 55.8 (6-OCH3), 55.3 (3′-OCH3); HR/MS (m/z): Calcd. for (M+H)+: 353.1137; Found: 353.1122; IR(cm−1): 3154(N–H), 1637(C=N), 1563(C–N), 1212, 1050(C–O).
(E)-N'-((6-methoxy-4-oxo-4H-chromen-3-yl)methylene)-4-fluorobenzohydrazide (8)
1H NMR (400 MHz, DMSO-d6) δ 11.92 (s, 1H, NH), 8.79 (s, 1H, H-2), 8.63 (s, 1H, N=CH), 8.01 (dd, 2H, H-2′, H-6′, J = 8.5, 5.6 Hz), 7.66 (d, 1H, H-8, J = 9.1 Hz), 7.46 (d, 1H, H-5, J = 3.0 Hz), 7.42 (dd, 1H, H-7, J = 9.1, 3.0 Hz), 7.35 (dd, 2H, H-3′, H-5′, J = 8.5, 8.5 Hz), 3.87 (s, 1H, 6-OCH3); 13C NMR (400 MHz, DMSO-d6) δ 174.7 (C-4), 164.2 (C-4′), 161.8 (NHC = O), 156.9 (C-6), 154.2 (C-2), 150.5 (C-9), 140.6 (N=CH), 130.3 (C-2′, C-6′), 129.6 (C-1′), 124.0 (C-10), 123.7 (C-7), 120.3 (C-8), 117.5 (C-3), 115.4 (C-3′, C-5′), 104.9 (C-5), 55.8 (6-OCH3); HR/MS (m/z): Calcd. for (M+H)+: 341.0938; Found: 341.0912; IR(cm−1): 3415(N–H), 1739(C=O), 1625(C=N), 1563(C–N), 1229, 1092(C–O).
(E)-N'-((6-methoxy-4-oxo-4H-chromen-3-yl)methylene)-4-methoxybenzohydrazide (9)
1H NMR (400 MHz, DMSO-d6) δ 11.79 (s, 1H, NH), 8.76 (s, 1H, H-2), 8.62 (s, 1H, N=CH), 7.92 (ddd, 2H, H-2′, H-6′, J = 8.8, 2.8, 1.8 Hz), 7.65 (d, 1H, H-8, J = 9.1 Hz), 7.45 (d, 1H, H-5, J = 3.1 Hz), 7.41 (dd, 1H, H-7, J = 9.1, 3.1 Hz), 7.04 (ddd, 2H, H-3′, H-5′, J = 8.8, 2.8, 1.8 Hz), 3.86 (s, 1H, 6-OCH3), 3.83 (s, 1H, 4′-OCH3); 13C NMR (400 MHz, DMSO-d6) δ 174.7 (C-4), 162.4 (NHC=O), 162.0 (C-4′), 156.9 (C-6), 153.9 (C-2), 150.5 (C-9), 139.8 (N=CH), 129.6 (C-2′, C-6′), 125.2 (C-1′), 124.0 (C-10), 123.6 (C-7), 120.3 (C-8), 117.6 (C-3), 113.7 (C-3′, C-5′), 104.9 (C-5), 55.8 (6-OCH3), 55.4 (4′-OCH3); HR/MS (m/z): Calcd. for (M+H)+: 353.1137; Found: 353.1162; IR(cm−1): 3398(N–H), 1739(C = O), 1628(C=N), 1563(C–N), 1250, 1028(C–O).
(E)-N'-((6-methyl-4-oxo-4H-chromen-3-yl)methylene)benzohydrazide (10)
1H NMR (400 MHz, DMSO-d6) δ 11.92 (s, 1H, NH), 8.79 (s, 1H, H-2), 8.63 (s, 1H, N=CH), 7.93 (d, 2H, H-2′, H-6, J = 8.0 Hz), 7.91 (m, 1H, H-5), 7.65 (m, 1H, H-7), 7.61 (m, 1H, H-8), 7.60 (m, 1H, H-4′), 7.52 (dd, 2H, H-3′, H-5′, J = 7.5, 7.2 Hz), 2.43 (s, 1H, 6-CH3); 13C NMR (400 MHz, DMSO-d6) δ 174.9 (C-4), 162.9 (NHC=O), 154.3 (C-2), 154.1 (C-9), 140.5 (N=CH), 135.7 (C-6), 135.6 (C-7), 133.2 (C-1′), 131.8 (C-4′), 128.5 (C-3′, C-5′), 127.6 (C-2′, C-6′), 124.4 (C-5), 123.0 (C-10), 118.5 (C-8), 118.2 (C-3), 20.4 (6-CH3); HR/MS (m/z): Calcd. for (M+H)+: 307.1083; Found: 307.1097; IR(cm−1): 3201(N–H), 1637(C=N), 1543(C–N), 1284, 1067(C–O).
(E)-N'-((6-methyl-4-oxo-4H-chromen-3-yl)methylene)nicotinohydrazide (11)
1H NMR (400 MHz, DMSO-d6) δ 12.05 (s, 1H, NH), 9.07 (d, 1H, H-2′, J = 1.7 Hz), 8.80 (s, 1H, H-2), 8.76 (dd, 1H, H-4′, J = 4.8, 1.4 Hz), 8.62 (s, 1H, N = CH), 8.26 (ddd, 1H, H-6′, J = 7.8, 1.7, 1.4 Hz), 7.90 (d, 1H, H-5, J = 1.7 Hz), 7.65 (dd, 1H, H-7, J = 8.6, 1.7 Hz), 7.60 (d, 1H, H-8, J = 8.6 Hz), 7.56 (dd, 1H, H-5′, J = 7.8, 4.8 Hz), 2.43 (s, 1H, 6-CH3); 13C NMR (400 MHz, DMSO-d6) δ 174.9 (C-4), 161.4 (NHC=O), 154.5 (C-2), 154.0 (C-9), 152.3 (C-4′), 148.6 (C-2′), 141.2 (N=CH), 135.7 (C-6), 135.6 (C-7), 135.4 (C-6′), 128.9 (C-1′), 124.4 (C-5), 123.5 (C-5′), 122.9 (C-10), 118.5 (C-8), 117.9 (C-3), 20.4 (6-CH3); HR/MS (m/z): Calcd. for (M+H)+: 308.1035; Found: not detected; IR(cm−1): 3248(N–H), 1671(C=N), 1540(C–N), 1311, 1108(C–O).
(E)-N'-((6-methoxy-4-oxo-4H-chromen-3-yl)methylene)isonicotinohydrazide (12)
1H NMR (400 MHz, DMSO-d6) δ 12.13 (s, 1H, NH), 8.84 (s, 1H, H-2), 8.78 (dd, 2H, H-3′, H-5′, J = 6.0, 1.5 Hz), 8.67 (s, 1H, N=CH), 7.84 (dd, 2H, H-2′, H-6′, J = 6.0, 1.5 Hz), 7.71 (d, 1H, H-8, J = 9.1 Hz), 7.50 (d, 1H, H-5, J = 3.0 Hz), 7.46 (dd, 1H, H-7, J = 9.1, 3.0 Hz), 3.88 (s, 1H, 6-OCH3); 13C NMR (400 MHz, DMSO-d6) δ 174.7 (C-4), 161.4 (NHC=O), 157.0 (C-6), 154.5 (C-2), 150.5 (C-9), 150.3 (C-3′, C-5′), 141.9 (N=CH), 140.2 (C-1′), 124.1 (C-10), 123.8 (C-7), 121.5 (C-2′, C-6′), 120.4 (C-8), 117.3 (C-3), 104.9 (C-5), 55.8 (6-OCH3); HR/MS (m/z): Calcd. for (M+H)+: 324.0984; Found: 324.0977; IR(cm−1): 3201(N–H), 1677(C=N), 1483(C–N), 1220, 1026(C–O).
(E)-N'-((6-methyl-4-oxo-4H-chromen-3-yl)methylene)isonicotinohydrazide (13)
1H NMR (400 MHz, DMSO-d6) δ 12.09 (s, 1H, NH), 8.78 (s, 1H, H-2), 8.77 (dd, 2H, H-3′, H-5′, J = 5.9, 1.5 Hz), 8.62 (s, 1H, N=CH), 7.87 (d, 1H, H-5, J = 1.9 Hz), 7.82 (dd, 2H, H-2′, H-6′, J = 5.9, 1.5 Hz), 7.63 (dd, 1H, H-7, J = 8.6, 1.9 Hz), 7.57 (d, 1H, H-8, J = 8.6 Hz), 2.41 (s, 1H, 6-CH3); 13C NMR (400 MHz, DMSO-d6) δ 174.9 (C-4), 161.4 (NHC=O), 154.6 (C-2), 154.0 (C-9), 150.3 (C-3′, C-5′), 141.9 (N=CH), 140.2 (C-1′), 135.8 (C-6), 135.7 (C-7), 124.4 (C-5), 122.9 (C-10), 121.5 (C-2′, C-6′), 118.5 (C-8), 117.9 (C-3), 20.4 (6-CH3); HR/MS (m/z): Calcd. for (M+H)+: 308.1035; Found: 308.1049; IR(cm−1): 3398(N–H), 1653 (C=N), 1481(C–N), 1234, 1067(C–O).
Inhibitory effect of 4-chromenone derivatives combined with N-acylhydrazone on clonogenicity of HCT116 cells
There are numerous methods to measure the anti-cancer activities of small compounds, including cytotoxicity assays in cancer cell lines. In this study, a long-term survival clonogenic assay was adopted (Fig. 1). The GI50 values ranged between 34.76 and 85.22 μM (Table 1 and Fig. 2). Seven of the derivatives (3–9) contained common substituents; a 6-methoxy group in the 4-chromenone moiety and a phenyl group in N-acylhydrazone. The different substituents of N-acylhydrazone appeared to have different effects on the GI50 values. More specifically, the 3-bromo (derivative 4), 3-fluoro (5), and 3-hydroxy (6) groups displayed the higher inhibitory activity of clonogenicity than the hydrogen (3), 3-methoxy (7), 4-fluoro (8), and 4-methoxy (9) groups. In compounds with a 4-methoxy group substituted to the phenyl ring of N-acylhydrazone, a 6-bromo group (derivative 2) on the 4-chromenone moiety displayed better activity than the 6-chloro (1) and 6-methoxy (9) groups. Derivatives 3 and 10 contained a phenyl group on the N-acylhydrazone moiety, yet derivative 10 displayed the higher inhibitory activity of clonogenicity than derivative 3. The three derivatives (11–13) that contained a pyridine group instead of a phenyl group on N-acylhydrazone appeared to cause relatively the higher inhibitory activity of clonogenicity.

Inhibitory effect of derivatives of 4-chromenone combined with N-acylhydrazone (1–13) on clonogenicity of HCT116 cells

The half-maximal inhibitory concentration (GI50) values of 4-chromenone derivatives combined with N-acylhydrazone on the clonogenicity of HCT116 cells
Inhibitory effect of 4-chromenone derivatives combined with N-acylhydrazone on the aurA kinase activity
Compounds containing an N-acylhydrazone moiety showed inhibitory effects on aurA [22]. To test whether the 13 derivatives synthesized here inhibit aurA, we conducted in vitro aurA kinase assay. The aurA inhibitor TCS7010 (2,4-bisanilinopyrimidine) with a reported IC50 value of 3.4 nM was used as a reference compound [33]. The IC50 values of the derivatives ranged from 1.2 to 54.1 μM (Table 1), suggesting that 4-chromenone derivatives combined with N-acylhydrazone exert inhibitory effect on aurA. Of these, we selected derivative 12, (E)-N'-((6-methoxy-4-oxo-4H-chromen-3-yl)methylene)isonicotinohydrazide, which showed the highest GI50 (34.8 μM) and the second-highest IC50 values (1.4 μM), and used for further experiments.
In silico molecular docking
To predict whether derivative 12 can directly bind to aurA, we analyzed the binding mode between aurA and derivative 12 using an in silico docking experiment. Of over 100 three-dimensional (3D) structures of aurA deposited in the protein data bank (PDB), the X-ray crystallographic structure 3uod.pdb [28] was selected as the 3D structure of aurA for in silico docking. This structure originated from Homo sapiens aurA expressed in Escherichia coli BL21(DE3), with a resolution of 2.50 Å. Its 3D structure contains the ligand 4-[(4-{[2-(trifluoromethyl)phenyl]amino}pyrimidin-2-yl)amino]benzoic acid (named as TPAP) (Additional file 1: Fig. S5), which is composed of two benzene rings and pyrimidine and has a molecular weight of 374 Da. Similarly, the thirteen derivatives synthesized here consist of three rings, and their molecular weights range between 322 and 401 Da. Human aurA consists of 403 amino acids and 3uod.pdb includes the majority of the protein, from Lys123 to Lys401. Importantly, it contains the protein kinase domain (133–383), making 3uod.pdb suitable to study in silico docking.
The 3D structure of derivative 12 was determined based on the previously reported X-ray crystallographic 3D structure of (E)-4-hydroxy-N’-(3-methoxybenzylidene)benzohydrazide (Additional file 1: Fig. S6) [29]. The apo-protein of aurA without a ligand was obtained using the Sybyl program and, after energy minimization, compared with the crystallographic structure revealing a root mean squared deviation of 0.65 Å. The binding site was determined based on published data and LigPlot program analysis as Glu260, Gly140, Leu139, Thr217, Val147, Leu263, Ala160, Glu211, Tyr212, Arg137, Gly216, Ala213, and Arg220. To confirm the validity of our in silico docking process, TPAP (the ligand contained in 3uod.pdb) was docked into the apo-protein of aurA, revealing that it docked inside the same binding site as in the crystal structure of aurA (3uod.pdb).
The Sybyl program provides the flexible docking method FlexX, and performs 30 iterations of the docking procedure so that 30 protein–ligand complexes were generated. The binding energy ranged between -21.2 and -26.9 kcal/mol. Because the first complex showed both the lowest binding energy and best docking pose, its complex was selected. As shown in Fig. 3a, derivative 12 was effectively docked into aurA in this complex. Analysis by the LigPlot program revealed the residues participating in the interaction (Additional file 1: Fig. S7). The three residues Tyr219, Leu263, and Ala213 showed hydrophobic interactions with derivative 12. The hydrogen atom of the amine group of Arg220 forms a hydrogen bond (H-bond) with the oxygen atom in the ketone group of the 4-chromenone moiety with a distance of 2.86 Å. The oxygen atom of Leu139 forms an H-bond with the hydrogen atom of the secondary amine of N-acylhydrazone (3.20 Å). In addition, the oxygen atom of the ketone group of N-acylhydrazone forms two H-bonds with two amine groups in Arg137 (2.69 Å and 2.96 Å). An image of the binding site of the derivative 12–aurA complex is shown in Fig. 3b, where three residues forming H-bonds with derivative 12 are marked. While the ligand TPAP interacts with 13 residues of aurA, derivative 12 appears to interact with six residues. The volumes of derivative 12 and TPAP were calculated using the Sybyl/MOLCAD module as 234.1 Å3 and 246.2 Å3, respectively. The larger volume of TPAP may, therefore, explain the difference in individual residue interactions mentioned above. However, while TPAP forms H-bonds with two residues of aurA, derivative 12 does so with three residues. Based on these data, it can be predicted that derivative 12 could directly bind to and inhibit the aurA kinase activity.

Images of the complex of derivative 12 and the apo-protein of aurora A kinase generated by the PyMol program (a) and its binding site (b), where Arg137, Leu139, and Arg220 of aurora A kinase form hydrogen bonds with derivative 12
Inhibitory effect of derivative 12 on phosphorylation of aurora kinases A
Phosphorylation at Thr288 in the activation loop of aurA is crucial for its activation [34, 35]. To validate whether derivative 12 could inhibit the phosphorylation of aurA at the cellular level, we treated HCT116 cells with various concentrations (0, 50, and 100 μM) of derivative 12. Phosphorylation of aurA on Thr288 was significantly reduced (p < 0.001, n = 3) in a dose-dependent manner (Fig. 4a). In contrast, phosphorylation of aurB on Thr232 and aurC on Thr198 were only slightly decreased at 100 μM. A time-course experiment confirmed that the addition of 50 μM derivative 12 significantly inhibited phosphorylation of aurA within 60 min, while the phosphorylation of aurB and aurC were relatively unaffected (Fig. 4b). These data suggest that derivative 12 preferentially inhibited aurA over aurB and aurC.

Effect of derivative 12 on the inhibition of aurora kinases. HCT116 cells were treated either with various concentrations of derivative 12 (0, 50, or 100 μM) for 60 min (a) or 50 μM derivative 12 over a time-course (0, 60, or 120 min) (b). Western blot analysis was performed using phospho-specific antibodies against aurA (Thr288)/aurB (Thr232)/aurC (Thr198). The anti-GAPDH antibody was used as an internal control. The band intensities of phosphorylated (p)-aurA, (p)-aurB, and (p)-aurC relative to GAPDH level were measured using ImageJ software. The data are presented as means ± SD (n = 3). NS, not significant; ***, P < 0.001 according to Dunnettt's multiple comparisons test
Effect of derivative 12 on the mitotic microtubule network
AurA is a mitotic kinase localized to the mitotic pole of spindle microtubules and has important functions in centrosome maturation, chromosome alignment, and mitotic spindle assembly [36, 37]. To investigate whether derivative 12 affects mitotic spindle assembly during mitosis, we analyzed the morphological features of mitotic spindles using immunofluorescent staining of α/β tubulin in mitotic cells. Phosphorylation of histone H3 on Ser10 plays a role in chromatin condensation during mitosis and is therefore used as a marker for mitotic cells [38]. Thus, we analyzed the mitotic spindle network in phospho-histone H3 (Ser10)-positive mitotic cells. In untreated cells, we observed that bipolar spindle microtubules were attached to chromosomes arranged along with the metaphase plate in phospho-H3 (Ser 10)-positive cells, exhibiting typical features of the metaphase stage of mitosis (Fig. 5, top panels). In contrast, treatment with derivative 12 induced a disorganized arrangement of microtubules and abnormal chromosome alignment in phospho-H3 (Ser 10)-positive cells (Fig. 5, bottom panels). These data suggest that inhibition of aurA by derivative 12 is functionally linked to aberrant mitotic progression.

Effect of derivative 12 on the disruption of mitotic spindle dynamics. HCT116 cells were treated with 50 μM for 24 h. The cells were fixed and incubated with antibodies specific to mouse anti-α/β-tubulin (1:500) and rabbit anti-phospho (p)-Histone H3 (Ser10; 1:500) for 2 h, followed by incubation with AlexaFluor 488-conjugated (green signal) and AlexFluor 555-conjugated (red signal) secondary antibodies for 30 min. Nuclear DNA was stained with 1 μg/mL Hoechst 33,258 for 10 min (blue signal). Fluorescent cells were examined under an EVOSf1 fluorescence microscope. Int, interphase cell; Mit, mitotic cell; Size bar, 50 mm. Arrows, Mitotic spindles attached to metaphase chromosomes
Here, we report that most of the derivatives exhibited moderate to high efficacy in inhibiting the clonogenicity of HCT116 colon cancer cells and in vitro aurA kinase activity. In particular, derivative 12, (E)-N'-((6-methoxy-4-oxo-4H-chromen-3-yl)methylene)isonicotinohydrazide, exhibited the suppression of clonogenicity of HCT116 cells with GI50 value of 34.8 μM and the inhibition of in vitro aurA kinase activity with IC50 value of 1.4 μM. In silico docking experiment predicted the interaction of derivative 12 with aurA. Furthermore, treatment of HCT116 cells with derivative 12 dose-dependently prevented the phosphorylation of aurA at Thr288 within 60 min. In addition, derivative 12 disrupted the mitotic spindle dynamics in HCT116 cells.
Lipinski’s rule of five is a method to evaluate the likelihood of a chemical compound being an orally active drug in humans based on its chemical and physical properties [39]. As listed in Table 1, all of the 13 derivatives studied here displayed logP values less than 5 and molecular weights less than 500 Da (ranging from 322 to 401 Da). In addition, the numbers of hydrogen bond donors and acceptors of the 13 derivatives were less than 5 and 10, respectively. Hence, all derivatives synthesized here satisfy Lipinski’s rule.
Collectively, these results suggest that derivatives of 4-chromenone combined with N-acylhydrazone could be considered as potential chemotherapeutic agents. However, there are limitations to this study, as we have focused on the design, characterization, and in vitro effects of these compounds. Future research should be directed towards evaluating the effectiveness of the 4-chromenone/N-acylhydrazone compounds in vivo, and ultimately clinical trials for the future benefit of cancer patients.
Availability of data and materials
The datasets used and analyzed in this study are available from the corresponding author on reasonable request.
Change history
28 May 2023
Missing funding information has been added
Abbreviations
- aur:
-
Aurora kinase
- DMF:
-
N,N-Dimethylformamide
- DMSO:
-
Dimethyl sulfoxide
- HR/MS:
-
High resolution/mass spectrometry
- NMR:
-
Nuclear magnetic resonance
- PDB:
-
Protein data bank
- UPLC-TOFMS:
-
Ultra-performance liquid chromatography-hybrid quadrupole-time-of-flight mass spectrometry
References
Krause T, Gerbershagen MU, Fiege M, Weißhorn R, Wappler F (2004) Dantrolene—a review of its pharmacology, therapeutic use and new developments. Anaesthesia 59:364–373
Corrêa JCR, Hiene MAC, Salgado HRN (2013) Physico-chemical characterization and analytical development for sodium azumolene, a potential drug designed to fight malignant hyperthermia. J Anal Bioanal Tech 5(1):1–6. https://doi.org/10.4172/2155-9872.1000177)
Begovic B, Ahmedtagic S, Calkic L, Vehabović M, Kovacevic SB, Catic T, Mehic M (2016) Open clinical trial on using nifuroxazide compared to probiotics in treating acute diarrhoeas in adults. Mater Sociomed 28:454–458
Passali GC, De Corso E, Bastanza G, Gennaro LD, HHT Gemelli Study Group (2015) An old drug for a new application: carbazochrome-sodium-sulfonate in HHT. J Clin Pharmacol 55:601–602
GuayDR, (2001) An update on the role of nitrofurans in the management of urinary tract infections. Drugs 61:353–364
Himmel DM, Sarafianos SG, Dharmasena S, Hossain MM, McCoy-Simandle K, Ilina T, Clark AD Jr, Knight JL, Julias JG, Clark PK, Krogh-Jespersen K, Levy RM, Hughes SH, Parniak MA, Arnold E (2006) HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl naphthyl hydrazone bound at a novel site. ACS Chem Biol 1:702–712
Ahmad MF, Alam I, Huff SE, Flanagan PJ, SA, Shewach D, Misko TA, Oleinick NL, Harte WE, Viswanathan R, Harris ME, Dealwis CG, (2017) Potent competitive inhibition of human ribonucleotide reductase by a nonnucleoside small molecule. Proc Natl Acad Sci USA 114:8241–8246
Carcelli M, Rogolino D, Gatti A, De Luca L, Sechi M, Kumar G, White SW, Stevaert A, Naesens L (2016) N-acylhydrazone inhibitors of influenza virus PA endonuclease with versatile metal binding modes. Sci Rep 6:31500
Prise VE, Honess DJ, Stratford MRL, Wilson J, Tozer GM (2002) The vascular response of tumor and normal tissues in the rat to the vascular targeting agent, combretastatin A-4-phosphate, at clinically relevant doses. Int J Oncol 21:717–726
Amaral DN, Cavalcanti BC, Bezerra DP, Ferreira PMP, Castro RP, Sabino JR, Machado CML, Chammas R, Pessoa C, Sant’Anna CMR, Barreiro EJ, Lima LM (2014) Docking, synthesis and antiproliferative activity of N-acylhydrazone derivatives designed as combretastatin A4 analogues. PLoS ONE 9(3):e85380
Rodrigues DA, Ferreira-Silva GA, Ferreira ACS, Fernandes RA, Kwee JK, Sant’Anna CMR, Ionta M, Fraga CAM (2016) Design, synthesis, and pharmacological evaluation of novel N-acylhydrazone derivatives as potent histone deacetylase 6/8 dual inhibitors. J Med Chem 59(2):655–670
Ma J, Zhang G, Han X, Bao G, Wang L, Zhai X, Gong P (2014) Synthesis and biological evaluation of benzothiazole derivatives bearing the ortho-hydroxy-N-acylhydrazone moiety as potent antitumor agents. Arch Pharm (Weinheim) 347(12):936–949
Chen X, Guo L, Ma Q, Chen W, Fan W, Zhang J (2019) Design, synthesis, and biological evaluation of novel N-acylhydrazone bond linked heterobivalent β-carbolines as potential anticancer agents. Molecules 24(16):2950
Imran M, Rauf A, Abu-Izneid T, Nadeem M, Shariati MA, Khan IA, Imran A, Orhan IE, Rizwan M, Atif M, Gondal TA, Mubarak MS (2019) Luteolin, a flavonoid, as an anticancer agent: A review. Biomed Pharmacother 112:108612
Choi HJ, Eun JS, Kim BG, Kim SY, Jeon H, Soh Y (2006) Vitexin, an HIF-1alpha inhibitor, has anti-metastatic potential in PC12 cells. Mol Cells 22(3):291–299
Feng S, Tian Y, Huo S, Qu B, Liu R, Xu P, Li Y, Xie Y (2020) Nobiletin potentiates paclitaxel anticancer efficacy in A549/T xenograft model: Pharmacokinetic and pharmacological study. Phytomedicine 67:153141
Mountzios G, Terpos E, Dimopoulo M (2008) Aurora kinases as targets for cancer therapy. Cancer Treat Rev 34:175–182
Jiang Y, Zhang Y, Lees E, Seghezzi W (2003) AuroraA overexpression overrides the mitotic spindle checkpoint triggered by nocodazole, a microtubule destabilizer. Oncogene 22:8293–8301
Qi W, Cooke LS, Liu X, Rimsza L, Roe DJ, Manziolli A, Persky DO, Miller TP, Mahadevan D (2011) Aurora inhibitor MLN8237 in combination with docetaxel enhances apoptosis and anti-tumor activity in mantle cell lymphoma. Biochem Pharmacol 81:881–890
Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, Su M, Golec JMC, Miller KM (2004) VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med 10:262–267
Keen N, Taylor S (2004) Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer 4:927–936
Koh D, Jung Y, Ahn S, Mok KH, Shin SY, Lim Y (2017) Synthesis and structure elucidation of polyphenols containing the N’-methyleneformohydrazide scaffold as aurora kinase inhibitors. Magn Reson Chem 55:864–876
Lee Y, Koh D, Lim Y (2018) 1H and 13C NMR spectral assignments of 25 ethyl 2-oxocyclohex-3-enecarboxylates. Magn Reson Chem 56:1188–1200
Ahn S, Shin SY, Jung Y, Jung H, Kim BS, Koh D, Lim Y (2016) 1H and 13C NMR spectral assignments of novel flavonoids bearing benzothiazepine. Magn Reson Chem 54:382–390
Shin SY, Yoon H, Ahn S, Kim D, Kim SH, Koh D, Lee YH, Lim Y (2013) Chromenylchalcones showing cytotoxicity on human colon cancer cell lines and in silico docking with aurora kinases. Bioorg Med Chem 21:4250–4258
Shin SY, Yoon H, Hwang D, Ahn S, Kim D, Koh D, Lee YH, Lim Y (2013) Benzochalcones bearing pyrazoline moieties show anti-colorectal cancer activities and selective inhibitory effects on aurora kinases. Bioorg Med Chem 21:7018–7024
Jung Y, Shin SY, Yong Y, Jung H, Ahn S, Lee YH, Lim Y (2015) Plant-derived flavones as inhibitors of aurora B kinase and their quantitative structure-activity relationships. Chem Biol Drug Des 85:574–585
Martin MP, Zhu J, Lawrence HR, Pireddu R, Luo Y, Alam R, Ozcan S, Sebti SM, Lawrence NJ, Schönbrunn E (2012) A novel mechanism by which small molecule inhibitors induce the DFG flip in Aurora A. ACS Chem Biol 7:698–706
Zhong Q, Hu S, Yan H (2016) Crystal structure of 1-benzyl-4-formyl-1H-pyrrole-3-carb-oxamide. Acta Crystallogr E Crystallogr Commun 72(Pt 2):133–135
Kim BS, Shin SY, Ahn S, Koh D, Lee YH, Lim Y (2017) Biological evaluation of 2-pyrazolinyl-1-carbothioamide derivatives against HCT116 human colorectal cancer cell lines and elucidation on QSAR and molecular binding modes. Bioorg Med Chem 25:5423–5431
Wallace AC, Laskowski RA, Thornton JM (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng 8:127–134
Lee JM, Lee MS, Koh D, Lee YH, Lim Y, Shin SY (2015) A new synthetic 2’-hydroxy-2,4,6-trimethoxy-5’,6’-naphthochalcone induces G2/M cell cycle arrest and apoptosis by disrupting the microtubular network of human colon cancer cells. Cancer Lett 354:348–354
Aliagas-Martin I, Burdick D, Corson L, Dotson J, Drummond J, Fields C, Huang OW, Hunsaker T, Kleinheinz T, Krueger E, Liang J, Moffat J, Phillips G, Pulk R, Rawson TE, Ultsch M, Walker L, Wiesmann C, Zhang B, Zhu B, Cochran AG (2009) A class of 2,4-bisanilinopyrimidine Aurora A inhibitors with unusually high selectivity against Aurora B. J Med Chem 52(10):3300–3307
Walter AO, Seghezzi W, Korver W, Sheung J, Lees E (2000) The mitotic serine/threonine kinase Aurora2/AIK is regulated by phosphorylation and degradation. Oncogene 19:4906–4916
Ohashi S, Sakashita G, Ban R, Nagasawa M, Matsuzuaki H, Murata Y, Taniguchi H, Shima H, Furukawa K, Urano T (2006) Phospho-regulation of human protein kinase Aurora-A: analysis using anti-phospho-Thr288 monoclonal antibodies. Oncogene 25:7691–7702
Fu J, Bian M, Jiang Q, Zhang C (2007) Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res 5:1–10
Barr AR, Gergely F (2007) Aurora-A: the maker and breaker of spindle poles. J Cell Sci 120:2987–2996
Hooser AV, Goodrich DW, Allis CD, Brinkley BR, Mancini MA (1998) Histone H3 phosphorylation is required for the initiation, but not maintenance, of mammalian chromosome condensation. J Cell Sci 111:3497–3506
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26
Funding
This study was supported by the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT, Republic of Korea (Grant no. NRF-2019R1F1A1058747). HR/MS was performed with the help of Dr. C. H. Lee at Konkuk University. This paper was supported by the KU Research Professor Program of Konkuk University.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare that there is no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Structures of (A) N-acylhydrazone, (B) dantrolene and azumolene, (C) nifroxiazide, (D) carbazochrome, (E) nitrofurantoin, (F) nitrofurazone, (G) N-(4-tert-butylbenzoyl)-2-hydroxy-1-naphthaldehyde hydrazone, and (H) 2-(3,4,5-trihydroxybenzylidene)hydrazinecarboxamide. Fig. S2. Structures of (A) combretastatin A4, (B) LASSBio-1593, (C) diphenyl-N-acylhydrazone, (D) ortho-hydroxy-N-acylhydrazone, and (E) heterobivalent β-carboline. Fig. S3. Structures of (A) luteolin(3′,4′,5,7-tetrahydroxyflavone), (B) vitexin (apigenin-8-C-glucoside), and (C) nobiletin(4′,5,5′,6,7,8-hexamethoxyflavone). Fig. S4. Structure of N-acylhydrazone combined with 4-chromenone. Fig. S5. Structure of 4-[(4-{[2-(trifluoromethyl)phenyl]amino}pyrimidin-2-yl)amino]benzoic acid. Fig. S6. Structure of (E)-4-hydroxy-N’-(3-methoxybenzylidene)benzohydrazide. Fig. S7. The residues participating in the binding site of derivative 12 and apo-protein of aurora A kinase complex analyzed using the LigPlot program.
Additional file 2.
The FT-IR spectra of the 13 derivatives.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shin, S.Y., Lee, J., Ahn, S. et al. Design, synthesis, and evaluation of 4-chromenone derivatives combined with N-acylhydrazone for aurora kinase A inhibitor. Appl Biol Chem 64, 21 (2021). https://doi.org/10.1186/s13765-021-00596-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-021-00596-4