
Abstract
The association between intestinal flora and ulcerative colitis (UC) was studied in order to provide a basis and method for clinical treatment. Fresh fecal samples were collected from 30 active UC patients and 10 healthy controls. The intestinal flora DNA from each sample was extracted and 16S rRNA gene sequencing was carried out using HiSeq platform to identify the intestinal flora in fecal samples. The richness and diversity of intestinal flora in UC patients were significantly lower than those in healthy control group (P < 0.05). Significant differences were observed between the intestinal flora-species of UC patients and healthy controls. Synergistetes (P < 0.01) and Firmicutes (P < 0.05), along with probiotics Veillonella (P < 0.01), Ruminococcus and Coprococcus (P < 0.05) in the UC patients were lower than that in the healthy controls significantly. Furthermore, compared with the control group, Tenericutes (P < 0.01) and intestinal pathogenic bacteria, including Bacteroides (P < 0.01), Escherichia and Sutterella (P < 0.05) were significantly increased. The incidence of UC is significantly associated with the changes in intestinal flora. Changes in intestinal flora may lead to a decrease in the diversity of intestinal flora or to the enrichment of a particular intestinal flora.
Similar content being viewed by others

Introduction
Ulcerative colitis (UC) is a chronic non-specific intestinal inflammatory disease of unknown etiology. Its main symptoms include abdominal pain, diarrhea and mucus, pus and blood. Some UC patients have extra-intestinal manifestations and complications, which affect their daily lives, and even their health (Whitehead 2016; Sykora et al. 2018). An epidemiological survey reported higher prevalence of UC among the people of 20–40 years, and higher prevalence in males than females (Lewin et al. 2019). The incidence rate of UC fluctuates from 5.5 to 24.3 per million people worldwide with significant regional and ethnic specificity. Statistically, the prevalence of UC in China is about 11.6 per million (Whitesides 1985; Molodecky et al. 2012; Ng et al. 2013; Cui et al. 2018). The pathogenesis of UC has been reported to be associated with environmental, genetic, immune and intestinal flora-factors (Cui et al. 2018). Among them, the barrier, metabolic and nutritive function of intestinal flora, immune system function, and the regulation of the balance of intestinal flora play a key role in the pathogenesis of UC.
The intestinal flora is currently considered to be a complex ecosystem, which is (Ahlawat et al. 2021b) composed of 500–1000 different species, accounting for 1014 bacterial cells, which is 10 times more than that of the total number of human cells (Domingo and Sanchez 2018). The genome of all intestinal microorganisms is known as “microbiome”, which is more than 100-times larger than the human nuclear genome (Huang et al. 2019). Intestinal microflora is increasingly recognized as a key factor of human health. Several studies have shown that many human chronic diseases are associated with intestinal microecological disorders (Tripathi et al. 2018; Zhang et al. 2018; Ma et al. 2019; Ahlawat et al. 2021a). The intestinal flora is an important environmental factor, which has been reported to be associated with a series of metabolic diseases, including obesity (Machiels et al. 2014) and diabetes (Yuan et al. 2018). It is crucial for human beings to maintain a good symbiotic relationship between human body and intestinal flora.
Although the specific pathogenesis of UC remains unclear, the interactions of genetic abnormalities, immune system dysfunction, intestinal barrier dysfunction and microbiological infection are thought to be the early risk factors of UC (Deng et al. 2019; Lewin et al. 2019; Lee et al. 2020). The microbiological factor has been recognized as the most potent environmental factor in the UC progression, which is possibly associated with the harmful mucosal invasion, activation of carcinogens, or inflammatory responses (Zheng et al. 2019). Moreover, the inflammatory cytokines and/or mixed inflammatory infiltrates in the intestinal mucosa may also be responsible for the development of UC (Kopecki et al. 2019). Therefore, a better understanding of the gastrointestinal microflora and inflammatory cytokines in the progression of UC has been of great significance.
This study provides a new basis and method for the clinical assessment of UC by analyzing the association between changes in intestinal flora and UC. It is of great importance to fully understand the changes of intestinal flora in UC. This study provides a theoretical basis for the regulation of intestinal flora in the treatment of UC and its complications.
Materials and methods
We have uploaded the data to the NCBI Sequence Read Archive database (Accession Number: PRJNA695366).
Materials
Agencourt AMPure XP magnetic beads and Elution Buffer were purchased from MGI Tech Co., Ltd. InhibitEX Buffer and Qubit® dsDNA BR Assay Kit were purchased from Shanghai Haoran Biotechnology Co., Ltd. Two wash buffers, including Buffer AW1 and Buffer AW2 were purchased from Shanghai Beinuo Biotechnology Co., Ltd. Buffer AL and Buffer ATE were purchased from Shanghai Limin Industrial Co., Ltd. KAPA HiFi HotStart DNA Polymerase was purchased from Shanghai HiFi Biotech Co., Ltd.
Sample collections and setting participants
According to the revised standards of “Consensus Opinions on Diagnosis and Treatment of Inflammatory Bowel Disease” formulated by the Inflammatory Bowel Disease Group of the Chinese Medical Association Gastroenterology Branch in 2018, 30 patients with ulcerative colitis were recruited from the First Affiliated Hospital, Harbin Medical University from January to December 2019. There were no gender restrictions. The inclusion criteria for the recruitment of patients included: (1) region, Heilongjiang province; (2) age, 30–65 years; (3) past history of ulcerative colitis, the ulcerative colitis active period; and (4) the patients are not treated with antibiotics and microecological agents or not treated with these drugs 4 weeks before the onset. The core standards included: (a) hematology tests, such as hemoglobin (Hb), white blood cells (WBC), erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP); (b) colonoscopy report showing ulcerative colitis; and (c) pathology report showing moderate to severe mucosal chronic inflammation or suggesting crypt abscess. The exclusion criteria of the UC patients included: (a) incomplete clinical data; (b) patients with severe heart, liver, lung, and kidney diseases; (c) patients with diabetes and severe infections; (d) patients with pregnancy and lactation; (e) patients with UC complications; (f) patients with extra-intestinal manifestations; and (g) patients with other autoimmune diseases. In addition, 10 healthy controls in the physical examination center of the First Affiliated Hospital, Harbin Medical University were recruited, regardless of gender. For the inclusion criteria of the recruitment of healthy controls, the region, age, and drugs usage were same as that of UC patients. Other inclusion criteria parameters included: (1) no history of chronic gastrointestinal diseases and routine physical examination and stool examination showed no abnormalities; and (2) information collection such as hematological tests, such as Hb, WBC, ESR, and CRP. The sample collection was reviewed by the Ethics Committee of the First Affiliated Hospital of Harbin Medical University.
Fresh stool samples of 1–10 g were collected in a medical stool collection vessels (the surface of the vessels had sample numbers) from the patients and healthy controls, and stored in a − 80 °C low-temperature freezer after one hour of sampling.
DNA extraction and sequencing
The intestinal floras from the fecal samples were homogenized and genomic DNA was extracted using QIAamp Fast DNA Stool Mini Kit (QIAGEN China (Shanghai) Co., Ltd.). The quality and integrity of the DNA extraction were evaluated using micro spectrophotometer and gel electrophoresis.
The extracted DNA samples and corresponding fusion primers were mixed in order to configure the PCR reaction system. The PCR reaction parameters were set for PCR amplification. Agencourt AMPure XP magnetic beads were used to purify the PCR amplification products, which were then dissolved in Elution Buffer, and labeled. The metagenomic library was prepared and the fragment range and concentration were analyzed using Agilent 2100 Bioanalyzer. The prepared colonies were selected for sequencing on HiSeq platform based on insert size.
Data filtering
The original sequencing data was processed to remove low-quality, low-complexity, pollution and N-terminal sequences and obtain clean data. FLASH (Fast Length Adjustment of Short reads, v1.2.11) software was used to sequence paired-end sequences using overlapping relationships The obtained paired-end sequences were assembled into a sequence to obtain the tags of hyper-variable region. USEARCH (v7.0.1090) software was used to perform clustering and splicing at 97% similarity. The spliced tags were clustered as OUT (Operational Taxonomic Units) and compared with the database and species annotations. The statistics table of OTU abundance for each sample was obtained, and the samples were analyzed on the basis of OTU and species annotation results.
Statistical analysis
Statistical analysis was carried out using Excel spreadsheets and SPSS statistical software. The differences in the species diversity and richness between the UC patients’ samples and that of healthy controls were analyzed. The rank sum test was used, and the results were analyzed using OTU Rank curve, alpha diversity box plot and beta diversity box plot. The similarity in the species of both the groups was analyzed by OTU PLS-DA (Partial Least Squares Discriminant Analysis) method. The RDP classifier Bayesian algorithm was used for species composition analysis and statistics. The histogram and heat map of species richness and the comparison histogram of key species difference were used for the analysis of species composition between two groups. U test was used to calculate the average relative abundance of species between the two groups and to analyze the significance of the difference test. The statistical differences of P < 0.05 and P < 0.01 were considered as significant and extremely significant, respectively. FastTree is used to construct evolutionary tree by selecting OTU or sequence corresponding to classification information at a certain level.
Results
Comparison of species diversity and richness
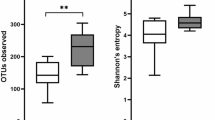
The comparison of species diversity between the UC patients and the healthy controls is shown in Figs. 1, 2, 3. Shannon index, simpson index and unweighted UniFrac Beta diversity box plot were used to reflect the diversity of bacterial community in the samples (Fig. 1). The species diversity in the UC patients was significantly lower than that in the health controls (P < 0.05). The comparison of species richness between the UC patients and the healthy controls is shown in Figs. 1, 3. The observed species index, chao index, and ACE index reflected the community richness between the two groups of samples. The species richness in the UC patients was significantly lower than that of the healthy controls (P < 0.05). The biodiversity statistics between the two groups are listed in Table 1.

Comparison of Alpha diversity box between disease group (UC patients) and normal group (healthy controls). Red and blue colored boxes show the richness of microflora in disease and control samples, respectively for all the statistical analysis mentioned with each graph. Clear significant differences were observed in all the statistical analyses

Unweighted UniFrac Beta diversity box and the histograms of species richness at phylum level, showing species diversity between UC patients and healthy controls (normal). A Cyan and yellow colored boxes show the diversity of microflora in disease and control samples. The species in the control samples were more diverse as compared to those in the disease samples. B Color identification of each phylum is shown in the side bar. The disease samples shows greater number of Bacteriodetes and Proteobacteria and lesser number of Firmicutes and Verrucomicrobia than the normal samples

OTU Rank graph showing the comparison of species richness between UC patients and healthy controls (normal). The side bar shows color identifications for 30 disease samples and 10 normal samples, denoted as D1-30 and N1-10, respectively. A clear divergence in the normal samples OTUs was observed
Comparison of species similarity
The species similarities between the samples of the UC patients and healthy controls were analyzed on the basis of two sets of OTU data and OTU PLS-DA analysis chart. As shown in Fig. 2B, the intestinal flora clustering of the UC patients and healthy controls showed distinct distribution from each other, without showing any intersection, which showed significant differences in the species diversity between the two sets of samples.
Species level composition analysis
The histogram and heat map of the species richness of the UC patients and healthy controls are shown in Figs. 4, 5, respectively. The average relative abundance and the significance of difference test are shown in Fig. 6 and provided in Additional file 1: Table S1. The probabilities of species distribution of the two groups are listed in Table 2. Synergistetes and Firmicutes were decreased in the UC patients as compared to the healthy control with statistical significance (P < 0.01 and P < 0.05), respectively. Compared with healthy controls, patients with UC showed a significant increase in Tenericutes (P < 0.01). There were no significant differences in Bacteroides, Proteobacteria, Actinomycetes, Fusobacteria, Verrucomicrobia, Lentisphaerae, Euryarchaeota between the UC patients and healthy controls.

Comparison of the histograms of species richness at phylum level between UC patients and healthy controls (normal). Color identification of each phylum is shown in the side bar. The disease samples shows greater number of Bacteriodetes and Proteobacteria and lesser number of Firmicutes and Verrucomicrobia than the normal samples

Heat map showing horizontal species richness between UC patients and healthy controls (normal). The relative abundance of bacteria is shown in terms of color intensities (denoted by the scale given in upper left corner). The differences in the color intensities between disease and normal samples show differences in the number of respective species

Histogram of key species showing differences between species in UC patients and healthy controls (normal). The species in disease and normal samples are denoted by red and grey bars, respectively, where the length of bars denotes the relative abundance of species
Genus level composition analysis
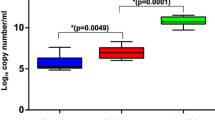
The average relative abundance of species in the UC patients and healthy controls is shown in Fig. 7 and provided in Additional file 1: Table S2. The beneficial bacteria Veillonella were significantly reduced in the UC patients as compared to the healthy controls, along with significant increase in harmful bacteria Bacteroides in the UC patients as compared to the healthy controls (P < 0.01). Firmicutes were also reduced in the UC patients as compared to the healthy controls with statistical significance of P < 0.05. The differences in Bacteroides, Proteobacteria, Actinomycetes, Fusobacteria, Verrucomicrobia, Lentisphaerae, Euryarchaeota between the UC patients and healthy controls were insignificant.

Histogram showing bacterial species composition in UC patients and healthy controls (normal). The species in disease and normal samples are denoted by red and grey bars, respectively, where the length of bars denotes the relative abundance of species
Phylogenetic analysis of species
The phylogenetic analysis of species level was studied as follows. Firmicutes and Proteobacteria were the two largest intestinal microbial species, which also the fastest growing species. Second, Actinobacteria and Bacteroidetes have more branches than other species. Euryarchaeota, Fusobacteria, Lentisphaerae, and Verrucomicrobia have less species evolution (Fig. 8).

The phylogenetic analysis of species level
Discussion
In this study, a case–control experimental method was adopted and 16S rRNA gene sequencing technology using Illumina HiSeq 2500 sequencing platform was applied to explore the relationship between changes in intestinal flora of UC patients and healthy controls. The results showed that the species richness and diversity of the intestinal flora in the UC patients were significantly lower than those of healthy controls. Consistent with the domestic and foreign studies, significant differences in the species diversity of the intestinal flora between UC patients and healthy controls were observed (Ruili et al. 2012; Min et al. 2015). It has been reported that the UC recurrence in the patients in UC remission period is associated with the aggravation of intestinal flora imbalance. It also found that the status of intestinal flora of UC patients is closely associated with the development of the disease by testing intestinal flora in the stool samples of UC active period, UC remission period and healthy controls (Min et al. 2015). Another study have also revealed that the diversity of intestinal flora in patients with active UC is lower than that in UC remission and healthy controls, analyzed by ERIC-PCR (enterobacterial repetitive intergenic consensus PCR) technology (Ruili et al. 2012).
It was found that the synergistetes and firmicutes decreased significantly, and the Soft-walled bacteria significantly increased in the UC patients as compared to healthy controls. The current research showed that the alterations in the intestinal flora of UC patients were mainly attributable to decrease in Firmicutes and increase in Proteobacteria. The trend of changes in Actinobacteria and Bacteroides has not been confirmed yet (Hold et al. 2014). Following these differences, the differences in the composition of intestinal flora in UC patients and healthy controls at genus level were analyzed, which showed that Escherichia and Sutterella were significantly increased in UC patients. The increase in Escherichia has been reported to disrupt the balance and integrity of intestinal environment and promote inflammation. A study reported that Helicobacter is a major flora in intestinal infections (Li et al. 2017). When the distribution of the flora changes, it can cause biliary infections, urinary tract infections, and intra-abdominal infections, and even life-threatening. Sutterella sp. may cause digestive disorders (Sartor and Wu 2017), which may be another factor that affect the aggravation and complications of UC patients. Coprococcus, a major flora found in the intestines of humans and animals, decreased significantly in the intestinal flora of UC patients, which can produce natural antibiotics, enhance intestinal function, and also benefit the body. Coprococcus, not only produce bacteriostatic substances, such as bacteriocins, that inhibit the growth of pathogenic bacteria, but they can also inhibit the reproduction of urease-producing bacteria and spoilage bacteria in the intestinal tract. Through these mechanisms it improves the protection of the intestinal microenvironment and reduces endotoxin and inflammation. Ruminococcus are the main fiber-degrading bacteria in gastrointestinal tract, which produce succinic acid, acetic acid, formic acid, ethanol and lactic acid, and build the intestinal microbial barrier alongside with other flora.
Our research showed a higher number of Bacteroides in UC patients than the healthy controls, which was consistent with a previous study (Furusawa et al. 2013). In our present study, the number of Faecalibacterium genus was relatively high as compared to the healthy controls, which was inconsistent with previous studies. One of the representative species of Clostridium prausnitzii is Faecalibacterium prausnitzii. Clostridium prausnitzii is a major probiotic in the gastrointestinal tract. It plays vital role in the formation of intestinal mucosal barrier, reduction of intestinal inflammation, competitive inhibition of pathogenic bacteria and reduction of pathogenic bacterial colonization. An early study showed that the number of Clostridium prasmoides in UC patients was significantly lower than healthy controls (Machiels et al. 2014). Man et al. (Man et al. 2013) compared the number of Bacteroides, Clostridium tenuiens and Bifidobacteria in UC patients and healthy controls. They showed an increased number of Bacteroides in UC patients, which could promote inflammation, and a decreased number of Clostridium tenuis and Bifidobacterium, which could inhibit inflammation (Man et al. 2013). We speculated that the UC patients who participated in this study did not take medication within 4 weeks. The accuracy of this experiment on the effect of medication was not studied in this study. The long course of UC patients, repeated symptoms and long-term use of aminosalicylic acid, biological agents, glucocorticoids, microecological agents, etc. might have impact on the results of this study. The degree of inflammation in UC patients is also an important factor that affect intestinal flora. This study did not strictly distinguish among patients having mild, moderate, and severe UC. Therefore, the role of these factors still needs to be studied in more detail. In addition, the age, gender, and living habits of the study participants can lead to different research conclusions. This requires the sample size to be expanded and refined and the sample variables to be specified as much as possible.
This study explored the feasibility of using intestinal flora to guide the treatment of UC by studying the intestinal flora of UC patients. Probiotics, such as Clostridium prasylvia, can positively regulate the stability of intestinal flora by increasing the permeability of intestinal mucosa and producing butyrate to reduce intestinal inflammation (Jinli et al. 2019); Lactobacillus regulates the balance of intestinal flora by promoting digestion and inhibiting the growth of potential pathogenic bacteria. Faecococcus can decompose nutrients to produce lactic acid, enhance intestinal function and inhibit the growth and reproduction of pathogenic bacteria. Fecal bacteria transplantation is another application of intestinal flora. By transplanting the intestinal flora of healthy people, it can reshape the intestinal flora barrier and can treat intestinal diseases. At present, there is evidence that the intestinal probiotics and intestinal fecal bacteria transplantation have a certain effect on the treatment of UC (Ding et al. 2019; Paramsothy et al. 2019). However, the therapeutic application of intestinal flora also has certain risks, especially in the immune-compromised people (such as patients with long-term use of glucocorticoids or immune-suppressants, patients with immunodeficiency, etc.). Therefore, the application of intestinal flora treatment for such people should be carefully considered. There may be risks of bacterial migration and the transmission of drug-resistant factors (Haiying et al. 2008). Thus, the extensive applications of intestinal flora still need a lot of clinical investigations.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its Additional file 1.
Abbreviations
- UC:
-
Ulcerative colitis
- Hb:
-
Hemoglobin
- WBC:
-
White blood cells
- ESR:
-
Erythrocyte sedimentation rate
- CRP:
-
C-reactive protein
References
Ahlawat S, Asha Y, Sharma K (2021a) Gut-organ axis: a microbial outreach and networking. Lett Appl Microbiol 72(6):636–668
Ahlawat S, Kumar P, Mohan H, Goyal S, Sharma K (2021b) Inflammatory bowel disease: tri-directional relationship between microbiota, immune system and intestinal epithelium. Crit Rev Microbiol 47(2):254–273
Cui H, Cai Y, Wang L, Jia B, Li J, Zhao S, Chu X, Lin J, Zhang X, Bian Y, Zhuang P (2018) Berberine regulates Treg/Th17 balance to treat ulcerative colitis through modulating the gut microbiota in the colon. Front Pharmacol 9:571
Deng F, He S, Cui S, Shi Y, Tan Y, Li Z, Huang C, Liu D, Zhi F, Peng L (2019) A molecular targeted immunotherapeutic strategy for ulcerative colitis via dual-targeting nanoparticles delivering miR-146b to intestinal macrophages. J Crohns Colitis 13(4):482–494
Ding X, Li Q, Li P, Zhang T, Cui B, Ji G, Lu X, Zhang F (2019) Long-term safety and efficacy of fecal microbiota transplant in active ulcerative colitis. Drug Saf 42(7):869–880
Domingo JJS, Sanchez CS (2018) From the intestinal flora to the microbiome. Rev Esp Enferm Dig 110(1):51–56
Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, Takahashi M, Fukuda NN, Murakami S, Miyauchi E, Hino S, Atarashi K, Onawa S, Fujimura Y, Lockett T, Clarke JM, Topping DL, Tomita M, Hori S, Ohara O, Morita T, Koseki H, Kikuchi J, Honda K, Hase K, Ohno H (2013) Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504(7480):446–450
Haiying Z, Xin L, Yuzhen L (2008) Safety and clinical use of microecological preparations. Adverse Drug React J 10(5): 340–346
Hold GL, Smith M, Grange C, Watt ER, El-Omar EM, Mukhopadhya I (2014) Role of the gut microbiota in inflammatory bowel disease pathogenesis: what have we learnt in the past 10 years? World J Gastroenterol 20(5):1192–1210
Huang X, Fan X, Ying J, Chen S (2019) Emerging trends and research foci in gastrointestinal microbiome. J Transl Med 17(1):67
Jinli H, Huiyun L, Huajun L (2019) Research of role and function of F. prausnitzii in intestinal microecology. Chin J Gastroenterol Hepatol 28(3):245–249
Kopecki Z, Yang G, Treloar S, Mashtoub S, Howarth GS, Cummins AG, Cowin AJ (2019) Flightless I exacerbation of inflammatory responses contributes to increased colonic damage in a mouse model of dextran sulphate sodium-induced ulcerative colitis. Sci Rep 9(1):12792
Lee HS, Vancamelbeke M, Verstockt S, Wilms T, Verstockt B, Sabino J, Ferrante M, Vermeire S, Cleynen I (2020) Molecular changes in the non-inflamed terminal ileum of patients with ulcerative colitis. Cells. https://doi.org/10.3390/cells9081793
Lewin SM, McConnell RA, Patel R, Sharpton SR, Velayos F, Mahadevan U (2019) Improving the quality of inpatient ulcerative colitis management: promoting evidence-based practice and reducing care variation with an inpatient protocol. Inflamm Bowel Dis 25(11):1822–1827
Li Q, Han Y, Dy ABC, Hagerman RJ (2017) The gut microbiota and autism spectrum disorders. Front Cell Neurosci 11:120
Ma Q, Li Y, Li P, Wang M, Wang J, Tang Z, Wang T, Luo L, Wang C, Wang T, Zhao B (2019) Research progress in the relationship between type 2 diabetes mellitus and intestinal flora. Biomed Pharmacother 117:109138
Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, Ballet V, Claes K, Van Immerseel F, Verbeke K, Ferrante M, Verhaegen J, Rutgeerts P, Vermeire S (2014) A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 63(8):1275–1283
Man J, Ping Y, Tao Y, Hongliang G, Lei W, Yongbo C (2013) Real-time fluorescence quantitative-PCR analysis of the variation of Bifidobacterium spp., Faecalibacterium prausnitzii and Bacteroides in patients with ulcerative colitis. Chin J Microecol 25(11):6
Min N, Tianbo S, Ruichun C, Yan D (2015) Analysis of intestinal flora distribution and detection of bacteria toxin gene in ulcerative colitis patients. J Zhengzhou Univ 4:514–518
Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, Kaplan GG (2012) Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 142(1):46-54.e42 (quiz e30)
Ng SC, Bernstein CN, Vatn MH, Lakatos PL, Loftus EV Jr, CTysk C, O’Morain C, Moum B, Colombel JF, Epidemiology and Natural History Task Force of the International Organization of Inflammatory Bowel Disease (2013) Geographical variability and environmental risk factors in inflammatory bowel disease. Gut 62(4):630–649
Paramsothy S, Nielsen S, Kamm MA, Deshpande NP, Faith JJ, Clemente JC, Paramsothy R, Walsh AJ, van den Bogaerde J, Samuel D, Leong RWL, Connor S, Ng W, Lin E, Borody TJ, Wilkins MR, Colombel JF, Mitchell HM, Kaakoush NO (2019) Specific bacteria and metabolites associated with response to fecal microbiota transplantation in patients with ulcerative colitis. Gastroenterology 156(5):1440-1454.e2
Ruili W, Ziqin S, Zhi W (2012) Itestinal flora structural features in the ulcerative colitis by ERIC-PCR fingerprinting. Chin J Gastroenterol Hepatol 21(9):798–801
Sartor RB, Wu GD (2017) Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology 152(2):327-339.e4
Sykora J, Pomahacova R, Kreslova M, Cvalinova D, Stych P, Schwarz J (2018) Current global trends in the incidence of pediatric-onset inflammatory bowel disease. World J Gastroenterol 24(25):2741–2763
Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, Knight R (2018) The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol 15(7):397–411
Whitehead L (2016) The impact of biological interventions for ulcerative colitis on health-related quality of life. Am J Nurs 116(11):21
Whitesides GM (1985) Applications of cell-free enzymes in organic synthesis. Ciba Found Symp 111:76–96
Yuan X, Ni H, Chen X, Feng X, Wu Q, Chen J (2018) Identification of therapeutic effect of glucagon-like peptide 1 in the treatment of STZ-induced diabetes mellitus in rats by restoring the balance of intestinal flora. J Cell Biochem 119(12):10067–10074
Zhang Y, Li T, Yuan H, Pan W, Dai Q (2018) Correlations of inflammatory factors with intestinal flora and gastrointestinal incommensurate symptoms in children with asthma. Med Sci Monit 24:7975–7979
Zheng JD, He Y, Yu HY, Liu YL, Ge YX, Li XT, Li X, Wang Y, Guo MR, Qu YL, Qin XF, Jiang MS, Wang XH (2019) Unconjugated bilirubin alleviates experimental ulcerative colitis by regulating intestinal barrier function and immune inflammation. World J Gastroenterol 25(15):1865–1878
Acknowledgements
Not applicable.
Funding
This study was supported by the General Program of Natural Science Foundation of Heilongjiang Province (No. D200826); and the General Projects of Scientific Research and Innovation Fund of the First Affiliated Hospital of Harbin Medical University (No. 2017Y006).
Author information
Authors and Affiliations
Contributions
YHT designed and wrote the manuscript; HCL and GS collected the data; TTW, YZ and LJS analyzed the data. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The sample collection was reviewed by the Ethics Committee of the First Affiliated Hospital of Harbin Medical University.
Consent for publication
Not applicable.
Competing interests
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Average relative abundance and significance of difference test of key species. Table S2. Composition of bacterial species in disease group and normal group.
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tang, Yh., Liu, Hc., Song, G. et al. A case–control study on the association of intestinal flora with ulcerative colitis. AMB Expr 11, 106 (2021). https://doi.org/10.1186/s13568-021-01267-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-021-01267-9