
Abstract
Background
The cereal cyst nematode (CCN, Heterodera avenae) is a devastating pathogen of wheat and barley crops in many countries. We aimed to prioritize genetic and molecular targets for H. avenae control via the powerful and integrative bioinformatics platform.
Results
Here, we sequenced mRNA isolated from Chinese H. avenae at pre-parasitic (consisting of egg, J1 and hatched-J2) stages and post-parasitic (consisting of parasitic-J2, J3, J4 and adults) stages. Total 1,066,719 reads of whole life cycle transcriptomes were assembled into 10,811 contigs with N50 length of 1754 bp and 71,401 singletons. Comparative analyses of orthologous among H. avenae and 7 other nematodes with various life-styles revealed the significance and peculiarity of neurological system for sedentary phytonematode. KEGG pathway enrichment demonstrated active crosstalk events of nervous system at pre-parasitic stages, and 6 FMRFamide-like neuropeptides were verified to display an expression peak at the hatched-J2 stage in H. avenae. Furthermore, multiple approaches were undertaken to mine putative effectors and parasitism-specific genes. Notably, H. avenae might represent the first phytonematode reported to possess the pioneer effectors with RxLR motif and potential effectors with homologies to Ant-5/Ant-34.
Conclusion
Our work provides valuable resources for in-depth understanding the parasitism and pathogenicity of H. avenae, as well as developing new targets-oriented strategies on effective managements.
Similar content being viewed by others
Background
As a worldwide plant pathogen, the cereal cyst nematode (CCN) Heterodera avenae is an essential threat to global food security. It is widely distributed in more or less temperate wheat-producing regions throughout the world; in some wheat fields, the losses caused by this nematode range from 30 to 100% [1, 2]. In China, the occurrence of H. avenae has distributed in 16 provinces, covering 80% of the total wheat growing regions (approximately 20 million ha), and has been reducing the yield in significant proportions every year [3].
Heterodera avenae is an obligate, sedentary endoparasite that has unique, biotrophic interactions with its host plants and reproduces by amphimixis. Its life cycle can be divided into pre-parasitic stages and post-parasitic stages. Life cycle of H. avenae starts with an egg present inside an encysted female. In pre-parasitic stages, the first stage larva (J1) develops within the egg, and the second stage juvenile (J2) hatches from cyst and migrates into the soil. Upon localizing host plant roots, the parasitic J2 invades the host tissue by its stylet and migrates intracellularly to select initial feeding cell for subsequent establishment of syncytial feeding site [4, 5]. Maintaining the enlarged syncytium is essential for the permanent nutrient sink during the sedentary J3 and J4 stages. It takes 3–6 weeks for developing into an adult male or an egg-laying female.
Based on the life style, the managements of H. avenae target at either pre-parasitic stages or post-parasitic stages. However, particular problems are caused by the ability of encysted H. avenae to survive for prolonged periods in the soil in the absence of a host, making control by rotations difficult [5]. Additionally, resistant cultivar is limited by the lack of dominant natural resistance genes and the long breeding period. Moreover, the use of chemical nematicides is withdrawn or severely limited by recent legislation due to potential adverse effects to both human health and natural environment [6]. Therefore, exploring novel approaches based on new gene targets is urgently needed for control of H. avenae.
Effectors, as ‘all pathogen proteins and small molecules that alter host-cell structure and function’, are considered valuable targets for novel management strategies of parasitic nematodes [7]. It is generally accepted that secretions produced by H. avenae are responsible for the successful parasitism. Particularly, the effectors expressed exclusively in the esophageal gland cells and secreted through the stylet play key roles in this process [8, 9]. Thus, effective strategies to discover effectors are presumed to rely on identifying nematode secretions directly by cDNA sequencing or proteomics approaches [10,11,12,13,14,15]. However, a more common and high throughput approach is to investigate the transcriptome or genome via bioinformatics tools to predict large putatively secreted proteins that are not limited to those from esophageal gland cells [16,17,18,19].
Recently, Kumar et al. reported the first transcriptomic analysis of two life stages of Indian population of H. avenae, including hatched-J2 and feeding female, and provided a friendly resource of database (HATdb) to help seeking the effectors [20]. However, the critical parasitism processes such as penetration, migration and feeding site initiation, establishment and maintenance were carried out from the early reaction of parasitism and during the life-long time pathogenesis of cyst nematodes [21]. For instance, the expression peaks of suppressors (Gr-UBCEP12 [22] and Gr-SPRYSEC-4/5/15/18/19 proteins [23]) to the plant defense response were present at parasitic J2 or J3 stage in potato cyst nematode Globodera rostochiensis. The perception of nematode CLAVATA3 (CLV3)/ESR (CLE)-like proteins by CLV2 and CRN was detected at the nematode-induced syncytia when the beet cyst nematode Heterodera schachtii developed into the parasitic-J2, J3 or J4 stages [24]. Thus, the genetic data of partial life stages might miss some effectors with low abundance or unique functions for suppressing the primary plant defense response or inducing and maintaining the syncytia by H. avenae during parasitism. More recently, with a focus on the early parasitic J2 stage (30 h, 3 and 9 days post infection), Zheng et al. reported 56 putative effectors among 681 parasitism genes of H. avenae during incompatible infection to Aegilops variabilis [25]. Nevertheless, it was arduous to fully separate the parasitic nematode genes from the plant-nematode mixed transcriptomes. Moreover, current available H. avenae gene set is not complete enough to analyze the whole life cycle of nematode invasion. Besides, these analyses might also lack genes specifically expressed in cyst stage to ensure the long-term surviving in the soil. Therefore, further expansion of the genetic resources of H. avenae and identification of pivotal parasitism genes as well as specific regulatory pathways of its whole life cycle were required.
In this study, we have sequenced transcriptomes of the pre-parasitic stages (including eggs, J1 and hatched-J2) and post-parasitic stages (including parasitic-J2, J3, J4, and adults) of Chinese H. avenae. In the context of other published nematode genetic datasets, we undertook multiple comparative analyses among nematodes with different life styles. Specific ESTs expressed in H. avenae were identified, and gene transcripts profiles have been compared between pre- and post-parasitic stages. Putative effectors, including pioneer effectors with RxLR motif and other potential effectors in H. avenae, were found through multiple strategies. These effectors highlighted important in-depth insights into the pivotal genes involved in suppressing plant defense and regulation of host plants for successful parasitism. Our work benefits the comprehensive elucidation of the molecular mechanisms underling parasitism and pathogenicity of H. avenae, and also provides valuable information on potential gene targets for its control.
Results
General overview of H. avenae transcriptome sequencing, assembly and annotations
Transcriptome sequencing using 454 FLX+/Roche platform was performed for pre-parasitic and post-parasitic stages of Chinese H. avenae (Fig. 1). This yielded 1,066,719 raw reads (551,935 reads from pre-parasitic stages library and 514,784 reads from post-parasitic stages library), for a total 520,863,572 bp, with an average length of 488 bp (Table 1). The comparable sequencing depth, reads length and quality (Fig. 2) allowed combined assembly of pre-parasitic and post-parasitic libraries for complete life cycle (Table 2). The final assembly resulted in total 82,212 expressed sequence tags (ESTs) consisting of 10,811 aligned contigs with an average length of 1442 bp and N50 of 1754 bp and 71,401 singletons with an average length of 430 bp (Table 2). From these contigs and singletons, we obtained 10,738 and 63,601 putative operons, respectively. Our pyrosequencing of complete life cycles of Chinese H. avenae produced both longer total transcripts and average ESTs in length, compared to that of the report from Kumar et al., which sequenced the two development stages (juvenile 2 and adult female) of Indian H. avenae (Tables 1 and 2).

Developmental stages of Heterodera avenae sampled for transcriptomic and gene expression analysis. Chinese H. avenae at pre-parasitic stages consisted of egg, J1 and hatched-J2 nematodes (on the up-left of the dotted curve) and post-parasitic stages consisted of parasitic-J2, J3, J4 and adult nematodes (on the down-right of the dotted curve)

The distribution of sequences lengths in the transcriptomes of H. avenae by 454 FLX+ sequencing. Histogram graphs show the distribution of reads lengths for the transcriptomes of H. avenae at pre-parasitic stages (a) and post-parasitic stages (b). c Bar graph shows the distribution of contigs lengths from the assembled transcriptome of whole life cycle H. avenae covering the above-mentioned two mixed stages
General annotation showed that 71.1% of contigs and 34.3% of singletons had homologous proteins from non-redundant GenBank proteins (NR) or Swiss-Prot/TrEMBL databases (BLASTX e-value <1e−6, Similarity >50%). Only 25,657 (30.9%) ESTs were successfully annotated by Gene Ontology (GO), of which 5816 (53.8%) contigs hit 21,633 GO terms while 19,841 (27.8%) singletons hit 129,349 GO terms as shown in Additional file 1: Figure S1a. Notably, 24,146 operons had KEGG orthologs (KO), of which 3338 were drawn on KEGG pathway maps (Additional file 1: Figure S1b). Among the 6803 contigs operons with KO, 1853 were involved in 287 biological pathways. Our subsequent analyses on pathway enrichment of significant biological categories were based on these 1853 contigs.
Phylogeny and ortholog analyses
The phylogeny and ortholog analyses were carried out by comparing 74,339 operons of H. avenae with other 7 published nematode genomes (Globodera pallida [26], Meloidogyne incognita [27], Meloidogyne hapla [28], Bursaphelenchus xylophilus [29], Caenorhabditis elegans [30], Pristionchus pacificus [31] and Ascaris suum [32]). This resulted in the identification of 12,703 orthologous families, of which 703 single-copy ortholog families were used to draw the phylogeny tree (Fig. 3a). Representing the varied life styles of these nematodes, the tree demonstrated a clear separation of plant parasitic nematodes (H. avenae, G. pallida, M. incognita, M. hapla and B. xylophilus), animal parasitic nematode (A. suum) and bacterivorous free-living nematodes (C. elegans and P. pacificus). While B. xylophilus represented migratory nematode, cyst nematodes (H. avenae, G. pallida) and root-knot nematodes (M. incognita, M. hapla) represented nematodes of sedentary endoparasites (Fig. 3a). As a satellite model of bacterivorous C. elegans, omnivorous P. pacificus can feed on not only various bacteria, but also fungi and even other nematodes [33]. Taking together, the relative proximity in the phylogeny tree well recapitulates the similarities of life styles among these nematodes.

Phylogeny and ortholog distribution of 8 nematodes with diverse life styles. a A genome-based phylogentic tree generated by 703 single ortholog families. Scale (0.1) represents the number of amino acid changes per site. b Distribution of orthologous identified in H. avenae and other 7 selected nematodes with published genomic data
The 11,345 operons from H. avenae, which belonged to 8086 orthologous families, were subjected to categorization by the distribution of orthologous among 8 nematodes. We subdivided these orthologous families into 6 groups (Fig. 3b). There were 2248 families, including 2879 H. avenae orthologous, shared by all the above 8 nematodes. Notably, 3070 orthologous specifically shared by H. avenae and other 5 plant-parasitic nematodes, among which 2699 only shared by sedentary nematodes and 1575 uniquely in cyst nematodes. Interestingly, 92 orthologous shared by H. avenae and omnivorous P. pacificus, but not by other phytonematodes, were also found in animal parasitic nematode A. suum. For a detailed insight on the comparisons between H. avenae and other nematodes, we performed GO enrichment analysis of the common H. avenae orthologous (Additional file 2: Figure S2).
As a premier biological and genetic model, C. elegans provides a critical reference for the investigation of biological pathways shared by nematodes [30]. The H. avenae orthologous that are corresponding to C. elegans genes involved in RNAi pathway (Additional file 3: Table S1a), immune response (Additional file 3: Table S1b), diapause pathway (Additional file 3: Table S1c), and neurotransmitter biogenesis pathways (Additional file 3: Table S1d) were extracted from 12,703 orthologous families and compared among other nematodes. These comparative analyses on orthologous in these biological pathways facilitate further characterization of H. avenae, as well as the understanding of molecular mechanisms involved in the nematodes-plant host interactions, thus assist the identification of new targets and development of novel strategies for control of H. avenae.
In order to find out unique genes in H. avenae, 10,811 contigs were compared with all the available 1,331,401 EST sequences of nematoda from NCBI and Nembase 4 (Additional file 4: Table S2) by reciprocal tblastx (cutoff score 50). As a result, 9145 contigs (84.6%) from H. avenae showed significant sequence similarity with other nematodes ESTs and were classified into three life styles in the venn diagram (Fig. 4a). The remaining 1666 contigs (15.4%) without obvious hit were considered as unique genes in H. avenae. Most of these unique ESTs did not have much annotation, and only 74 of them had GO assigns involved in biological progress (Fig. 4b), cellular component (Fig. 4c) and molecular function (Fig. 4d).

Summary and classification of ESTs in H. avenae transcriptome. a The contigs identified in H. avenae were compared with known ESTs from nematodes with three distinct life styles by Venn diagram. PPN plant parasitic nematodes, APN animal parasitic nematodes, FLN free-living nematodes. Functional classification of H. avenae-specific transcripts using Gene Ontology (GO) terms in the categories of biological progress (b), cellular component (c) and molecular function (d)
Differential expression analyses between pre- and post-parasitic stages
In order to obtain the differential expression profiling between the pre- and post-parasitic stages, we calculated gene expression from the assembled reads of two libraries, using 10,811 assemble contigs of H. avenae full life cycle as reference. The pre-parasitic library, consisting of 10,176 contigs, exhibited similar expression abundance and distribution profiling with the post-parasitic library, which was composed of 9510 contigs (Additional file 5: Figure S3a, b). There were 8877 common contigs and 3378 significantly differentially expressed contigs (FDR < 0.001, |log2(fold change)| > 1) between these two libraries, of which 1779 contigs were highly expressed in pre-parasitic stages and 1598 contigs in post-parasitic stages, calculated by DEGseq with MA-plot-based method (Additional file 5: Figure S3c).
Furthermore, we generated 330 biologic pathways gene-sets from 1853 H. avenae contigs assigned by KEGG pathway, of which 569 contigs exhibited significantly differential expression before or after parasitism. To discern telltale biological themes, the 569 differential expressed contigs were ranked from the expression datasets and examined for the statistically significant, concordant differences by Gene Set Enrichment Analysis (GSEA) with a weighted resampling based pathway enrichment test. As demonstrated in the layout visualization by Enrichment Map, there were 30 crosstalk pathways significantly enriched at the cut off level of p values <0.01, FDR < 0.25, similarity >0.5, of which 16 pathways were actively involved in pre-parasitic stages and 14 pathways in post-parasitic stages (Fig. 5). For pre-parasitic stages, 16 nodes linked by 45 edges indicated the crosstalk events. The grouped big clique represented the active pathways in digestive system, circulatory system, human disease, nervous/sensory system and environmental information processing (Fig. 5a). However, for H. avenae at the post-parasitic stages, significantly enriched biological processes were focused on metabolism, genetic information processing, cell cycle and cell growth and death, which were denoted by three clique-groups consisting of 14 nodes linked by 12 edges (Fig. 5b). The heat map depicting the expression profiles of crosstalk genes relevant to these pathways is shown in Additional file 6: Figure S4. The differentially expressed genes in each node and the normalized quantification were listed in Additional file 7: Table S3.

Crosstalks of significantly enriched KEGG pathways before and after parasitism in H. avenae. Crosstalks of significantly enriched KEGG pathways from H. avenae at pre-parasitic (a, red nodes) and post-parasitic (b, blue nodes) stages. Node, a KEGG pathway gene-set, and the node sizes positively correlate with the number of genes in the pathway; the darker the color is, the more significantly the gene-sets are enriched (P < 0.01, FDR < 0.25). Edge, the relevance of two gene-sets, and the more relevant (similarity >0.5), the more genes shared by two gene-sets; the darkness and thickness of lines positively correlate with amount of genes and similarity of two gene-sets, respectively. The normalized enrichment score (NES) is the primary statistic for evaluating gene set enrichment by GSEA
High expression of neuropeptides at pre-parasitic stages
Acting as neuromodulators and primary transmitters, neuropeptides have a diverse role in the development and function of nervous system in nematodes [34,35,36]. The main role of neuropeptides is as modulators of synaptic activity in a range of processes including sensory perception, locomotion, development, egg-laying and dauer formation [26]. FMRFamide-like peptides (FLPs), named from the sequence Phe-Met-Arg-Phe-NH2, are essential neuropeptides with diverse physiological functions [37]. Interestingly, we found a significant decline of genes coding for FLPs in our H. avenae transcriptome data after parasitism (Fig. 6a). These neuropeptides were identified as CCN-FLP-1, CCN-FLP-6, CCN-FLP-12, CCN-FLP-14, CCN-FLP-16 and CCN-FLP-18. Consistent with this notion, the quantitative PCR analysis further confirmed that all of these FLPs exhibited remarkably highest expression at the hatched-J2 stage (Fig. 6b). Although a peak expression was observed at J3 stage for each FLP, the expression level at J3 stage was substantially lower than that at pre-J2 stage. Notably, even lower expression of each FLP was found at the J4 and adult female stages. Our results support the notion of vital roles of enriched FLPs genes for nervous system before invasion, providing highly valued targets for pharmacological interference prior to the parasitism of H. avenae.

Differential expressions of FMRFamide-like peptides before and after parasitism in H. avenae. a Comparison of expression levels of indicated FLPs at pre-parasitic and post-parasitic stages by analyzing transcriptome-sequencing data. * Log2(fold change) pre/post, normalized Log2(pre-parasitic RPKM/post-parasitic RPKM), calculated by DEGseq. b Real-time PCR results for the expression of indicated FLPs genes in five different developmental stages. The FLPs expression levels in H. avenae at hatched-J2, parasitic-J2, J3, J4 and female adult stages were normalized to that of egg stage. Statistic significance was determined by one way analysis of variance (ANOVA) test. ***p < 0.001
Potential effectors of H. avenae
Decades of research demonstrates that numerous proteinaceous effectors are synthesized in the esophageal gland cells with N-terminal signal peptides, which direct the effectors to the classic secretory pathway [7]. However, effectors without signal peptides are also identified from the extracellular culture by protein mass spectrometry [11]. We identified total 4229 putative secretory proteins, from our H. avenae transcriptomes, as the proposed effectors repertory. This repertory consisted of 3985 signal peptide-marked proteins and 244 operons with secretory homologues. Notably, 59 published phytonematodes key effectors (without CAZymes) that had homologues in H. avenae were listed in Additional file 8: Table S4a.
Carbohydrate-active enzymes (CAZymes) secreted by nematodes to modify plant cell walls, are essentially considered as critical factors for successful parasitism of plant host [7]. We identified 261 H. avenae operons with 270 CAZymes modules classified into 87 families by searching dbCAN database. The H. avenae ESTs coding for CAZy enzymes with homologies to that of other phytonematodes were listed in Additional file 8: Table S4b, and interestingly, the majority had an expression bias at pre-parasitic stages. This notion was supported by the RT-PCR analysis of cellulases (Ha-eng-2 and Ha-eng-3) reported by Long et al. [38]. These two CAZymes had the most transcriptions in the hatched-J2s, medium transcriptions in the early parasitic-J2s, and almost undetectable transcriptions at the late parasitic stages of H. avenae [38]. It is possible that the accumulation and stock of mRNA for these CAZymes might be a prerequisite for initial infection of host plant, because a large number of templates for protein translations are urgently needed upon invading the host tissue.
RxLR effectors facilitate the transportation of cytoplasm proteins from malaria parasite to the host and assist suppression of the defense of plant-host triggered by microbe pathogens [39, 40]. They are characterized by a highly conserved region defined by the invariant RxLR motif sequence (R, Arg; L, Leu; x, any residue) within the N-terminal 30–60 amino acids after secretory signal peptides [41]. Surprisingly, we also found 61 predicted secretory peptides with RxLR motif within the N-terminal 100 amino acids in our H. avenae transcriptome as putative RxLR effectors. Among them, 10 candidates had RxLR-[E/D/Q] motif, which was considered as host-targeting motif aiding effector export from malaria parasite [39, 42]. We listed 15 putative RxLR/RxLR-[E/D/Q] effectors with homologues in NR database (blastp, e-value <1e−5) (Table 3). Our results imply the possibility of existence for pioneer RxLR effectors in H. avenae.
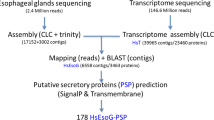
Effectors secreted during parasitism play vital roles in establishing and maintaining feeding site of a permanent source of nutrients, as well as in response to immune defense of plant hosts [7, 9, 43]. Based on the available transcriptome sequencing data of H. avenae, we designed a strategy to mine the potential effectors highly expressed during parasitism (Fig. 7). The raw sequencing reads of 4 libraries from differed samples, including H. avenae at the hatched-J2 stage [20], adult female stage [20], pre-parasitic mixed-stages (egg, J1, hatched-J2) and parasitic mixed-stages (parasitic-J2, J3, J4, female, male) were mapped to the reference transcripts of whole life H. avenae (derived from our 10,811 configs), respectively. The statistic numbers of mapped reads revealed the expression of transcripts from each sample. After strict screening, the 45 transcripts that significantly enriched in post-parasitic mixed-stages (FDR < 0.001) but displayed no read mapped from neither hatched-J2 nor adult female stages were considered as potentially key factors at the earlier stage of parasitism phase. Among these 45 transcripts, there were 3 transcripts that did not have operon, and 13 transcripts had operons with predicted secretory signal peptides. Markedly, we found 16 transcripts that possess obvious homologous by searching NR database (blastx, e-value <1e−5) (Table 4). Interestingly, 3 H. avenae transcripts that share strong sequence and structural resemblances with Toxocara canis ANT-5/ANT-34 and flavivirus NS2/NS5 were not identified in any other plant nematodes.

The schematic representation of the screening pipeline for highly expressed parasitism-specific genes in H. avenae. The gene contigs significantly enriched in post-parasitic stages [FDR < 0.001, Normalized log2(fold change) >1] but without expression in neither hatched-J2 nor adult female stages are considered potentially parasitism-specific genes
Discussion
Without genomic reference, we chose the relatively more costing platform of 454 FLX+/Roche to sequence the transcriptomes of H. avenae at pre-parasitic mix-stages (including eggs, J1 and hatched-J2) and post-parasitic mix-stages (including parasitic-J2, J3, J4, and adults) (Table 1). Our de novo assembly of 82,212 expressed sequence tags (ESTs) that covered the entire life cycle of this parasite was benefited from the long reads of combined libraries. Among these ESTs, 10811 contigs had notable lengths with N50 of 1754 bp, while 71,401 singletons had an average length of 430 bp (Table 2). Besides the transcriptomes of two life stages (hatched-J2 and feeding female) sequenced by Kumar et al. and the parasitism genes reported by Zheng et al. during incompatible invasion [20, 25], our data substantially enriched the genetic information of the cereal pathogen H. avenae.
Orthologs are genes in different species that evolved from a common ancestral gene by speciation, normally retaining the same function in the course of evolution [45]. For reliable prediction of gene function, we identified and compared orthologs from the putative translated products of H. avenae trancriptomes and other completely sequenced genomes of 7 nematodes, which are representative species with different life styles. As expected, there are great correlations between the similarity of life styles and the proximity of compared nematodes in phylogenetic tree generated from single copy ortholog families (Fig. 3). The closest species to H. avenae is G. pallida, which agrees with the report from Kumar et al., because they belong to cyst genus and have the most similar parasitic life style. In addition, our GO enrichment analyses show that the organonitrogen compound metabolic process is unique in the cyst nematodes (Additional file 2: Figure S2D). Moreover, the GO terms on single-organism cellular process and neurological system process are shared only by sedentary nematodes (Additional file 2: Figure S2c). Like other sedentary phytonematodes, H. avenae has evolved a special sensorial processing system that is distinguished from migratory parasites during obligate sedentary parasitism. Surprisingly, three H. avenae ESTs have been identified with sequence similarity to Ants, which are only shared by animal parasitic nematode and H. avenae (Fig. 4a). The potential role of Ants as novel functional effectors is discussed below.
Currently, RNAi technology has become one of the predominant tools for studying gene function in phytonematode, as well as an effective anti-parasite strategy. Nevertheless, some indispensable factors for the RNAi pathway in C. elegans (Additional file 3: Table S1a), such as sid-1 [46, 47] and sid-2 [48], which are required for the uptake of dsRNA in the intestine, are not identified in H. avenae and any other phytonematodes. In fact, repeated reports of successful silencing of target genes by feeding dsRNA using socking stimulant or host-derived delivery [49, 50] imply that phytonematode like H. avenae might have alternative signaling pathway for up taking of dsRNA in the intestine. The variance on dsRNA processing between H. avenae and C. elegans remains a subject for further investigation, and the better understanding will definitely illuminate new ideas for the control of H. avenae.
Like C. elegans [51] and G. pallida [26], H. avenae relies tightly on innate immunity to defend the infection and infiltration of different microbial pathogens. As compared with other sedentary nematodes, we found that the overwhelming majority of immune signaling pathways appeared to be highly conserved between H. avenae and C. elegans, except ikb-1 in the toll-signaling pathway (Additional file 3: Table S1b). The relative conservation of innate immune defense system in H. avenae and C. elegans highlights the potentials of disrupting the immune system to increase the susceptibility of H. avenae to microbial infection for biocontrol.
Diapause under hot dry conditions in summer is one of the unique habits of H. avenae. However, the facultative diapause ability of the populations of Chinese H. avenae is distinct from that of the populations in Mediterranean climates. Their dormancy is also promoted after chilling in winter, which ensures the emergence of infective juveniles upon temperature rising in spring [52,53,54]. This facultative diapause of H. avenae in China, regulated by different thermal conditions, is the same case in the laboratory [55]. Thus, the characterization of the signaling pathways that control the developmental decision to enter and leave the dauer stage is essential to manipulate the life cycle of H. avenae for control. In C. elegan, many mutants with high temperature induction of dauer (Hid) phenotype have defects in synaptic transmission, highlighting the neuroendocrine nature of dauer regulation [56,57,58]. Although the sensory of Hid couples downstream to insulin-like and TGFβ-like signaling pathways in C. elegan [57, 59, 60], we did not identify these two intact dauer regulatory pathways from both H. avenae and G. pallida, Only Guanylyl cyclase pathway was conserved in both H. avenae and G. pallida with reference to the genome of C. elegan. 8-bromo-cGMP (8Br-cGMP) functions in this pathway to regulate dauer arrest in both C. elegans and G. pallida. While 8Br-cGMP rescued the phenotype of constitutive dauer in daf-11 mutants [61], study in G. pallida also demonstrated that 8Br-cGMP facilitated the release of diapause by activation of hatching [62]. Due to the functional conservation of guanylyl cyclase pathway between C. elegans and cyst nematodes (Additional file 3: Table S1c), it is deducible that manipulation of this pathway might be effective for controlling the hatching of H. avenae.
To provide novel insights into the differences of H. avenae at stages before and after parasitism, 3378 significantly differentially expressed contigs were gained from the comparison of transcriptomes. The divergence of enrichment KEGG pathways of these differential expression genes indeed reflects distinct demands for survival before and after parasitism. In the pre-parasitic stages, the active highly socialized pathways, which consisted of environmental information processing system, nervous system, digestive system, circulatory system and human disease (Fig. 5a), benefit the adaptability and motion of H. avenae in soil and facilitate the perception of complex ambient stresses and invasion of host plant. From the above crosstalk pathways, nervous system probably play a signaling or integrative role in the pre-parasitism activities of H. avenae, such as the defense against the microbial pathogens [63] in soil, temperature induction of dauer [60] and seeking for the host plant. After successful parasitism, metabolism related pathways, including amino acid metabolism, biosynthesis of other secondary metabolites, carbohydrate metabolism and lipid metabolism, were predominant to coordinate the production of endocrine hormone, cellular proliferation and differentiation, as well as cell apoptosis and death (Fig. 5b). Probably the secretory molecules in these pathways that can be delivered into host plant cell and further regulate host plant-nematode interaction are putative effectors, which essentially benefit the initiation and sustaining of parasitic life for H. avenae.
As demonstrated previously, there is significant neurological system in sedentary nematodes (Additional file 2: Figure S2c) and an obvious bias of active biosynthesis of neurotransmitters towards the pre-parasitic stages of H. avenae (Fig. 5a). Substantially, neurotransmitters contribute to the complex and subtle behaviors of nematodes, such as recognition of hosts, defense to pathogens and response to environmental stresses [26, 64]. Thus, the better characterization of the synthesis, transportation and metabolism of neurotransmitters may help exploiting particular targets for rational chemical control methods. As compared to G. pallida, another well characterized cyst nematode, H. avenae had the same genes profile in the majority of signaling pathways like acetylcholine and dopamine (Additional file 3: Table S1d). However, serotonin biosynthetic enzyme coding gene tph-1 was absent in H. avenae. It is worth noting that the octopamine, glutamate, GABA signalling pathways were highly conserved in all the nematodes, which implies the universality of existed and novel strategies targeting these neurotransmitter pathways. For instance, octopamine is used to stimulate ingestion of dsRNA in invitro RNAi experiments by increasing the pharyngeal pumping of plant nematodes [65]. On the other hand, since octopamine displays roles in inhibition of egg laying in C. elegans, investigators have adopted octopamine as an effect approach in the control of phytonematodes [66, 67]. Our greater understanding of the available target molecules will considerably help in the rational design of new biological nematicide, as well as in the development of next-generation chemical nematicide with minimal adverse effects.
In addition, neuropeptides may also function as primary neurotransmitters. The FMRFamide-like peptides (FLPs) are broadly conserved neuropeptides in nematodes, while the profiling of FLPs varies among phytonematode and other nematodes in terms of structure, copy, expression location and developmental expression pattern [37, 68]. For example, N-terminal variations of FLP-12 exist among the animal parasites, plant parasites Heteroderidae spp. and Meloidogyne species [37]. The FLP-6 expression was observed in the circum pharyngeal nerve ring (main response pharyngeal pumping) of potato cyst nematode of G. pallida, whereas FLP-6 in bacterivorous C. elegans was reported solely in the ASE head neurons [68]. Paul McVeigh et al. reported that there is a general expression bias of FLPs towards Clade V species (such as C. elegans, P. Pacificus), appearing to be over represented at L3 stage juveniles and female worms, according to the analysis of EST database sequences [37]. However, our investigation demonstrated that H. avenae, as a Clade VI plant parasite, had highest expression of a series of FLPs at hatched J2 stage before parasitism in plant host (Fig. 6). These distinctions of FLPs among nematodes indicate that the special roles of FLPs are inextricably linked to parasite motor and sensory function, particularly in plant nematodes.
Disruption of parasite FLP signals may be considered as an approach for developing new specific nematocides. Since our identified FLPs (CCN-FLP-1, CCN-FLP-6, CCN-FLP-12, CCN-FLP-14, CCN-FLP-16 and CCN-FLP-18) in H. avenae had major expression peak at hatched-J2 and minor expression peak at post-parasitic J3 stages (Fig. 6b), they have obvious potentials as targets for parasite control on both ecto- and endoparasitic phases. In deed, significant reduction of infection and multiplication of M. incognita was observed in the genetically engineered tobaccos using plant-mediated RNAi of nematode FLP-14 or FLP-18 [69]. We still know relatively little about the downstream signaling processes through which the effects of these FLPs are exerted. However, on the other hand, the signaling molecules and receptors involved in these pathways could also be the targets for pharmacological interference and the control of phytonematodes.
Numbers of putative effectors were mined from H. avenae transcriptomes. The key genes identical from phytonematodes reported effectors can be categorized into the following groups [9]: (1) effectors for invasion, extension and degradation of host tissues. This group is dominated by Cazymes, such as cellulase, pectate lyase, calreticulin, expansin; (2) effectors for regulating plant cell division and differentiation to maintain the integrity of syncytium for the feeding site, such as CLE-like proteins; (3) effectors that protect nematodes from anti-pathogen compounds in host defenses, such as fatty acid and retinol binding protein, peroxiredoxin; (4) effectors that target plant signalling pathways and suppress host defenses, such as annexin, venom allergen like protein, transthyretin-like protein, ubiquitin extension proteins; (5) effectors required for feeding efficiency, such as cathepsin; (6) putative effectors with unknown function, such as 14-3-3b protein and some putative secretory proteins identified in esophageal gland cells (Additional file 8: Table S4).
The RxLR-[E/D/Q] motif, serving as an export signal at the N-terminus of cargo proteins, was firstly explored from the virulence proteins of Plasmodium falciparum [39]. While a family of host-translocated effectors with same structure, produced by oomycete pathogens, are termed RxLR effectors and may manipulate host physiological and biochemical events in host cells [41]. Previous studies reveal that RxLR effectors of oomycete may suppress callose deposition and cell death triggered by multiple elicitors, impair plant immunity and facilitate bacterial colonization [40]. From these results, the major function of the secretory RxLR-[E/D/Q] motif bearing proteins is concluded to be host-translocated effectors and suppress the host immunity. Notably, a SPRY domain containing protein (ISOTIG07167) was identified from our exploiting of the pioneer effectors with RxLR-[E/D/Q] in H. avenae. The secreted SPRY domain (SPRYSEC) family proteins from the potato cyst nematode G. rostochiensis, were reported to suppress plant hypersensitive response (HR) induced by effector-triggered immunity [69, 70]. The pro-apoptotic protein BAX can also trigger plant programmed cell death (BT-PCD), physiologically resembling what associated with plant HR [71]. The ability to suppress defense-associated BT-PCD has proven to be a valuable initial screening method for microbial plant-pathogen effectors [72, 73]. Interestingly, ISOTIG17370, as a candidate without pentameric motif RxLR-[E/D/Q] but as a homolog of esophageal gland cell secretory protein 4 in Heterodera glycines (a soybean cyst nematode) [15], could suppress BT-PCD in our preliminary experiments. It is possible that some RxLR effectors, even without RxLR-[E/D/Q] core, might be still actively involved in inhibition of plant immunity (like oomycete), although the underlying mechanisms on how they enter host are still unknown.
From the exploring by combinational analyses of our and another reported transriptomes for H. avenae, we captured 16 highly expressed candidate key genes that putatively play essential roles during parasitism (Fig. 7). Five of these genes were suggested to be regulators and participators of RNA transcription, such as translation initiation factor IF-3, transcription termination/anti-termination protein NusA, DNA-directed RNA polymerase and ribosomal protein (Table 4). ANTs (abundant novel transcripts) are one group of highly enriched transcripts initially identified from the infective stage of Toxocara canis in nematoda [74]. It is worth noting that three putative parasitism-specific ESTs in H. avenae share sequence similarity to ANT-5/ANT-34 or NS2/NS5. They were also considered as the inhibitors of transcription in both Toxocara canis and flaviviruses [75]. To our knowledge, genes homologous to ANTs only exist in H. avenae and T. canis, among all the nematoda with available sequencing data. ANT-5 and ANT-34 are homologous of NS2 and NS5 in flaviviruses, and share strong sequence and structural resemblances of NSP1 and NSP2 in a tick-borne virus-Jingmen tick virus [76]. ANT-5 shows distant similarity to RNA regulatory protein and RNA-dependent RNA polymerase, while the 3′UTR of ANT-34 greatly reduces reporter gene expression, inhibiting both transcription and translation [75]. Anti-ANT-34 antibody had been found in the sera of hosts with Toxocara canis infection via high-affinity immune assays [75]. These points to the possibility that ANTs interact with the host and play roles in the extracellular environment although they could be RNA regulatory proteins and without identifiable N-terminal secretory signal peptides. Thus, it is plausible that ANTs from H. avenae could also be secreted and transported into host plants to function as effectors. We did not find the expression of secretory signal peptides for 32 putative effector ESTs. This is perhaps due to the lack of intactness in N-terminus of sequenced ESTs. However, it is also possible that these effectors share a non-canonical secretory pathway, just like ANTs.
Conclusion
Taking together, we present the transcriptomic data of H. avenae at pre- and post-parasitisitic stages. To our knowledge, this is the first report on de novo analysis of transcriptomes covering the complete life stages of Chinese H. avenae. The comparative analyses of transcriptomes for H. avenae and other nematodes facilitate the molecular characterization of H. avenae in parasitism, development, metabolism, immune defense and life style, providing guidance of multiple control approaches. Our data on in-depth mining of the potential pioneer effectors should be valuable resources not only for better understanding of the unique biology of H. avenae, but also for further developing possible targets-orientated strategies on effective control of H. avenae.
Methods
Collection and mass rearing of H. avenae
Heterodera avenae nematodes were collected from the field of Baoding city in Hebei province of China, identified by morphology and internal transcribed spacer (ITS)-ribosomal (r)DNA [77], and propagated on wheat (Aikang 58) from initial single cyst in an artificial environment. In this study, H. avenae of pre-parasitic stages (Fig. 1) included embryo eggs and the first stage of juveniles (J1s) gathered from crushed newly formed cysts or cysts incubated at 4 °C for 45 days, and the hatched-J2s collected by hatching cysts at 15 °C. The post-parasitic stages (Fig. 1) included parasitic J2, J3, J4, and adult female and male worms were isolated from the infected wheat roots digested by 6% cellulose, and distinguished under dissecting microscope as described previously [78]. Both pre-parasitic and post-parasitic H. avenae samples were frozen at −80 °C for later RNA isolation.
RNA isolation, cDNA library preparation and sequencing
Frozen nematodes of various developmental stages were pooled and categorized to be the pre-parasitic and post-parasitic samples. The mRNA was isolated by Dynabeads mRNA Direct Kit (Invitrogen, USA) according to the manufacture’s instructions. Further purification was performed using MicroPoly(A) Purist Kit (Ambion, USA) after removal of trace amounts of DNA contamination with DNase I (Ambion, USA). NanoDrop and Agilent 2100 Bioanalyzer assessed the quantity and quality of purified mRNA, respectively. For 454 pyosequencing, the first-strand cDNA was synthesized from mRNA reverse transcription with Superscript II Reverse Transcriptase (Invitrogen, USA) and GsuI-oligo dT primer, and biotinylated at 5′ cap structure. The biotinylated first-strand cDNAs were released from mRNA/cDNA complexes by alkaline lysis and purified with Dynal M280 microbeads (Invitrogen, USA). Second strand cDNA was synthesized by Ex Taq polymerase (Takara, Japan) after adding 5′ adaptors with DNA ligase (Takara, Japan). The cDNA was purified by Ampure beads (Agencourt, USA) after removing polyA and 5′cap using Gsu I enzyme (Fermentas, USA).
Single stranded template DNA library was prepared by GS DNA Library Preparation Kit (Roche Applied Science, USA), and subsequently amplified by GS emPCR kit (Roche Applied Science, USA). The resultant two libraries of pre- and post- parasitic mix-stages cDNA pools were sequenced using Roche 454 Genome Sequencer FLX at Chinese National Human Genome Center at Shanghai, China.
De novo transcriptome assembly and annotation
The reads of two libraries were combined and assembled using Roche official software Newbler 2.7 (454 Life sciences, Branford, CT, USA) at the default setting. The open reading frame (ORF) prediction was identified by using an in-house developed program based on ‘GetORF’ from EMBOSS [79]. To identify homologues of H. avenae transcripts in public databases, BLASTx searching was performed against Swiss-Prot/TrEMBL (http://www.ebi.ac.uk/) and non-redundant GenBank proteins (NR) (http://www.ncbi.nlm.nih.gov/) databases at an E value <1e−5. For function annotation, Gene Ontology (GO, http://www.geneontology.org/) analysis was performed with Blast2GO [80].
The KEGG pathway was constructed using the KEGG Automatic Annotation Server (KAAS) with BBH (bi-directional best hit, http://www.genome.jp/tools/kaas/) method [81].
Phylogenetic and comparative analysis of orthologous families
Based on all-to-all blast (E value <1e−6) of predicted proteins from Globodera pallida [26], Meloidogyne incognita [27], Meloidogyne hapla [28], Bursaphelenchus xylophilus [29], Caenorhabditis elegans [30], Pristionchus pacifics [31] and Ascaris suum [32] genomes and H. avenae transcriptomes, orthologous families were constructed by orthAgogue [82] (default parameters) and MCL [83] (I = 1.5). The 703 single-copy ortholog families of all 8 nematodes were subjected to multiple global alignment by MUSLE [84] and trimming by trimAI [85]. The phylogenetic tree was inferred by MEGA6 [86] using neighbor-joining method with 1000 bootstraps replicates. The GO term enrichment analyses of H. avenae genes of different orthologous families groups were performed using ontologizer with term-to-term or parent–child-intersection approach and Bongerroni correction [87]. Genome datasets and affiliated annotations were downloaded from Wormbase WS245 (http://www.wormbase.org/). Sequences for G. pallida were from website (ftp://ftp.sanger.ac.uk/). The reciprocal tblastx (Score bits >50) strategy was used in the nematode ESTs comparisons. All the nematoda ESTs, consisting of 1,331,401 ESTs sequences from 45 genuses and 75 species clustered into three life styles, were from NCBI and Nembase 4 [88] databases (Additional file 4: Table S2).
Comparison of gene homologues in critical biological pathways
The genes involved in critical biological pathways of C. elegans, including immune response pathway [51], diapause pathway [60], neurotransmitter synthesis, transportation and metabolism pathway [89], and RNAi pathway [90] were well defined by previously published literatures. The orthologous of genes in other nematodes were identified based on the orthAgogue results and manual confirmation through blast, largely following the methods used for analyses of G. pallida and M. incognita genome datasets [26, 27].
Analyses of differential gene expression and KEGG pathway enrichment
For each contig of H. avenae, the number of reads was extracted from the raw assembly file (ACE file) corresponding to two parasitic stages and transformed into RPKM (reads per kilo bases per million reads) [80]. Differently expressed contigs were identified statistically by DEGseq package using Fisher’s exact test with MARS (MA-plot-based method with Random Sampling model) method [91]. We used “FDR ≤ 0.001 and the absolute value of log2(fold change) ≥1” as the threshold to judge the significance of contig expression differences.
A total of 330 pathways and 1853 contigs were collected from KEGG assigned H. avenae contigs. To avoid pathways defined for too specific or too general biological processes, we selected those with at least 15 and at most 500 genes, resulting in 184 valid pathways for our subsequent analysis. The normalized log2(fold change) value calculated by DEGseq was considered as expression data for H. avenae contigs. Among 1853 contigs, 569 differential expressed contigs were ranked and examined for significant over- or down- representation using the threshold-free technique of Gene Set Enrichment Analysis (GSEA) with a weighted resampling Kolmogorov–Smirnov-like statistic. The enrichment result was visualized by Enrichment Map (P value <0.01, FDR < 0.25 and similarity <0.5) as a plugin of Cytoscape [92]. The heatmap matrix of genes expression profile that represented the enriched crosstalk edges was drawn by Pheatmap of R package [93].
Mining of putative candidate effectors
To gain the secretome repertory of H. avenae, we filtered the predicted products of operons with signal peptides lacking transmembrane domains by SignalP 4.0 [94] and TMHMM 2.0 [95], and searched the notable homologues from a collection of published secretomes of other nematodes with reciprocal blast (E value <1e−5, and minimal overlap >60%). The putative H. avenae homological effectors for published plant parasitic key genes were selected after manual validation. Genes encoding putative carbohydrate-active enzymes (CAZymes) were identified by HMM profiles searching as described previously [18]. RxLR effectors were acquired by structure scanning with ps_scan.pl of ScanProsite modular [96].
For the mining of potential effectors genes highly expressed during parasitism, the raw Illumina reads pairs of H. avenae transcriptomes by Kumar et al. [20]. were mapped to the assembled ESTs contigs of the whole life stages of H. avenae in this study by Bowtie2 [97]. Statistics of contigs without mapping reads from hatched J2 and female stages were calculated by SAMtools [98]. The Illumina Genome Analyzer II data of H. avenae transcriptomes by Kumar et al. were downloaded from NCBI SRA (ERR414136 for hatched J2 stage and ERR414137 for female stage).
Quantitative analysis of neuropeptides expression
Total RNA of different life stages of H. avenae was extracted using the RNeasy Plus Micro Kit (Qiagen, Germany), which included genomic DNA eliminator columns to remove DNA. The subsequent quantitation of mRNA for FMRFamide-like peptides (FLPs) was performed by quantitative real-time PCR (qPCR) as previously described [78]. Briefly, a SYBR Green based qPCR assay was used to quantify the expression of candidate FLPs after cDNA preparation by QuantiTect Whole Transcriptome Kit (Qiagen, Germany). qPCR was performed using a SYBR Premix Ex Taq (TaKaRa, Japan) in an ABI Prism 7000 instrument (Applied Biosystems, USA). Triplicate PCR reactions for each cDNA sample were completed, and the assay consisted of three technical replicates. Data were analyzed using the 2−ΔΔCt method with Gapdh as the endogenous control. The sequences for qPCR primers are as following: CCN-Flp-1, forward 5′-ACGGGCACCTTACGCCAATG-3, reverse 5′-GTTCGGCTCAGAAGCGGACA-3′; CCN-Flp-6, forward 5′-TCTTTGCCGTCTGTTCC-3, reverse 5′-ATTTCGTCGCATCGTC-3′; CCN-Flp-12, forward 5′-TTTCCCTTCCGTCGTTCTC-3, reverse 5′-ACAGCAGTGCCAAAGTCATC-3′; CCN-Flp-14, forward 5′-TGCTTCATCTGCCTCCC-3, reverse 5′-CAATGCCTTCGTTCACCA-3′; CCN-Flp-16, forward 5′-ACCTTCTCCTGCGTTTCG-3, reverse 5′-AAGTTGGGTGACGGTTTGA-3′; CCN-Flp-18, forward 5′-TCCAAAGAATCGCCACA-3, reverse 5′-TACGCCGTTGAGAAAGT-3′; CCN-Gapdh, forward 5′-AGCGGCACAGAACATCATCC-3, reverse 5′-GGTCCTCCGTGTAGCCCAAA-3′.
Abbreviations
- ANTs:
-
abundant novel transcripts
- BT-PCD:
-
BAX triggered plant programmed cell death
- CAZymes:
-
carbohydrate-active enzymes
- CCN:
-
cereal cyst nematode
- cDNA:
-
complementary DNA
- dsRNA:
-
double stranded RNA
- EST:
-
expressed sequence tag
- FDR:
-
false discovery rate
- FLPs:
-
FMRFamide-like peptides
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- KO:
-
KEGG orthologs
- NR:
-
non-redundant GenBank proteins database
- ORF:
-
open reading frame
- RNAi:
-
RNA interference
- RPKM:
-
reads per kilo bases per million reads
- UTR:
-
untranslated region
References
Nicol JM, Turner SJ, Coyne DL, Nijs LD, Hockland S, Maafi ZT. Current nematode threats to World agriculture. In: Jones J, Gheysen G, Fenoll C, editors. Genomics and molecular genetics of plant-nematode interactions. Dordrecht: Springer; 2011. p. 21–43.
Bonfil DJ, Dolgin B, Mufradi I, Asido S. Bioassay to forecast cereal cyst nematode damage to wheat in fields. Precis Agric. 2004;5:329–44.
Qiao F, Luo L, Peng H, Luo S, Huang W, Cui J, Li X, Kong L, Jiang D, Chitwood DJ, Peng D. Characterization of three novel fatty acid- and retinoid-binding protein genes (Ha-far-1, Ha-far-2 and Hf-far-1) from the cereal cyst nematodes Heterodera avenae and H. filipjevi. PLoS ONE. 2016;11:e0160003.
Bohlmann H, Sobczak M. The plant cell wall in the feeding sites of cyst nematodes. Front Plant Sci. 2014;5:89.
Jones JT, Haegeman A, Danchin EG, Gaur HS, Helder J, Jones MG, Kikuchi T, Manzanilla-Lopez R, Palomares-Rius JE, Wesemael WM, Perry RN. Top 10 plant-parasitic nematodes in molecular plant pathology. Mol Plant Pathol. 2013;14:946–61.
Ntalli NG, Caboni P. Botanical nematicides: a review. J Agric Food Chem. 2012;60:9929–40.
Mitchum MG, Hussey RS, Baum TJ, Wang X, Elling AA, Wubben M, Davis EL. Nematode effector proteins: an emerging paradigm of parasitism. New Phytol. 2013;199:879–94.
Davis EL, Hussey RS, Mitchum MG, Baum TJ. Parasitism proteins in nematode-plant interactions. Curr Opin Plant Biol. 2008;11:360–6.
Haegeman A, Mantelin S, Jones JT, Gheysen G. Functional roles of effectors of plant-parasitic nematodes. Gene. 2012;492:19–31.
Wang XR, Moreno YA, Wu HR, Ma C, Li YF, Zhang JA, Yang C, Sun S, Ma WJ, Geary TG. Proteomic profiles of soluble proteins from the esophageal gland in female Meloidogyne incognita. Int J Parasitol. 2012;42:1177–83.
Bellafiore S, Shen Z, Rosso MN, Abad P, Shih P, Briggs SP. Direct identification of the Meloidogyne incognita secretome reveals proteins with host cell reprogramming potential. PLoS Pathog. 2008;4:e1000192.
Gao B, Allen R, Maier T, Davis EL, Baum TJ, Hussey RS. The parasitome of the phytonematode Heterodera glycines. Mol Plant Microbe Interact. 2003;16:720–6.
Rutter WB, Hewezi T, Abubucker S, Maier TR, Huang G, Mitreva M, Hussey RS, Baum TJ. Mining novel effector proteins from the esophageal gland cells of Meloidogyne incognita. Mol Plant Microbe Interact. 2014;27:965–74.
Huang G, Gao B, Maier T, Allen R, Davis EL, Baum TJ, Hussey RS. A profile of putative parasitism genes expressed in the esophageal gland cells of the root-knot nematode Meloidogyne incognita. Mol Plant Microbe Interact. 2003;16:376–81.
Wang X, Allen R, Ding X, Goellner M, Maier T, de Boer JM, Baum TJ, Hussey RS, Davis EL. Signal peptide-selection of cDNA cloned directly from the esophageal gland cells of the soybean cyst nematode Heterodera glycines. Mol Plant Microbe Interact. 2001;14:536–44.
Haegeman A, Joseph S, Gheysen G. Analysis of the transcriptome of the root lesion nematode Pratylenchus coffeae generated by 454 sequencing technology. Mol Biochem Parasitol. 2011;178:7–14.
Nicol P, Gill R, Fosu-Nyarko J, Jones MG. de novo analysis and functional classification of the transcriptome of the root lesion nematode, Pratylenchus thornei, after 454 GS FLX sequencing. Int J Parasitol. 2012;42:225–37.
Li X, Yang D, Niu J, Zhao J, Jian H. De novo analysis of the transcriptome of Meloidogyne enterolobii to uncover potential target genes for biological control. Int J Mol Sci. 2016;17:1442.
Eves-van den Akker S, Lilley CJ, Jones JT, Urwin PE. Identification and characterisation of a hyper-variable apoplastic effector gene family of the potato cyst nematodes. PLoS Pathog. 2014;10:e1004391.
Kumar M, Gantasala NP, Roychowdhury T, Thakur PK, Banakar P, Shukla RN, Jones MG, Rao U. De novo transcriptome sequencing and analysis of the cereal cyst nematode Heterodera avenae. PLoS ONE. 2014;9:e96311.
Kong LA, Wu DQ, Huang WK, Peng H, Wang GF, Cui JK, Liu SM, Li ZG, Yang J, Peng DL. Large-scale identification of wheat genes resistant to cereal cyst nematode Heterodera avenae using comparative transcriptomic analysis. BMC Genom. 2015;16:801.
Chronis D, Chen S, Lu S, Hewezi T, Carpenter SC, Loria R, Baum TJ, Wang X. A ubiquitin carboxyl extension protein secreted from a plant-parasitic nematode Globodera rostochiensis is cleaved in planta to promote plant parasitism. Plant J. 2013;74:185–96.
Ali S, Magne M, Chen S, Obradovic N, Jamshaid L, Wang X, Belair G, Moffett P. Analysis of Globodera rostochiensis effectors reveals conserved functions of SPRYSEC proteins in suppressing and eliciting plant immune responses. Front Plant Sci. 2015;6:623.
Replogle A, Wang J, Bleckmann A, Hussey RS, Baum TJ, Sawa S, Davis EL, Wang X, Simon R, Mitchum MG. Nematode CLE signaling in Arabidopsis requires CLAVATA2 and CORYNE. Plant J. 2011;65:430–40.
Zheng M, Long H, Zhao Y, Li L, Xu D, Zhang H, Liu F, Deng G, Pan Z, Yu M. RNA-Seq based identification of candidate parasitism genes of cereal cyst nematode (Heterodera avenae) during incompatible infection to Aegilops variabilis. PLoS ONE. 2015;10:e0141095.
Cotton JA, Lilley CJ, Jones LM, Kikuchi T, Reid AJ, Thorpe P, Tsai IJ, Beasley H, Blok V, Cock PJ, et al. The genome and life-stage specific transcriptomes of Globodera pallida elucidate key aspects of plant parasitism by a cyst nematode. Genome Biol. 2014;15:R43.
Abad P, Gouzy J, Aury JM, Castagnone-Sereno P, Danchin EG, Deleury E, Perfus-Barbeoch L, Anthouard V, Artiguenave F, Blok VC, et al. Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nat Biotechnol. 2008;26:909–15.
Opperman CH, Bird DM, Williamson VM, Rokhsar DS, Burke M, Cohn J, Cromer J, Diener S, Gajan J, Graham S, et al. Sequence and genetic map of Meloidogyne hapla: a compact nematode genome for plant parasitism. Proc Natl Acad Sci USA. 2008;105:14802–7.
Kikuchi T, Cotton JA, Dalzell JJ, Hasegawa K, Kanzaki N, McVeigh P, Takanashi T, Tsai IJ, Assefa SA, Cock PJ, et al. Genomic insights into the origin of parasitism in the emerging plant pathogen Bursaphelenchus xylophilus. PLoS Pathog. 2011;7:e1002219.
Consortium CES. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998;282:2012–8.
Dieterich C, Clifton SW, Schuster LN, Chinwalla A, Delehaunty K, Dinkelacker I, Fulton L, Fulton R, Godfrey J, Minx P, et al. The Pristionchus pacificus genome provides a unique perspective on nematode lifestyle and parasitism. Nat Genet. 2008;40:1193–8.
Jex AR, Liu S, Li B, Young ND, Hall RS, Li Y, Yang L, Zeng N, Xu X, Xiong Z, et al. Ascaris suum draft genome. Nature. 2011;479:529–33.
Kroetz SM, Srinivasan J, Yaghoobian J, Sternberg PW, Hong RL. The cGMP signaling pathway affects feeding behavior in the necromenic nematode Pristionchus pacificus. PLoS ONE. 2012;7:e34464.
Li C, Nelson LS, Kim K, Nathoo A, Hart AC. Neuropeptide gene families in the nematode Caenorhabditis elegans. Ann NY Acad Sci. 1999;897:239–52.
Kimber MJ, Fleming CC. Neuromuscular function in plant parasitic nematodes: a target for novel control strategies? Parasitology. 2005;131(Suppl):S129–42.
Brownlee DJ, Fairweather I, Holden-Dye L, Walker RJ. Nematode neuropeptides: localization, isolation and functions. Parasitol Today. 1996;12:343–51.
McVeigh P, Leech S, Mair GR, Marks NJ, Geary TG, Maule AG. Analysis of FMRFamide-like peptide (FLP) diversity in phylum Nematoda. Int J Parasitol. 2005;35:1043–60.
Long H, Peng D, Huang W, Peng H, Wang G. Molecular characterization and functional analysis of two new β-1,4-endoglucanase genes (Ha-eng-2, Ha-eng-3) from the cereal cyst nematode Heterodera avenae. Plant Pathol. 2013;62:953–60.
Hiller NL, Bhattacharjee S, van Ooij C, Liolios K, Harrison T, Lopez-Estrano C, Haldar K. A host-targeting signal in virulence proteins reveals a secretome in malarial infection. Science. 2004;306:1934–7.
Xiang J, Li X, Wu J, Yin L, Zhang Y, Lu J. Studying the mechanism of Plasmopara viticola RxLR effectors on suppressing plant immunity. Front Microbiol. 2016;7:709.
Morgan W, Bos J, Bruce C, Lee M, Liu H-Y, Oh S-K, Song J, Win J, Young C, Kamoun S. Structure and function of RXLR effectors of plant pathogenic oomycetes. In: Gustafson JP, Taylor J, Stacey G, editors. Genomics of Disease. New York: Springer New York; 2008. p. 161–71.
Boddey JA, Hodder AN, Gunther S, Gilson PR, Patsiouras H, Kapp EA, Pearce JA, de Koning-Ward TF, Simpson RJ, Crabb BS, Cowman AF. An aspartyl protease directs malaria effector proteins to the host cell. Nature. 2010;463:627–31.
Hewezi T, Baum TJ. Manipulation of plant cells by cyst and root-knot nematode effectors. Mol Plant Microbe Interact. 2013;26:9–16.
Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–5.
Fitch WM. Distinguishing homologous from analogous proteins. Syst Zool. 1970;19:99–113.
Feinberg EH, Hunter CP. Transport of dsRNA into cells by the transmembrane protein SID-1. Science. 2003;301:1545–7.
Winston WM, Molodowitch C, Hunter CP. Systemic RNAi in C. elegans requires the putative transmembrane protein SID-1. Science. 2002;295:2456–9.
Britton C, Murray L. Using Caenorhabditis elegans for functional analysis of genes of parasitic nematodes. Int J Parasitol. 2006;36:651–9.
Le X, Wang X, Guan T, Ju Y, Li H. Isolation and characterization of a fatty acid- and retinoid-binding protein from the cereal cyst nematode Heterodera avenae. Exp Parasitol. 2016;167:94–102.
Lilley CJ, Davies LJ, Urwin PE. RNA interference in plant parasitic nematodes: a summary of the current status. Parasitology. 2012;139:630–40.
Ewbank JJ. Signaling in the immune response (January 23, 2006). WormBook, ed. 2006.
Jing BX, He Q, Wu Hai Y, De Peng L. Seasonal and temperature effects on hatching of Heterodera avenae(Shandong population, China). Nematology. 2014;16:1209–17.
Rivoal R, Nicol JM. Past research on the cereal cyst nematode complex and future needs. Addis Ababa: International Maize and Wheat Improvement Centre (CIMMYT); 2009. p. 3–10.
Zhang Dongsheng PD, Qi Shuhua. Factors influencing hatching of the cereal cyst nematode from the Northern Part of HuaBei Plain. Acta Phytopathologica Sinica. 1995;26:158 (in Chinese).
Zheng J, Cheng H, Fang Z. Hatching characteristics of cereal cyst nematode, Heterodera avenae in Shanxi, China. J Zhejiang Agric Univ. 1997;23(6):667–71 (in Chinese).
Ailion M, Inoue T, Weaver CI, Holdcraft RW, Thomas JH. Neurosecretory control of aging in Caenorhabditis elegans. Proc Natl Acad Sci USA. 1999;96:7394–7.
Ailion M, Thomas JH. Dauer formation induced by high temperatures in Caenorhabditis elegans. Genetics. 2000;156:1047–67.
Mahoney TR, Liu Q, Itoh T, Luo S, Hadwiger G, Vincent R, Wang ZW, Fukuda M, Nonet ML. Regulation of synaptic transmission by RAB-3 and RAB-27 in Caenorhabditis elegans. Mol Biol Cell. 2006;17:2617–25.
Apfeld J, Kenyon C. Regulation of lifespan by sensory perception in Caenorhabditis elegans. Nature. 1999;402:804–9.
Hu PJD. Dauer. The C. elegans Research Community. WormBook; 2007.
Birnby DA, Link EM, Vowels JJ, Tian H, Colacurcio PL, Thomas JH. A transmembrane guanylyl cyclase (DAF-11) and Hsp90 (DAF-21) regulate a common set of chemosensory behaviors in Caenorhabditis elegans. Genetics. 2000;155:85–104.
Palomares-Rius JE, Jones JT, Cock PJ, Castillo P, Blok VC. Activation of hatching in diapaused and quiescent Globodera pallida. Parasitology. 2013;140:445–54.
Engelmann I, Pujol N. Innate immunity in C. elegans. Adv Exp Med Biol. 2010;708:105–21.
Chase DL, Koelle MR. Biogenic amine neurotransmitters in C. elegans. WormBook; 2007.
Urwin PE, Lilley CJ, Atkinson HJ. Ingestion of double-stranded RNA by preparasitic juvenile cyst nematodes leads to RNA interference. Mol Plant Microbe Interact. 2002;15:747–52.
Masler EP. Responses of Heterodera glycines and Meloidogyne incognita to exogenously applied biogenic amines. Nematology. 2008;10:911–7.
Alkema MJ, Hunter-Ensor M, Ringstad N, Horvitz HR. Tyramine Functions independently of octopamine in the Caenorhabditis elegans nervous system. Neuron. 2005;46:247–60.
McVeigh P, Geary TG, Marks NJ, Maule AG. The FLP-side of nematodes. Trends Parasitol. 2006;22:385–96.
Papolu PK, Gantasala NP, Kamaraju D, Banakar P, Sreevathsa R, Rao U. Utility of host delivered RNAi of two FMRF amide like peptides, flp-14 and flp-18, for the management of root knot nematode, Meloidogyne incognita. PLoS ONE. 2013;8:e80603.
Postma WJ, Slootweg EJ, Rehman S, Finkers-Tomczak A, Tytgat TO, van Gelderen K, Lozano-Torres JL, Roosien J, Pomp R, van Schaik C, et al. The effector SPRYSEC-19 of Globodera rostochiensis suppresses CC-NB-LRR-mediated disease resistance in plants. Plant Physiol. 2012;160:944–54.
Lacomme C, Santa Cruz S. Bax-induced cell death in tobacco is similar to the hypersensitive response. Proc Natl Acad Sci USA. 1999;96:7956–61.
Abramovitch RB, Kim YJ, Chen S, Dickman MB, Martin GB. Pseudomonas type III effector AvrPtoB induces plant disease susceptibility by inhibition of host programmed cell death. EMBO J. 2003;22:60–9.
Jamir Y, Guo M, Oh HS, Petnicki-Ocwieja T, Chen S, Tang X, Dickman MB, Collmer A, Alfano JR. Identification of Pseudomonas syringae type III effectors that can suppress programmed cell death in plants and yeast. Plant J. 2004;37:554–65.
Tetteh KK, Loukas A, Tripp C, Maizels RM. Identification of abundantly expressed novel and conserved genes from the infective larval stage of Toxocara canis by an expressed sequence tag strategy. Infect Immun. 1999;67:4771–9.
Callister DM, Winter AD, Page AP, Maizels RM. Four abundant novel transcript genes from Toxocara canis with unrelated coding sequences share untranslated region tracts implicated in the control of gene expression. Mol Biochem Parasitol. 2008;162:60–70.
Qin XC, Shi M, Tian JH, Lin XD, Gao DY, He JR, Wang JB, Li CX, Kang YJ, Yu B, et al. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proc Natl Acad Sci USA. 2014;111:6744–9.
Yan G, Smiley RW. Distinguishing Heterodera filipjevi and H. avenae using polymerase chain reaction-restriction fragment length polymorphism and cyst morphology. Phytopathology. 2010;100:216–24.
Chen C, Liu S, Liu Q, Niu J, Liu P, Zhao J, Jian H. An ANNEXIN-like protein from the cereal cyst nematode Heterodera avenae suppresses plant defense. PLoS ONE. 2015;10:e0122256.
Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16:276–7.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8.
Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–60.
Ekseth OK, Kuiper M, Mironov V. orthAgogue: an agile tool for the rapid prediction of orthology relations. Bioinformatics. 2014;30:734–6.
van Dongen S, Abreu-Goodger C. Using MCL to extract clusters from networks. Methods Mol Biol. 2012;804:281–95.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Bauer S, Grossmann S, Vingron M, Robinson PN. Ontologizer 2.0—a multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics. 2008;24:1650–1.
Parkinson J, Whitton C, Schmid R, Thomson M, Blaxter M. NEMBASE: a resource for parasitic nematode ESTs. Nucleic Acids Res. 2004;32:D427–30.
Loer C. Neurotransmitters in Caenorhabditis elegans. WormAtlas; 2010.
Dalzell JJ, McVeigh P, Warnock ND, Mitreva M, Bird DM, Abad P, Fleming CC, Day TA, Mousley A, Marks NJ, Maule AG. RNAi effector diversity in nematodes. PLoS Negl Trop Dis. 2011;5:e1176.
Wang L, Feng Z, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–8.
Isserlin R, Merico D, Voisin V, Bader GD. Enrichment map—a cytoscape app to visualize and explore OMICs pathway enrichment results. F1000Research. 2014;3:141.
Kolde R. Pheatmap: pretty heatmaps. R package version 1.0.8. 2015. http://cran.r-project.org/package=pheatmap.
Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–6.
Sonnhammer EL, von Heijne G, Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol. 1998;6:175–82.
Gattiker A, Gasteiger E, Bairoch A. ScanProsite: a reference implementation of a PROSITE scanning tool. Appl Bioinformatics. 2002;1:107–8.
Langdon WB. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 2015;8:1.
Ramirez-Gonzalez RH, Bonnal R, Caccamo M, Maclean D. Bio-samtools: ruby bindings for SAMtools, a library for accessing BAM files containing high-throughput sequence alignments. Sour Code Biol Med. 2012;7:6.
Authors’ contributions
DY and HJ conceived and designed the experiments; DY and CC performed the experiments; DY analyzed the data; CC and QL contributed reagents/materials/analysis tools; DY wrote the paper and HJ revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We appreciate the editorial assistance from Dr. Fei Tang.
Competing interests
The authors declare that they have no financial competing interests. The datasets on transcriptome sequencing of H. avenae are only available from the corresponding author on reasonable request, due to potential conflicts of interests.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its Additional files. The datasets on original sequencing of H. avenae are not publicly available due to potential conflicts of interests, but are available from the corresponding author on reasonable request.
Funding
The current research was financially supported by the National Basic Research Program of China (No. 2013CB127501), the Special Fund for Agro-scientific Research in the Public Interest China (No. 201503114) and Chinese Universities Scientific Fund (No. 2015NX005).
Author information
Authors and Affiliations
Corresponding author
Additional files
13578_2017_138_MOESM1_ESM.pdf
Additional file 1: Figure S1. The distribution of categories of GO (A) and KEGG pathways (B) annotations for H. avenae ESTs (GO terms: Level <3, Percent of genes >5%).
13578_2017_138_MOESM2_ESM.pdf
Additional file 2: Figure S2. Hierarchical layout of significantly enriched GOs of orthologous identified in H. avenae and 7 other nematodes. a Enriched GOs of orthologous present in H. avenae and all other 7 nematodes (Globodera pallida, Meloidogyne incognita, Meloidogyne hapla, Bursaphelenchus xylophilus, Caenorhabditis elegans, Pristionchus pacifics and Ascaris suum). b Enriched GOs of orthologous shared only by H. avenae and other plant-parasitic nematodes (G. pallida, M. incognita, M. hapla and B. xylophilus). c Enriched GOs of orthologous shared only by H. avenae and other sedentary plant-nematodes (G. pallida, M. incognita and M. hapla). d Enriched GOs of orthologous present only in cyst nematodes H. avenae and G. pallida. Light color represents significant enrichment (p < 0.1) and darker color represents more significant enrichment (p < 0.05).
13578_2017_138_MOESM3_ESM.pdf
Additional file 3: Table S1. Comparative analyses of orthologous identified in biological pathways. Orthologous in RNAi pathway a, immune responses b, dauer/diapause pathway c and the synthesis, transportation and metabolism of neurotransmitters d were compared among C. elegans, H. avenae and other 6 nematodes with published genomic data. Cel, Caenorhabditis elegans; CCN, Cereal cyst nematode H. avenae; Gpa, Globodera pallida; Mha, Meloidogyne hapla; Min, Meloidogyne incognita; Bxy, Bursaphelenchus xylophilus; Asu, Ascaris suum; Ppa, Pristionchus pacifics.
13578_2017_138_MOESM4_ESM.pdf
Additional file 4: Table S2. Datasets of the nematoda ESTs used in this study. The datasets from NCBI and Nembase 4 databases consist of 1,331,401 ESTs sequences from 45 genuses and 75 species clustered into three life styles. * Life style classification: FLN, free living nematode; PPN, plant parasitic nematode; APN, animal parasitic nematode.
13578_2017_138_MOESM5_ESM.pdf
Additional file 5: Figure S3. Transcriptomes comparison of H. avenae at pre-parasitic and post-parasitic stages. Dot plots show the distributions of contig lengths and abundances in transcriptomes of H. avenae before (a) and after (b) parasitism. c MA-plot shows differential expression of genes before and after parasitism. The M is the reads number ratio and the A is the average. Red dots, genes with significantly higher expression at pre-parasitic stages; Blue dots, genes with significantly higher expression at post-parasitic stages (FDR < 0.001). The dotted lines indicate the M values as -1, 0 and 1.
13578_2017_138_MOESM6_ESM.pdf
Additional file 6: Figure S4. Heat map depicting the expression profiles of crosstalks genes relevant to the KEGG pathways significantly enriched before and after parasitism in H. avenae. The first box of each row marks the enrichment of crosstalk at either pre-parasitic stages (purple) or post-parasitic stages (green). Each red box denotes a single gene with higher expression at pre-parasitic stages; each blue box denotes a single gene with higher expression at post-parasitic stages. The darkness of the color is positively correlated with the absolute value of fold change, which is calculated as normalized log2(pre-parasitic RPKM/post-parasitic RPKM) with DEGseq.
13578_2017_138_MOESM7_ESM.pdf
Additional file 7: Table S3. The list of differentially expressed genes in each node from the H. avenae KEGG pathways enriched crosstalks. The ‘Log2(fold change) Pre/Post’ means the normalized Log2(pre-parasitic RPKM/post-parasitic RPKM) of each contig. Both Log2(fold change) and Q value threshold were calculated by DEGseq. Some contigs might be presented in multiple crosstalk nodes.
13578_2017_138_MOESM8_ESM.pdf
Additional file 8: Table S4. Putative phytonematode effectors identified in H. avenae. a Putative key genes based on blastx searching against other phytonematode effectors. b Carbohydrate-active enzymes with homology to those of other phytonematodes. * # CCN ESTs, number of ESTs in Cereal cyst nematode H. avenae. ‘Pre’ means significantly high expression in pre-parasitic stage; ‘Pre*’ means coding by singleton in pre-parasitic library. The singleton might be the gene with low expression and was sequenced only once. CBM, carbohydrate binding module; GH, glycoside hydrolase; GT, glycosyl transferase; PL, polysaccharide lysate. NR means non-redundant GenBank proteins database.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yang, D., Chen, C., Liu, Q. et al. Comparative analysis of pre- and post-parasitic transcriptomes and mining pioneer effectors of Heterodera avenae . Cell Biosci 7, 11 (2017). https://doi.org/10.1186/s13578-017-0138-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-017-0138-6