
Abstract
Background
Xq duplication is a rare condition with a very variable phenotype, which could mimic other genetic syndromes involving the long arm of chromosome X. Sometimes short stature and diminished ovarian reserve (DOR) may be present. Treatments with rGH (Recombinant growth Hormon) or with fertility preservation strategies have not been previously described.
Case presentation
We present the case of a female with a novel de novo Xq partial duplication (karyotype: 46,Xder(X)(qter→q21.31::pter→qter) confirmed by array-CGH analysis. She presented with short stature, Nonverbal Learning Disability, developmental delay during childhood, severe scoliosis, spontaneous onset of menarche and irregular menstrual cycles. AMH (Anti-Müllerian Hormone) allowed detection of a preserved but severely diminished ovarian reserve with a POI (Premature Ovarian insufficiency) onset risk. She was effectively subjected to fertility preservation strategies and rGH therapy. We also reviewed other published cases with Xq duplication, reporting the main clinics characteristics and any adopted treatment.
Conclusions
rGH treatment and cryopreservation in a multidisciplinary approach are good therapeutic strategies for Xq duplication syndrome with short stature and premature ovarian failure.
Similar content being viewed by others


Background
Duplications of the long arm of chromosome X (Xq) include intrachromosomal duplications and partial disomies/trisomies resulting from unbalanced translocations with an autosome or with a chromosome Y [1]. Complex and extended, cytogenetically visible duplications of Xq are rare and more often involve the distal Xq27-qter region [2]. To date, CGH array techniques allow even the detection of smaller imbalances. Prevalence of duplications of the distal part of chromosome Xq is yet unknown; in particular, duplications that include MECP2 gene in Xq28, responsible for neurologic impairment in these patients, are of major interest with a few hundred cases reported in literature [14, 20].
While males with dup (Xq) usually present short stature, mental retardation, feeding problems, microcephaly, facial dysmorphism, hypotonia, and hypoplastic genitalia, females with dup (Xq) may show some phenotypic abnormalities that could include short stature, developmental delay, facial dysmorphism, and gonadal dysgenesis [1, 7]. This condition could be defined as Xq Duplication Syndrome [7]. The phenotypic characteristics vary significantly in expression and severity depending on the size of duplication, the sex of the affected person, and on the genes of the chromosome duplicated segment [15]. In general, males are more likely to be severely affected by this syndrome than females bearing the same duplication, since the duplicated X chromosome is usually inactivated [1, 5].
Moreover, it is well known that the integrity and the correct functionality of the long arm of the X chromosome are required to maintain fertility. This is demonstrated by Turner Syndrome (TS), a clinical conditions with complete or partial loss of X chromosome, which is the main genetic cause of POI (Premature Ovarian Insufficiency) [21].
Our study will presents for the first time the case of a woman with a de novo duplication of Xq21.31- > qter with short stature and affected by Nonverbal Learning Disability, responsive to rGH therapy and having a preserved, even if severely compromised, ovarian activity. We also reviewed other female Xq duplication cases reported in literature in the last 35 years, highlighting the main clinical features and treatments adopted.
Case presentation
Our patient is a 18 years old woman, the second child of a healthy and unrelated couple. Both parents were 32 years old at her birth. Her older sister was healthy. She was born small for gestational age (SGA) at 35 gestational weeks by vaginal delivery. Birth weight was 1100 g and body length was 41 cm. Developmental delay with short stature and low body weight, moderate scoliosis and a very mild intellectual disability with delayed speech have been noticed since her childhood. Spontaneous thelarche occurred at age of 11 years and menarche at the age of 14 years. The menses were always present even if irregular in rhythm. Daily subcutaneous rGH therapy at dose of 0.2 mg/kg/week was started at the age of 4 years because of SGA (clonidine GH stimulation test was: GH peak value of 11 ng/ml; arginine GH stimulation test was: GH peak value of 16 ng/mL); at that time, her weight was 11 kg (− 2.7 SD score) and her height was 90 cm (− 2.9 SD score) with a target height of 169.5 cm (+ 1.25 SD score). The 3 years follow-up growth parameters did not reveal any improvement; therefore at the age of 7 years the dose was increased to 0.3 mg/kg/week, as Variant of Turner Syndrome.
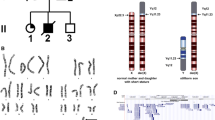
Metaphase chromosome analysis from peripheral blood lymphocytes culture and GTG banding was performed using standard protocol. Cytogenetic analysis showed a female karyotype containing additional material on one X chromosome. Fluorescence in situ hybridization (FISH) with arm-specific probes (chromoprobe, Cytocell) and subtelomeric Xp and Xq probes (Vysis) allowed detection of the Xp and Xq regions on the duplicated X chromosome. The karyotype was then described as 46,X dup(X) (q21.31 → q28) (Fig. 1a and b). In order to better define the exact extent of duplication and to deepen the origin of the developmental delay and the short stature, she performed oligonucleotide-based array comparative genomic hybridization which showed partial trisomy and tetrasomy Xq [arr Xq21.31(89,594,027-89,837,847) × 3,Xq21.31(89,853,144-90,222,617) × 4,Xq21.31q28(90,254,419-155,257,082) × 3] with a 65 Mb duplication.

a Patient’s G banded karyotype shows one normal X chromosome and one aberrant X chromosome that contains a duplicated segment Xq 21.31- > qter on Xp extremity. b Fluorescence in situ hybridization using specific chromoprobe for Xp (green probe) and Xq arms (red probe)
When she was 18 years old the weight was 42,7 Kg (− 2 DS score) and her height was 153,2 cm (− 1 DS score): physical examination showed general facial hypotonia and nistagmus, long face with tall chin, hypotrophy of the lower extremities, widely spaced teeth, complete pubertal development (Tanner stage 5) and a moderate scoliosis for which surgery was recommended. Her follow-up shows improvement in her growth parameters (Fig. 2 a and b).

a Imagine of the patient with scoliosis, short stature and long face. b Growth curve with response to GH therapy. (Patient’s one is shown by the dot line)
A formal cognitive evaluation was performed for the first time at 15 years old: the Wechsler Intelligence Scale for Children (WISC-III) revealing a total IQ of 77 (Verbal IQ: 81, Performance IQ: 79). Further neuropsychological evaluations highlighted the presence of a nonverbal learning disability. By age of 18 neurological evaluation remarked only a slight motor clumsiness. An electroencephalogram recorded at the same age both in wakefulness and sleep did not reveal any abnormalities. Overall, the patient’s cognitive level is sufficient to guarantee satisfactory daily activity and learning.
Laboratory test at 3rd day of menstrual cycle showed: FSH 7,8 mIU/mL, (normal 1.5–11 mIU/ml), LH 4.51 mIU/ml (normal 1–25), estradiol 104 ng/L (normal 70–530) TSH 1.28 mIU/mL (normal 0.35–3 mIU/mL), AMH 0,39 ng/dl (abnormal < 1 ng/dl) and IGF1 50 nmol/L (normal 33–62). Ultrasound examination performed by transabdominal approach showed the presence of regular uterus and ovaries with an AFC (Antral Follicle Count) of 3.
Hormonal and ultrasound examinations were compatible with a severely reduced ovarian reserve, with AMH and AFC both <5th centile for age. Considering ovarian reserve results, a counselling about available fertility preservation strategies was done. Since both the patient and her parents showed interest in this opportunity, further multidisciplinary evaluations were performed before to proceed. Risks, methods and limits of the procedure were explained, especially regarding the paucity of data about success, genetic transmission risks and the possibility to perform a Preimplantation Genetic Diagnosis (PGD) in the future. Finally, patient’s psycho-intellectual adequacy and her understanding of the proposed procedure were confirmed, and adequate time for decision was given to patient.
A cardiological examination with no contraindications detected, at time of evaluation, for future pregnancies was also performed.
The patient and her parents decided to proceed. After controlled ovarian stimulation (COS) three oocytes were obtained; two of them were in Metaphase II and were cryopreserved with vitrification. Further COS cycles are planned according to patient’s and her parents’ wills, in order to increase the number of oocytes to be preserved.
Results of these literature findings are summarized in the discussion. Our aim is to evaluate a possible correlation between genetic test results and clinic outcomes as well as to suggest possible management strategies for fertility, genetic and endocrinology specialists.
Discussion and conclusions
In this report, we describe a patient SGA with short stature, developmental delay, mild facial dysmorphism, spontaneous puberty, DOR and Nonverbal Learning Disability. Oligonucleotide-based array comparative genomic hybridization demonstrated partial trisomy and tetrasomy Xq.
The abnormal phenotype in female with dup (X) based on selective inactivation of the aberrant dup (X) chromosome is not predictable; this phenotypic diversity may be caused by functional disomy limited to the duplicated X region, an inter-individual difference in X inactivation pattern, a tissue-dependent X activation pattern and possible incomplete inactivation of the duplicated X chromosomal segment.
Zhang et al. [22] analyzed genes that escape X-inactivation showing that the majority of them are located in the short arm and in the distal portion of the long arm of the X chromosome (a region where various genes responsible of intellectual deficit are located). Distribution of escaped genes is particularly important in polyX karyotypes because genes that are more likely to escape inactivation determine clinical symptoms related to the presence of more copies of the gene itself, while inactivated genes remain silent [22, 23] .
Although similar cases have been previously reported, clinical features of patients with Xq Duplication Syndrome are still not well defined.
Tachdjian et al. [5] reported the case of a girl with duplication Xq21.1-q25 and abnormal phenotype with growth retardation, hypotonia, and nystagmus. Carozzo et al. [15] reported a more severe case with congenital anomalies and Pelizaeus-Merzbacher disease given by PLP gene duplication. Other Authors reported cases of Xq duplication of variable extension with different phenotypic characteristics and with the presence, among others, of short stature, premature menopause and mental retardation [3, 16]. Scientific literature reported few cases with GH deficiency in Xq duplication, but all were associated to hypothyroidism or empty sella [24, 25]. No cases are reported about benefits derived from treatment with rGH in patients with Xq duplication syndrome and short stature without GH deficiency.
Our patient manifested short stature since the age of 4 years old with normal levels of IGF 1 and GH. She also had moderate scoliosis and irregular menstrual cycles with incipient ovarian failure. Scoliosis could be part of Xq duplication syndrome [8], but as in patients with TS or Prader–Willi syndrome (PWS), even in the presence of scoliosis GH therapy can be initiated or continued [26, 27]. Our patient started GH therapy and during the treatment she has always been subjected to strict medical controls of scoliosis.
Mechanisms involved in short stature are not clear and the specific genes in the duplicated region that determine height are unknown; conversely, we know that the patient did not present thyroid abnormalities or GH deficiency and that SHOX gene on Xp was normally present in two copies.
Eventually, there might be a positive correlation between the position of the X chromosome duplicated region and the growth deficit. A possible explanation is that the rearranged X chromosome with Xq duplication, could, as well as a deletion, cause alteration of chromatin structure and induce inactivation process of some genes on the X chromosome that are normally active (with possible involvement of the PAR 1 region where the SHOX gene is located and the result of its haploinsufficiency) [28].
Moreover, the number of chromosomes (or genes copies) can also affects height outcome [29]; as a matter of fact, our patient has partial trisomy and tetrasomy Xq.
In our patient, the X chromosome duplicated region contains more than 900 genes; more than 150 of them, if impaired, are responsible for known pathological pictures and many of them could determine premature ovarian failure [30, 31] and intellectual disability [32].
Among all, FMR1 and FMR2 have a double role because the expansion of their trinucleotide repetition may cause both intellectual disability and POI. Other important genes associated with ovarian failure are DACH2 (in Xq21.2), DIAPH2 (in Xq21.33) and PGRMC1 (in Xq24) [30, 33, 34]; as a consequence, it can be assumed that Xq21-Xq24 is a putative region for ovarian reserve.
Despite the presence of spontaneous menstrual cycles and normal values of FSH and LH, ovarian reserve markers such as AMH and AFC allowed detection of a compromised, even if still present, ovarian activity. At our knowledge, this is the first case of Xq duplication reporting assessment of AMH value, which is actually recognized to be the best marker of ovarian reserve in general population [35]. For these reasons, it has been proposed as a marker for early detection of ovarian failure in particular conditions such as, among others, Turner Syndrome [36,37,38]. Therefore, because of her risk of POI onset, a close monitoring of ovarian activity in this patient is needed [38]. Periodically evaluation recording menses characteristics and hormonal dosages are required for early detection and treatment with hormonal replacement therapy for endogenous estrogenic deficiency. As regards fertility issues, an adequate counselling is needed as well: nowadays, fertility preservation strategies such as oocyte cryopreservation and PGD are available and, even if still experimental, could be recommended in selected situations after ethical concerns have been taken into consideration and an adequate psyco-intellectual maturity [36, 39] has been assessed.
Moreover, available literature describes many cases of fertility preservation in Turner Syndrome, which could be considered a similar clinical condition [21, 39].
In conclusion, our patient presents essentially typical facies, mild intellectual disability and some features of Turner Syndrome such as reduced ovarian reserve, scoliosis and short stature. Since the latter is a very frequent condition in girls with dup Xq, it is useful to treat the patient with intensive GH therapy, even in the presence of laboratory data indicative for no GH deficiency.,
We here described for the first time a favourable response to rGH in a girl with Xq duplication. We performed an up-to date review of literature for female with Xq duplication (results are reported in Table 1). Phenotypic features reported are very variable: ovarian insufficiency and short stature are often described in these patients but AMH assessment for ovarian reserve is never reported and no patient was ever treated with rGH or preservation of fertility (Table 1).
We can conclude that females affected by Xq duplication could present very variable clinical features that must be evaluated case by case. Our patient, despite IQ at the lower limits and a Non Verbal Learning disability, carries on a challenging course of study with adequate support. For the short stature, despite absence of GH deficiency, she has been treated intensively with rGH, reaching an acceptable social height. As regards reproductive issues, she is still carefully monitored in order to promptly detect the onset of POI.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study. Moreover, Authors can confirm that all clinical relevant data are included in the article and/or its additional files.
Abbreviations
- AFC:
-
Antral Follicle Count
- AMH:
-
Anti Müllerian Hormone
- COS:
-
Controlled ovarian stimulation
- DOR:
-
Diminished ovarian reserve
- PGD:
-
Preimplantation genetic diagnosis
- POF:
-
Premature ovarian Failure (Table 1)
- POI:
-
Premature Ovarian Insufficiency
- rGH:
-
Recombinant growth hormone
- SGA:
-
Small for gestational age (Table 1)
- TS:
-
Turner Syndrome
References
Salaville D, Schluth-Bolard C, Turleau C. Distal Xq duplication and functional Xq disomy. Orphanet J Rare Dis. 2009;20:4–4. https://doi.org/10.1186/1750-1172-4-4.
Bijlsma EK, Collins A, Papa FT, Tejada MI, Wheeler P, Peeters EA, et al. Xq28 duplications including MECP2 in five females: expanding the phenotype to severe mental retardation. Eur J Med Genet. 2012;55(6–7):404–13. https://doi.org/10.1016/j.ejmg.2012.02.009.
Chen CP, Su XN, Lin HH, Chern SR, Tsai FJ, Wu PC, et al. De novo duplication of Xq22.1->q24 with a disruption of the NXF gene cluster in a mentally retarded woman with short stature and premature ovarian failure. Taiwan J Obstet Gynecol. 2011;50:339–344. Doi: https://doi.org/10.1016/j.tjog.2011.01.018
Donnelly DE, Jones J, McNerlan SE, McGratta P, Humphreys M, McKee S. Growth retardation, developmental delay and dysmorphic features in a girl with a partial duplication of Xq. Clin Dysmorphol. 2011;20:82–5. https://doi.org/10.1097/MCD.0b013e328340b7be.
Tachdjian G, Aboura A, Benkhalifa M, Creveaux I, Foix-Hélias L, Gadisseux JF, et al. De novo interstitial direct duplication of Xq21.1q25 associated with skewed X-inactivation pattern. Am J Med Genet A. 2004;131(3):273–80.
Lachlan KL, Collinson MN, Sandford RO, Van Zyl B, Jacobs PA, Thomas NS. Functional disomy resulting from duplications of distal Xq in four unrelated patients. Hum Gene Ther. 2004;115(5):399–408 Epub 2004 Aug 24.
Armostrong L, McGowan-Jordan J, Brierley K, Allanson JE. De novo dup (X) (q22.3q26) in a girl with evidence that functional disomy of X material is the cause of her abnormal phenotype. Am J Med Genet A. 2003;116A(1):71–6.
Tihi F, Lemyre E, Lemieux N, Dallaire L. De novo dup(X)(q22.1q25) in a girl with an abnormal phenotype. Am J Med Genet. 1999;87(4):302–5.
Correa-Cerro L, Garcia-Cruz D, Ruiz MX, Sanchez-Corona J. De novo duplication xq22-q23 in a girl with short stature and gonadal dysgenesis. Ann Genet. 1999;42(1):41–4.
Monaghan KG, Van Dike DL, Fedelman GL. Prader-Willi-like syndrome in a patient with an Xq23q25 duplication. Am J Med Genet. 1998 Nov 16;80(3):227–31.
Garcia Heras J, Martin JA, Day DW, Scacheri P, Witchel SF. “De novo” duplication Xq23-->Xq26 of paternal origin in a girl with a mildly affected phenotype. Am J Med Genet. 1997;70(4):404–8.
Aughton DJ, AlSaadi AA, Johnson JA, Transue DJ, Trock GL. Dir dup(X) (q13-->qter) in a girl with growth retardation, microcephaly, developmental delay, seizures, and minor anomalies. Am J Med Genet. 1993;46(2):159–64.
Van Dyke DL, Millr MJ, Weiss L. The origin of inverted tandem duplications, and phenotypic effects of tandem duplication of the X chromosome long arm. Am J Med Genet. 1983;15(3):441–50.
Yi Z, Pan H, Li L, Wu H, Wang S, Ma Y, Qi Y. Chromosome Xq28 duplication encompassing MECP2: clinical and molecular analysis of 16 new patients from 10 families in China. Eur J Med Genet. 2016;59(6–7):347–53. https://doi.org/10.1016/j.ejmg.2016.05.004.
Carrozzo R, Arrigo G, Rossi E, Bardoni B, Cammarata M, Gandullia P, et al. Multiple congenital anomalies, brain hypomyelination, and ocular albinism in a female with dup(X)(pter➔q24::q21.32➔qter) and random X inactivation. Am J Med Genet. 1997;72(3):329–34.
Volleth M, Stumm M, Mohnike K, Kalscheuer VM, Jakubiczka S, Wieacher P, et al. Preferential inactivation of a dupX(q23->q27-28) chromosome in a girl with mental retardation and dysmorphy. Hum Hered. 2001;52:177–82.
El Chehadeh S, Touraine R, Prieur F, Reardon W, Bienvenu T, Chantot-Bastaraud S, et al. Xq28 duplication including MECP2 in six unreported affected females: what can we learn for diagnosis and genetic counselling? Clin Genet. 2017;91(4):576–88. https://doi.org/10.1111/cge.12898 Epub 2017 Feb 16.
Maurin ML, Arfeuille C, Sonigo P, Rondeau S, Vekemans M, Turleau C, et al. Large duplications can be benign copy number variants: a case of a 3.6-Mb Xq21.33 duplication. Cytogenet Genome Res. 2017;151(3):115–8. https://doi.org/10.1159/000460278 Epub 2017 Mar 9.
Rajangam S, Lincoln S, Tilak P, Thomas IM. Primary amenorrhea with Xq duplication. Indian J Med Sci. 1999 Feb;53(2):49–52.
Miguet M, Faivre L, Amiel J. N M, Touraine R, Prieur F, et al. further delineation of the MECP2 duplication syndrome phenotype in 59 French male patients, with a particular focus on morphological and neurological features. J Med Genet. 2018;55(6):359–71. https://doi.org/10.1136/jmedgenet-2017-104956.
Oktay K, Bedoschi G, Berkowitz K, Bronson K, Kashani B, McGovern P, et al. Fertility preservation in women with Turner syndrome: a comprehensive review and practical guidelines. J Pediatr Adolesc Gynecol. 2016;29(5):409–16. https://doi.org/10.1016/j.jpag.2015.10.011.
Zhang Y, Castillo-Morales A, Jiang M, Zhu Y, Hu L, Urrutia AO, et al. Genes that escape X-inactivation in humans have high intraspecific variability in expression, are associated with mental impairment but are not slow evolving. Mol Biol Evol. 2013;33(1):302. https://doi.org/10.1093/molbev/msv180.
Disteche CM, Berletch JB. X chromosome inactivation and escape. J Genet. 2015;94:591–9.
Shapira M, Dar H, Bar-El H, Even L, Borochowitz Z, et al. Inherited inverted duplication of X chromosome in a male: report of a patient and review of the literature. Am J Med Genet. 1997;72:409–14.
Yokoyama Y, Narahara K, Tsuji K, Moriwake T, Kanzaki S, Murakami M, et al. Growth hormone deficiency and empty sella syndrome in a boy with dup (X) (q13.3---q21.2). Am J Med Genet. 1992;42:660–664.
Ricotti S, Petrucci L, Carenzio G, Klersyc C, Calcaterra V, Larizza D, et al. Prevalence and incidence in scoliosis in Turner syndrome: a study in 49 girls follow-up for 4 years. Eur J Phys Rehabil Med. 2011;47(3):447–53 Epub 2011 Apr 20.
Allen D B, Backeljauw P, Bidlingmaier M, Biller BMK, Boguszewski M, Burman P, et al. GH safety workshop position paper: a critical appraisal of recombinant human GH therapy in children and adults. Eur J Endrocrinol 2015;174(2): P1-P9. doi: https://doi.org/10.1530/EJE-15-0873.
Hj V d K, Kant SG, Ruivenkamp CA, Gijsbers AC, Haring D, Ostdijk W. Pseudoisodicentric Xp Chromosome [46,X,psu idic(X)(q21.1)] and its effect on growth and pubertal development. Horm Res in Paediat r. 2014;81:416–21. https://doi.org/10.1159/000357141.
Ottesen AM, Aksqlaede L, Garn I, Tartaglia N, Tassone F, Gravholt CH, et al. Increased number of sex chromosomes affects height in a nonlinear fashion: a study of 305 patients with sex chromosome aneuploidy. Am J Med Genet A. 2010;152A(5):1206–12. https://doi.org/10.1002/ajmg.a.33334.
Pelosi E, Farabosco A, Schlessinger D. Genetics of the ovarian reserve. Front Genet. 2015;6:308. https://doi.org/10.3389/fgene.2015.00308.
Orlandini C, Regini C, Vellucci FL, Petraglia F, Luisi S. Genes involved in the pathogenesis of premature ovarian insufficiency. Minerva Ginecol. 2015;67:421–30.
Niranjan TS, Skinner C, May M, Turner T, Rose R, Stevenson R, et al. Affected kindred analysis of juman X chromosome exomes to identify novel X linked intellectual disability genes. PLoS One. 2015;10:e0116454. https://doi.org/10.1371/journal.pone.0116454.
Quin Y, Jiao X, Simpson JL, Chen ZJ. Genetics of primary ovarian insufficiency: new developments and opportunities. Hum Reprod Update. 2015;21:787–808. https://doi.org/10.1093/humupd/dmv036.
Bione S, Rizzolio F, Sala C, Ricotti R, Goegan M, Manzini MC, et al. Mutation analysis of two candidate genes for premature ovarian failure, DACH2 and POF1B. Hum Reprod. 2004;19:2759–66. https://doi.org/10.1093/humrep/deh502.
Broer SL, Broekmans FJ, Laven JS, Fauser BC. Anti-Mullerian hormone ovarian reserve testing and its potential clinical implications. Hum Reprod Update. 2014;19:2759–66. https://doi.org/10.1093/humupd/dmu020.
Gravholt CH, Andersen NH, Conway GS, dekkers OM, Geffner ME, Klein KO, et al. International Turner Syndrome Consensus Group. Clinical Practice guidelines for the care of girls and women with Turner Syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177:G1–G70. https://doi.org/10.1530/EJE-17-0430.
Lunding SA, Aksglaede L, Anderson RA, Main KM, Juul A, Hagen CP, Pedersen AT. AMH as predictor of premature ovarian insufficiency: a longitudinal study of 120 Turner syndrome patients. J Clin Endocrinol Metab. 2015;100:E1030–8. https://doi.org/10.1210/jc.2015-1621.
Hamza RT, Mira MF, Hamed AI, Ezzat T, Sallam MT. Anti-Müllerian hormone levels in patients with turner syndrome: relation to karyotype, spontaneous puberty, and replacement theraphy. Am J Med Genet A. 2018;176:1929–34. https://doi.org/10.1002/ajmg.a.40473.
Talaulikar VS, Conway GS, Pimblett A, Davies MC. Outcome of ovarian stimulation for oocyte cryopreservation in women with Turner syndrome. Fertil Steril. 2018;111:505–9. https://doi.org/10.1016/j.fertnstert.2018.11.010.
Acknowledgements
Not applicable.
Funding
No funding sources to declare.
Author information
Authors and Affiliations
Contributions
All Authors meet the criteria for authorship according to the International Committee of Medical Journal Editors (ICMJE). The Authors’ contribution were as follows: RG, FP and MP: design of the work, acquisition, analysis, and interpretation of data, drafting the work and final approval of final version. RDP, GC, EM, OZ, ES: acquisition of data, revising the work critically for important intellectual content and final approval of final version. MF, SZ, PC, FA and GP: interpretation of data for the work, revising the work critically for important intellectual content and final approval of final version. All the Authors decided to submit the paper for publication and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
In accordance with Verona University and University of Verona Institutional review Board (IRB) policies, case reports are not subjected to IRB review and are thus except from formal IRB approval for publication. All procedures followed were conducted ethically in accordance with the World Medical Association Declaration of Helsinki.
Consent for publication
A written consent from the patient for publication of personal, imagine and genetic data was obtained.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Parissone, F., Pucci, M., Meneghelli, E. et al. A novel de novo partial xq duplication in a girl with short stature, nonverbal learning disability and diminished ovarian reserve - effect of growth hormone treatment and fertility preservation strategies: a case report and up-to-date review. Int J Pediatr Endocrinol 2020, 1 (2020). https://doi.org/10.1186/s13633-019-0071-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13633-019-0071-z