
Abstract
Proteus mirabilis is a commensal bacterium dwelling in the gastrointestinal (GI) tract of humans and animals. Although New Delhi metallo-β-lactamase 1 (NDM-1) producing P. mirabilis is emerging as a threat, its epidemiology in our society remains largely unknown. LHPm1, the first P. mirabilis isolate harboring NDM-1, was detected from a companion dog that resides with a human owner. The whole-genome study revealed 20 different antimicrobial resistance (AMR) genes against various classes of antimicrobial agents, which corresponded to the MIC results. Genomic regions, including MDR genes, were identified with multiple variations and visualized in a comparative manner. In the whole-genome epidemiological analysis, multiple phylogroups were identified, revealing the genetic relationship of LHPm1 with other P. mirabilis strains carrying various AMR genes. These genetic findings offer comprehensive insights into NDM-1-producing P. mirabilis, underscoring the need for urgent control measures and surveillance programs using a “one health approach”.
Similar content being viewed by others


Introduction
Proteus mirabilis is a ubiquitous bacterium found not only in soil, water, and sewage environments but also as a commensal bacterium in the gastrointestinal (GI) tract of humans and animals [1]. P. mirabilis is one of the most significant pathogens responsible for urinary tract infections (UTIs) in health care settings, with a suspected origin of the GI tract [1, 2]. P. mirabilis is also responsible for various clinical conditions, including respiratory tract infections, neonatal meningoencephalitis, empyema, and osteomyelitis [3]. Several broad-spectrum antibiotic agents, such as ampicillin, amoxicillin-clavulanate, ceftriaxone, ciprofloxacin, levofloxacin, trimethoprim-sulfamethoxazole (TMP-SMX), piperacillin-tazobactam, and carbapenems, are important treatment options for clinical P. mirabilis infections. Therefore, the emergence of carbapenemase-producing P. mirabilis could dramatically reduce treatment options for these clinical infections [4].
Multidrug-resistant (MDR) bacterial infection, especially that caused by New Delhi metallo-β-lactamase 1 (NDM-1)-producing gram-negative bacteria has already become a major threat to the nosocomial environment and public health [5,6,7]. Carbapenemase-producing strains acquire resistance to more than 2–3 classes of antibiotics, including carbapenems [8], which makes it incredibly difficult to treat these MDR pathogens in clinical situations. With a major association with mobile gene elements such as conjugative plasmids and integrative conjugative elements (ICEs) [9,10,11], blaNDM-1 is rapidly mutating and disseminating worldwide.
Carbapenem usage in animals is not allowed globally, yet unauthorized adjustments of the drugs in animal clinical environments remain, leading to the dissemination of carbapenemase-producing strains as an unaddressed threat. Therefore, the unidentified dissemination of carbapenemase producers among companion animals in our society should be considered a major threat to public health [12, 13]. Carbapenemase-producing bacteria are not a concern limited to human health; they require novel and urgent control measures and should not be neglected. However, there is only a limited amount of investigative data on carbapenemase-producing strains in animals in our society.
In this study, we employed molecular epidemiological approaches to investigate a carbapenemase producing P. mirabilis isolate. Because this was the first identification of an NDM-1 harboring P. mirabilis isolated from a companion dog in South Korea, a molecular approach in a whole-genome manner was performed to obtain detailed information about the genomic characteristics of the identified strain. The aim of our study was to gain a comprehensive understanding of future control measures in order to achieve the “one health approach” in our society.
Materials and methods
Bacterial strain isolation, carbapenemase gene detection and minimum inhibitory concentration determination
Swabbed samples obtained from rectal and nasal swabs of dogs and cats visiting an animal clinical hospital in Seoul, South Korea, were subjected to screening on meropenem-impregnated (1 μg/mL) MacConkey (MIM) agar for carbapenem-resistant strain identification. The total bacterial DNA was isolated using the Wizard Genomic DNA purification kit (Promega, Madison, WI, USA). For the identification of carbapenemase genes, PCR screening using previously described primers [14] was initially performed, and the confirmed strains were subjected to minimum inhibitory concentration (MIC) level identification and next-generation sequencing (NGS). The identified microbial species were determined with matrix-assisted laser desorption ionization–time of flight-mass spectrometry (MALDI-TOF-MS; Bruker Daltonik GmbH, Bremen, Germany).
The MICs of the isolates were determined for 14 antimicrobial agents using E-test (Biomerieux, Marcy L’Étoile, France) technique in accordance with the manufacturer’s instructions. E. coli strain ATCC 25922 was used as a quality control strain and only drugs with quality standards were used in the test.
Illumina/MinION sequencing and characterization
For whole genome sequencing, purified DNA acquired from a Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) was used. In summary, two independent genomic DNA libraries were prepared for both short and long read systems and sequenced. Long read genomic sequencing with Oxford Nanopore (Oxford Nanopore Technologies, Oxford, UK) platforms was corrected using Illumina NovaSeq 6000 (Illumina, San Diego, CA, USA) following a paired-end 2 × 150-bp protocol.
The DNA library was prepared according to the Illumina TruSeq DNA PCR-Free Library Prep protocol (Cat. 20015963). For sample library preparation, 2 μg of 550 bp inserts of high molecular weight genomic DNA was randomly sheared to yield DNA fragments using the Covaris S2 system. The fragments were blunt ended and phosphorylated, and a single “A” nucleotide was added to the 3’ ends of the fragments in preparation for ligation to an adapter that has a single-base “T” overhang. Adapter ligation at both ends of the genomic DNA fragment conferred different sequences at the 5’ and 3’ ends of each strand in the genomic fragment. The quality of the libraries was verified by capillary electrophoresis (Bioanalyzer, Agilent).
The ONT library was constructed by using a Ligation Sequencing Kit (SQK-LSK109). This library was sequenced with a Ligation Sequencing Kit (SQK-LSK109), a Flow Cell Priming Kit (EXP-FLP002), and a Flowcell (FLO-MIN106). All runs were performed on the MinION sequencer.
After quantification using the QX200 Droplet Digital PCR System (Bio-Rad), we combined libraries that were index tagged in equimolar amounts in the pool. WGS sequencing was performed using the Illumina NovaSeq 6000 system according to the protocol provided for 2 × 150 sequencing.
Reads were trimmed and filtered for long and high-quality reads using FiltLong v0.2.0 [15]. Filtered long read data were prepared using Flye v2.8.3 to proceed de novo assembly [16] and checked to determine whether they were circular or linear for the contig produced as a result of assembly using Circlator v1.5.5 [17]. Data polishing was carried out with Pilon v1.23 [18] for the contigs whose structural shape was identified, and evaluation of the assembly results was carried out through BUSCO v4.1.2 [19]. After polishing, structural annotation was performed using Prokka v1.14.6 [20] for the contigs to determine the location, length and number of CDS, rRNA and tRNA. Functional annotation was performed with DIAMOND v 0.9.26 [21] to process the file, and then Blast2GO v4.1.9 [22] was used to perform Gene Ontology analysis.
Genetic structure analysis and bioinformatic comparison
For genomic structure visualization of the whole chromosome, CGView [23] was employed. Easyfig 2.2.3 was used for the pairwise BLASTn alignment of genomic structures, including characteristic transposons.
A total of 98 whole-genome datasets of P. mirabilis strains were downloaded from the National Center for Biotechnology Information [24] for comparison with the sequence in this study by bioinformatic manners. The assembled genomes were screened for comparison of the resistance genes and plasmid types on the Center for Genomic Epidemiology (CGE) server [25] in silico utilizing ResFinder 4.1 and PlasmidFinder 2.1. The whole-chromosome structure map was generated using the CGView server [23], with P. mirabilis HI4320 (GenBank accession no. AM942759.1) applied as a backbone. Whole-chromosome single nucleotide polymorphism (SNP) datsets were generated [26], and a concatenated alignment was created using CSI Phylogeny [27] with standard settings and using P. mirabilis HI4320 (GenBank accession no. AM942759.1) as a reference. MEGA 11 software [28] was employed for reliable maximum likelihood (ML) tree construction with 1000 bootstrap replicates. The epidemiological visualization was generated on iTOLs [29] for comparison of the characteristic information heatmaps along with the whole-genome phylogenetic tree.
This study describes the first carbapenemase-producing P. mirabilis strain isolated from a companion animal in South Korea. The identified whole-genome structure revealed 20 different AMR genes, including 2 tandem copies of blaNDM-1, which contributed to the MDR capacity of the isolate. Multiple variations in MDR gene regions and phylogenetic relatedness were identified by whole-genome analysis. The findings from the carbapenemase-producing P. mirabilis strain from a companion animal indicate that the problem of AMR is not limited to human health and should be addressed from the perspective of the “one health approach”.
Results
Characterization of carbapenemase-producing P. mirabilis and MIC determination
Proteus mirabilis strain LHPm1 was isolated on 9 April 2021 from a companion dog at a clinical animal hospital in Seoul, South Korea. The isolate was identified from a rectal swab of 6 year-old neutralized female poodle, without any characteristic clinical symptoms. Rectal swabs were performed along with nasal swabs for the purpose of screening and identifying carbapenem-resistant Enterobacterales (CRE). LHPm1 was isolated as a result of rectal swab screening on meropenem impregnated (1 µg/mL) MacConkey (MIM) agar.
As a result of PCR identification, the carbapenem-resistant isolate was found to carry the carbapenemase-producing gene blaNDM. Subsequently, the isolate underwent MIC evaluation and whole-genome sequencing. The MIC was evaluated against 14 different antimicrobial agents (Table 1) and was confirmed to have high (MIC value higher than 32 µg/mL) meropenem resistance. The isolate was found to have resistance against β-lactam agents, such as amoxicillin/clavulanic acid (penicillin), ceftazidime and ceftriaxone (3rd generation cephalosporin), ceftolozane/tazobactam (4th generation cephalosporin), and piperacillin/tazobactam (ureidopenicillin). Tetracycline, chloramphenicol, gentamicin, and polymyxins were also found to be unreliable options. Reliable options against LHPm1 included aztreonam (monobactam), amikacin (aminoglycoside), tigecycline (glycylcyclines), and nalidixic acid (quinolone).
Sequenced and assembled whole-genome datasets
As a result of the whole genome sequencing and assembly, a high-quality sequence was generated (Additional file 1). A 4000428 base-pair long chromosome was identified, without any identifiable plasmid (Table 2).
Whole-genome structure and characteristic gene visualization
The whole-genome structure was visualized (Figure 1) using CGView, with the P. mirabilis HI4320 (GenBank accession no. AM942759.1) sequence as a reference. A total of 20 different antimicrobial resistance (AMR) genes were identified from the whole genome of LHPm1. Genomic positions of characteristic genes, such as AMR genes, mobile genes and CDS, are depicted with distinguishable colored arrows in the whole chromosomal map.

Identification of the whole chromosome structure of LHPm1. The whole chromosome of NDM-1-carrying P. mirabilis (4 000 428 bp) was identified in this study and visualized. The chromosomal map of P. mirabilis HI4320 (4 063 606 bp, GenBank accession no. AM942759.1) was utilized as the backbone for visualization and is depicted as a black circle. The characteristic gene positions, such as that of resistance genes and mobile gene elements, are additionally highlighted in the outermost 2 circles. The circular map was generated by CGView.
Two genomic regions were identified with concentrated AMR genes and mobile genes. Comparative visualization (Figure 2) revealed the variated structure of the genomic regions, from previously reported datasets.

Genomic comparison of the identified novel variants in the MDR region. Multiple AMR genes and mobile genes were found to be concentrated in particular gene regions and visualized via linear comparison. The transposable elements of LHPm1 were identified and compared with the Tn6450 part discovered in P. mirabilis strain SNYG17 (GenBank accession no. MF805806.1) and the Tn6765 part in strain SCBX 1.1 (GenBank accession no. MT503200.1). Transposons and integron structures are indicated with black arrowheads. Two sites of blaNDM-1 were identified from LHPm1 in a variated form and comparatively visualized with corresponding structures identified from E. coli strain Y5 (GenBank accession no. CP013483.1) and P. mirabilis strain PMBJ023-2 (GenBank accession no. CP065145.1). Gray shading was adjusted to indicate corresponding shared regions with more than 96% gene identity. Easyfig 2.2.3 was used for the pairwise BLASTn alignment comparison.
A 50,640-base pair-long Tn7-like transposon structure with a transposition module of tnsA-tnsB-tnsC-△tnsD was identified, with novel variations (Figure 2A). The MDR gene region was found to harbor various classes of AMR genes, namely, aminoglycoside (aac(6’)-lb, aac(3)-IV, aph(4)-la, aph(3’)-la and aadA1), rifampin (arr-3), sulfonamide (sul1 and sul2), florfenicol and chloramphenicol (floR), macrolide (mphE and msrE), trimethoprim (dfrA1) and lincosamide (linF) AMR genes. The novel variation of the Tn7-like region was identified in comparison to Tn6450 (GenBank accession no. MF805806.1) and Tn6765 (GenBank accession no. MT503200.1). In particular, a class I integron was identified to be interrupted with a reversed gene region of Tn21, consequently leading to the deletion of the blaDHA-1 gene.
Two sites of the blaNDM-1 gene were identified in LHPm1 and associated with multiple mobile gene elements (Figure 2B). Both blaNDM-1 genes had base-pair substitution sites at C621A. A particular blaNDM-1 gene sequence contained an additional mutated 107T insertion and G108A substitution, speculatively resulting in a frame shift of the amino acid sequence and marked (*) in Figure 2B. The blaNDM-1 gene was coupled with mobile genes and multiple AMR genes, such as sul1, arr-3, aadA12 and dfrA27. The 41,315-base pair-long gene structure between the two sites of ISVsa5 was different from previously reported gene structures and was comparatively visualized with corresponding similar genes identified in E. coli strain Y5 (GenBank accession no. CP013483.1) and P. mirabilis strain PMBJ023-2 (GenBank accession no. CP065145.1). While the MDR regions, including sul1, arr-3, bleMBL and blaNDM-1, were similar, the outer structure was different among the strains.
Epidemiologic gene characterization in comparison with worldwide datasets
A total of 98 P. mirabilis whole-genome datasets available from the National Center for Biotechnology Information [24] were employed for whole-genome comparison (Additional file 2). The strains were isolated between 1933 and 2021 and from 20 different countries (66 strains from China). The strains were isolated from various sources, including 15 different host species (48 strains from humans) and environmental samples. Pathogenic bacterial strains identified from human clinical infections were also included in the study. As a result of whole genome database screening, a total of 80 heterogeneous AMR genes were identified from the whole-genome datasets of 99 P. mirabilis isolates (Additional file 3). PlasmidFinder 2.1 analysis revealed 3 types of plasmids, namely Col3M, IncC and IncQ1, identified in 6 P. mirabilis whole-genome datasets (Additional file 4). Of these, 5 strains carried Col3M (% identity 98.09) whereas P. mirabilis strain LB UEL H-11 (GenBank accession no. CP086377.1) carried IncC and IncQ1.
Whole chromosome SNPs were identified using the CSI Phylogeny pipeline, with P. mirabilis HI4320 (GenBank accession no. AM942759.1) as a reference gene for pairwise SNP difference measurement. The pairwise SNP difference (Additional file 5) ranged from 0 (between C74 and C55) to 45,353 (between MPE0156 and CCUG70746). The P. mirabilis strain XH1568 (GenBank accession no. CP049941.1) was revealed to have the smallest pairwise SNP difference (6302 positions) from LHPm1.
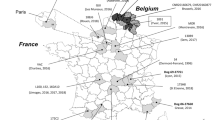
Subsequently, a whole chromosome SNP-based ML tree was constructed based on the pairwise SNP difference. To gain a clearer epidemiological perspective of LHPm1, epidemiological information, such as isolative sources, location, year and the AMR gene identity heatmap, was visualized with the whole chromosome phylogenetic tree (Figure 3). As a result of the whole genome epidemiology of the P. mirabilis datasets, 5 phylogroups were distinguishable based on phylogenetic relatedness, isolative source or resistance gene distribution pattern. The identified phylogroups are depicted in Figure 3 with colored backgrounds. LHPm1 was grouped in phylogroup C (green background), which is a clade that can be characterized by highly distributed AMR genes.

Epidemiological comparison of the whole genome datasets of 99 P. mirabilis strains. The whole-genome SNP identification using P. mirabilis HI4320 (GenBank accession no. AM942759.1) as a reference was adjusted to construct a maximum likelihood (ML) tree and the results were visualized by iTOLs. The LHPm1 strain identified in this study is highlighted with a black background. The years that each strain was isolated are listed in the square boxes and colored differently according to their location of origin. The isolation source, whether from humans, animals, or the environment, is displayed using colored boxes. The AMR gene identities were displayed by heatmaps, colored differently according to the antimicrobial classes. The identified phylogroups (A–E) are depicted in distinguishable colored backgrounds.
Discussion
In this study, carbapenemase NDM-1 carriage was revealed from the common rectal screen of companion animals, which are close members of human society yet outside of the major surveillance system of our community. Featuring high mortality and morbidity especially in nosocomial environments, most of the infections caused by MDR Enterobacterales are likely derived from the gastrointestinal (GI) tract [30, 31]. P. mirabilis is a commensal strain, and it has been suspected to be responsible for UTI infections originating from the GI tract [1, 2]. However, the host dog of the isolate in this study was healthy to the best of our knowledge, and showed no special symptoms or clinical situation. This is not strange because P. mirabilis is known as a commensal strain in the GI tract of humans and animals [1]. In human investigations of Korea, P. mirabilis was reported as the 5th most prevalent (6.5%) urine isolate and 5th most prevalent (2.8%) species among CREs [32,33,34]. Among carbapenemase-producing P. mirabilis isolates, the most prevalent carbapenemase type was NDM-1, consistent with the findings in this study. The NDM-1-producing P. mirabilis isolate in this study was the first strain detected in a dog in our country. In a nationwide surveillance study on dogs [35], P. mirabilis was reported as the 4th most isolated species from stools (4.4%), 5th from skin (3.7%), and 2nd from urine (20.0%). There has been no clear report supporting direct dissemination or infection of P. mirabilis between humans and animals. However, considering the increasing role of companion dogs in human society, the discovery of NDM-1 harboring P. mirabilis in the GI tract of a companion dog should be taken seriously.
Although LHPm1 was isolated from a dog without any identified clinical symptoms, the clinical potential of MDR P. mirabilis as a pathogen should also be considered. Several key virulence factors of P. mirabilis, including iron acquisition systems, lipopolysaccharide, hemolysins, proteases, and flagella, are known to confer pathogenicity to the isolates [36, 37]. Although it is still unclear, swarming capability on surface is suspected to be correlated with urease and crystalline biofilm formation [38]. Fimbriae formation is also known to be responsible for adhering to the uroepithelium, localizing in the urinary tract, and forming biofilms [39, 40]. There are 17 important virulence factors reported to affect fimbriae formation, including the well-known mannose-resistant Proteus-like (MR/P) fimbria [41]. The genomic variations identified in this study (Figure 2) did not include any differences of known virulence factors of P. mirabilis. Interestingly, pathogenic factors such as swarming motility, mutual growth, and biofilm formation were reported to be correlated with the AMR capacity of P. mirabilis [42]. MDR P. mirabilis strains exhibited higher mutual growth and biofilm formation but lower swarming motility. However, LHPm1 was identified in a dog without any clinical complaints, and the clinical potential of the strain was not assessed in this study. Therefore, potential clinical impact of the LHPm1 should not be neglected, and further investigation into its potential pathogenicity should be undertaken.
The result of whole-genome resistance gene identification using a public database was consistent with the MIC values. The MIC evaluation revealed resistance to multiple classes of antibiotics, including β-lactam agents such as cephalosporins, penicillins and carbapenems, aminoglycosides, chloramphenicol, macrolides and polymyxins. NDM-1 is a well-known carbapenemase, that confers resistance against β-lactam agents by hydrolyzing antibiotics containing β-lactam rings [43]. The resistance level of LHPm1 against meropenem, cephalosporins (ceftazidime and ceftriaxone), and penicillin (amoxicillin) seems to be due to the presence of NDM-1. Various aminoglycoside genes were identified in the chromosome of LHPm1, namely aac(3)-Iid, aac(3)-IV, aac(6’)-Ib-cr, aadA1, aadA16, aph(3’)-Ia, aph(3’’)-Ib, aph(3’)-Via, aph(4)-Ia and aph(6)-Id [44]. These genes are speculated to be responsible for the resistance of LHPm1 to gentamicin. The loss of mobile gene elements in the genomic region (Figure 2), such as ISCR1, the istAB operon, and IS1353, indicates that the AMR genes linked with these genes would be stable in the chromosome of the LHPm1 [45]. The variations in the genomic region could stabilize the AMR genes and drug resistance ability derived from this region, such as aminoglycosides, rifampicin, and sulfonamides, potentially making treatment using these drugs difficult. The resistance of the isolate to doxycycline seems to be due to the presence of the tet(J). The MIC determination also revealed the resistance of the isolate to chloramphenicol, which is believed to be due to the existence of the resistance genes catB3 and floR. Similarly, qnrA1 is suspected to be responsible for the resistance to nalidixic acid [46].
Tn7-like transposons contribute to the horizontal transfer of AMR genes among bacterial species via transposases [47]. However, the role of Tn7-like transposons in MDR isolates in our society remains largely unexplored. In this study, multiple genetic variations were discovered in LHPm1 and comparatively visualized with the Tn7-like structures Tn6450 (GenBank accession no. MF805806.1) and Tn6765 (GenBank accession no. MT503200.1).
The identification of MDR genes associated with multiple mobile genes from companion dogs is highly worrisome, considering the increasing role of animals in our society. Furthermore, MDR genes coupled with multiple mobile gene elements were identified with two tandem copies of blaNDM-1, which could confer an even broader spectrum of MDR capacity to the strain. Two tandem copies of blaNDM-1 have been reported from multiple species of bacterial chromosomes in previous reports [48,49,50], including P. mirabilis. Isolates with multiple copies of blaNDM-1 are known to show elevated carbapenem resistance [50]. In this study, 2 copies of blaNDM-1 were identified in P. mirabilis, yet one of them seems to have undergone a frame shift of the amino acid sequence. Although the other copy of blaNDM-1 seems to be enough to confer carbapenem resistance for the isolate (MIC value higher than 64 µg/mL), the underlying reason for the identified mutation remains unknown.
The whole-genome epidemiological study has reconfirmed that P. mirabilis is a ubiquitous bacterial species that can be isolated from humans, animals (both wildlife and domestic) and the environment. The whole-genome study revealed various classes of AMR genes in P. mirabilis datasets. Tetracycline resistance genes, mostly tet(J), were identified in all of the investigated P. mirabilis strains. Among the datasets, the largest number of AMR genes was identified in FZP3105, with a total of 28 different AMR genes. Although only whole-genome datasets of significant isolates tend to be accessible in GenBank, it is clear that P. mirabilis is capable of carrying various AMR genes. In the whole genome phylogeny, 5 phylogroups were identified. Interestingly, isolates grouped in an identical phylogroup had a similar resistance gene distribution pattern. Isolates of phylogroup A originated from various animal species in China and carried either blaNDM-1 or blaOXA-1. Phylogroup B, C and E were characterized by a high distribution of aminoglycoside, amphenicol, and folate pathway antagonist resistance genes, relative to other phylogroups. Phylogroup D was identified with extended-spectrum β-lactamase (ESBL) and carbapenemase genes, yet other AMR genes, such as aminoglycoside, macrolide and quinolone resistance genes were relatively less prevalent. LHPm1 was included in phylogroup C, which is capable of colonizing both humans and animals, and was found to carry various AMR genes.
In phylogroup C, four P. mirabilis strains were closely grouped with LHPm1, raising concerns about serious clinical situations for humans. The four isolates of phylogroup C included two strains isolated from humans and the other two from animals. P. mirabilis strain XH1568 was identified from a gallbladder sample collected in China [51], from a patient diagnosed with gallbladder carcinoma. An ESBL blaCTX-M-65 producing isolate FZP2826 was also isolated in human patient [52]. P. mirabilis strain CC15031 was isolated from a canine diarrhea sample [53] and characterized to demonstrate high pathogenicity and antimicrobial resistance. An MDR P. mirabilis strain ChSC1905 was from nasal swab of a diseased pig of swine farm [54]. Given the host range of phylogroup C and its proximity to LHPm1, the potential for upcoming human infections must be taken seriously.
Therefore, novel control measures, from the perspective of the “one health approach”, are needed for public health, based on the findings from the whole-genome epidemiology.
This study describes the first carbapenemase-producing P. mirabilis strain isolated from a companion animal in South Korea. The identified whole-genome structure revealed 20 different AMR genes, including 2 tandem copies of blaNDM-1, which contributed to the MDR capacity of the isolate. Multiple variations in MDR gene regions and phylogenetic relatedness were identified by whole-genome analysis. The findings from the carbapenemase-producing P. mirabilis strain from a companion animal indicate that the problem of AMR is not limited to human health and should be addressed from the perspective of the “one health approach”.
Availability of data and materials
Publicly available datasets were analyzed in this study. This data can be found in Additional file 2 for all accession numbers.
Change history
29 April 2024
A Correction to this paper has been published: https://doi.org/10.1186/s13567-024-01317-7
Abbreviations
- AMR:
-
Antimicrobial resistance
- CGE:
-
Center for Genomic Epidemiology
- CLSI:
-
Clinical and laboratory standards institute
- CRE:
-
carbapenem-resistant enterobacterales
- ESBL:
-
extended-spectrum β-lactamase
- GI:
-
gastrointestinal
- ICEs:
-
integrative conjugative elements
- MALDI-TOF–MS:
-
matrix-assisted laser desorption ionization–time of flight-mass spectrometry
- MDR:
-
multi-drug resistance
- MIC:
-
minimum inhibitory concentration
- MIM:
-
meropenem impregnated MacConkey
- ML:
-
maximum likelihood
- NDM-1:
-
New Delhi metallo-β-lactamase 1
- NGS:
-
next generation sequencing
- SNP:
-
single nucleotide polymorphism
- TMP-SMX:
-
trimethoprim-sulfamethoxazole
- UTIs:
-
urinary tract infections
References
O’Hara CM, Brenner FW, Miller JM (2000) Classification, identification, and clinical significance of Proteus, Providencia, and Morganella. Clin Microbiol Rev 13:534–546
Mathur S, Sabbuba N, Suller M, Stickler D, Feneley R (2005) Genotyping of urinary and fecal Proteus mirabilis isolates from individuals with long-term urinary catheters. Eur J Clin Microbiol Infect Dis 24:643–644
Schaffer JN, Pearson MM (2015) Proteus mirabilis and urinary tract infections. Microbiol Spectr 3:10.1128/microbiolspec.uti-0017-2013
Kanzari L, Ferjani S, Saidani M, Hamzaoui Z, Jendoubi A, Harbaoui S, Ferjani A, Rehaiem A, Boubaker IBB, Slim A (2018) First report of extensively-drug-resistant Proteus mirabilis isolate carrying plasmid-mediated blaNDM-1 in a Tunisian intensive care unit. Int J Antimicrob Agents 52:906–909
Yong D, Toleman MA, Giske CG, Cho HS, Sundman K, Lee K, Walsh TR (2009) Characterization of a new metallo-β-lactamase gene, blaNDM-1, and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agent Chemother 53:5046–54
Walsh TR, Weeks J, Livermore DM, Toleman MA (2011) Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: an environmental point prevalence study. Lancet Infect Dis 11:355–362
Dortet L, Poirel L, Nordmann P (2014) Worldwide dissemination of the NDM-type carbapenemases in Gram-negative bacteria. Biomed Res Int 2014:249856
Deshpande LM, Rhomberg PR, Sader HS, Jones RN (2006) Emergence of serine carbapenemases (KPC and SME) among clinical strains of Enterobacteriaceae isolated in the United States medical centers: report from the MYSTIC Program (1999–2005). Diagn Microbiol Infect Dis 56:367–372
Wu W, Feng Y, Tang G, Qiao F, McNally A, Zong Z (2019) NDM metallo-β-lactamases and their bacterial producers in health care settings. Clin Microbiol Rev 32:e00115-18
Dong D, Li M, Liu Z, Feng J, Jia N, Zhao H, Zhao B, Zhou T, Zhang X, Tong Y (2019) Characterization of a NDM-1-encoding plasmid pHFK418-NDM from a clinical Proteus mirabilis isolate harboring two novel transposons, Tn6624 and Tn6625. Front Microbiol 10:2030
Kong L-H, Xiang R, Wang Y-L, Wu S-K, Lei C-W, Kang Z-Z, Chen Y-P, Ye X-L, Lai Y, Wang H-N (2020) Integration of the blaNDM-1 carbapenemase gene into a novel SXT/R391 integrative and conjugative element in Proteus vulgaris. J Antimicrob Chemother 75:1439–1442
Shaheen BW, Nayak R, Boothe DM (2013) Emergence of a New Delhi metallo-β-lactamase (NDM-1)-encoding gene in clinical Escherichia coli isolates recovered from companion animals in the United States. Antimicrob Agent Chemother 57:2902–2903
Stolle I, Prenger-Berninghoff E, Stamm I, Scheufen S, Hassdenteufel E, Guenther S, Bethe A, Pfeifer Y, Ewers C (2013) Emergence of OXA-48 carbapenemase-producing Escherichia coli and Klebsiella pneumoniae in dogs. J Antimicrob Chemother 68:2802–2808
Poirel L, Walsh TR, Cuvillier V, Nordmann P (2011) Multiplex PCR for detection of acquired carbapenemase genes. Diagn Microbiol Infect Dis 70:119–123
Wick R, Menzel P (2019)
Kolmogorov M, Yuan J, Lin Y, Pevzner PA (2019) Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol 37:540–546
Hunt M, Silva ND, Otto TD, Parkhill J, Keane JA, Harris SR (2015) Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol 16:294
Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069
Buchfink B, Reuter K, Drost H-G (2021) Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Method 18:366–368
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Stothard P, Wishart DS (2005) Circular genome visualization and exploration using CGView. Bioinformatics 21:537–539
O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44:D733-45
Center for Genomic Epidemiology (CGE) server (2011)
Kaas RS, Leekitcharoenphon P, Aarestrup FM, Lund O (2014) Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS One 9:e104984
Price MN, Dehal PS, Arkin AP (2010) FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490
Tamura K, Stecher G, Kumar S (2021) MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38:3022–3027
Letunic I, Bork P (2021) Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucl Acids Res 49:W293-96
Patel G, Huprikar S, Factor SH, Jenkins SG, Calfee DP (2008) Outcomes of carbapenem-resistant Klebsiella pneumoniae infection and the impact of antimicrobial and adjunctive therapies. Infect Control Hosp Epidemiol 29:1099–1106
Yamamoto M, Pop-Vicas AE (2014) Treatment for infections with carbapenem-resistant Enterobacteriaceae: what options do we still have? Crit Care 18:229
Park S-H, Kim J-S, Kim H-S, Yu J-K, Han S-H, Kang M-J, Hong C-K, Lee S-M, Oh Y-H (2020) Prevalence of carbapenem-resistant Enterobacteriaceae in Seoul, Korea. J Bacteriol Res 50:107–116
Park S-H, Park S-H, Kim J-S, Yu J-K, Kim J-K, Suh H-S, Kwon EY, Park KA, Cha EK, Shin JM (2022) Genetic distribution of carbapenem-resistant Enterobacteriaceae in Seoul Korea, 2018–2020. J Bacteriol Virol 52:28–38
Kwak B, Hong J, Bae HG, Park YS, Lee MK, Lee K, Lee KR (2022) Microorganisms isolated from urine cultures and their antimicrobial susceptibility patterns at a commercial laboratory during 2018–2020. Korean J HealthcAssoc Infect Control Prev 27:51–58
Moon D-C, Choi J-H, Boby N, Kang H-Y, Kim S-J, Song H-J, Park H-S, Gil M-C, Yoon S-S, Lim S-K (2022) Bacterial prevalence in skin, urine, diarrheal stool, and respiratory samples from dogs. Microorganisms 10:1668
Rózalski A, Sidorczyk Z, Kotełko K (1997) Potential virulence factors of Proteus bacilli. Microbiol Mol Biol Rev 61:65–89
Cestari SE, Ludovico MS, Martins FH, da Rocha SPD, Elias WP, Pelayo JS (2013) Molecular detection of HpmA and HlyA hemolysin of uropathogenic Proteus mirabilis. Curr Microbiol 67:703–707
Norsworthy AN, Pearson MM (2017) From catheter to kidney stone: the uropathogenic lifestyle of Proteus mirabilis. Trend Microbiol 25:304–315
Manos J, Belas R (2006) The genera Proteus, Providencia, and Morganella. Prokaryotes 6:245–269
Jacobsen SM, Shirtliff ME (2011) Proteus mirabilis biofilms and catheter-associated urinary tract infections. Virulence 2:460–465
Armbruster CE, Mobley HL, Pearson MM (2018) Pathogenesis of Proteus mirabilis infection. EcoSal Plus. https://doi.org/10.1128/ecosalplus.ESP-0009-2017
Filipiak A, Chrapek M, Literacka E, Wawszczak M, Głuszek S, Majchrzak M, Wróbel G, Łysek-Gładysińska M, Gniadkowski M, Adamus-Białek W (2020) Pathogenic factors correlate with antimicrobial resistance among clinical Proteus mirabilis strains. Front Microbiol 11:579389
Nordmann P, Naas T, Poirel L (2011) Global spread of carbapenemase-producing Enterobacteriaceae. Emerg Infect Dis 17:1791–1798
Shaw K, Rather P, Hare R, Miller G (1993) Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol Rev 57:138–163
Chen Y-P, Lei C-W, Kong L-H, Zeng J-X, Zhang X-Z, Liu B-H, Li Y, Xiang R, Wang Y-X, Chen D-Y (2018) Tn6450, a novel multidrug resistance transposon characterized in a Proteus mirabilis isolate from chicken in China. Antimicrob Agents Chemother 62:02192–17
Poirel L, Leviandier C, Nordmann P (2006) Prevalence and genetic analysis of plasmid-mediated quinolone resistance determinants QnrA and QnrS in Enterobacteriaceae isolates from a French university hospital. Antimicrob Agent Chemother 50:3992–3997
He J, Li C, Cui P, Wang H (2020) Detection of Tn7-like transposons and antibiotic resistance in Enterobacterales from animals used for food production with identification of three novel transposons Tn6813, Tn6814, and Tn6765. Front Microbiol 11:2049
Jovčić B, Lepsanović Z, Begović J, Rakonjac B, Perovanović J, Topisirović L, Kojić M (2013) The clinical isolate Pseudomonas aeruginosa MMA83 carries two copies of the blaNDM-1 gene in a novel genetic context. Antimicrob Agent Chemother 57:3405–3407
Shen P, Yi M, Fu Y, Ruan Z, Du X, Yu Y, Xie X (2017) Detection of an Escherichia coli sequence type 167 strain with two tandem copies of blaNDM-1 in the chromosome. J Clin Microbiol 55:199–205
Yang L, He H, Chen Q, Wang K, Lin Y, Li P, Li J, Liu X, Jia L, Song H (2022) Nosocomial outbreak of carbapenemase-producing Proteus mirabilis with two novel Salmonella genomic island 1 variants carrying different blaNDM–1 gene copies in China. Front Microbiol 12:800938
Hua X, Zhang L, Moran RA, Xu Q, Sun L, Van Schaik W, Yu Y (2020) Cointegration as a mechanism for the evolution of a KPC-producing multidrug resistance plasmid in Proteus mirabilis. Emerg Microb Infect 9:1206–1218
Li Y, Liu Q, Qiu Y, Fang C, Zhou Y, She J, Chen H, Dai X, Zhang L (2022) Genomic characteristics of clinical multidrug-resistant Proteus isolates from a tertiary care hospital in southwest China. Front Microbiol 13:977356
Hu R, Wang X, Muhamamd I, Wang Y, Dong W, Zhang H, Wang Y, Liu S, Gao Y, Kong L (2020) Biological characteristics and genetic analysis of a highly pathogenic Proteus mirabilis strain isolated from dogs in China. Front Vet Sci 7:589
Song Z, Lei C-W, Zuo L, Li C, Wang Y-L, Tian Y-M, Wang H-N (2022) Whole genome sequence of Proteus mirabilis ChSC1905 strain harbouring a new SXT/R391-family ICE. J Glob Antimicrob Resist 30:279–281
Acknowledgements
The authors would like to thank veterinarians working in animal hospitals in Seoul, Republic of Korea, for collecting samples used in this study. We also express our appreciation to the Research Institute for Veterinary Science, Seoul National University, Seoul, Republic of Korea, for their support of this research.
Funding
This study was supported by National Research Foundation (NRF- 2020R1A2C200879414), BK21 FOUR Future Veterinary Medicine Leading Education and Research Center and Research Institute for Veterinary Science, Seoul National University, Seoul, Republic of Korea.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: SMK and HSY. Performed the experiments: SMK, XX, and JHL. Analyzed the data: SMK and EL. Wrote the paper: SMK. Reviewed and edited the paper: HSY. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Handling editor: Marcelo Gottschalk
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: the authors informed us about a spelling in the title.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kyung, S.M., Lee, J.H., Lee, ES. et al. Emergence and genomic characterization of Proteus mirabilis harboring blaNDM-1 in Korean companion dogs. Vet Res 55, 50 (2024). https://doi.org/10.1186/s13567-024-01306-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13567-024-01306-w