
Abstract
Sex-related differences in the occurrence of autoimmune diseases is well documented, with females showing a greater propensity to develop these diseases than their male counterparts. Sex hormones, namely dihydrotestosterone and estrogens, have been shown to ameliorate the severity of inflammatory diseases. Immunologically, the beneficial effects of sex hormones have been ascribed to the suppression of effector lymphocyte responses accompanied by immune deviation from pro-inflammatory to anti-inflammatory cytokine production. In this review, we present our view of the mechanisms of sex hormones that contribute to their ability to suppress autoimmune responses with an emphasis on the pathogenesis of experimental autoimmune encephalomyelitis.
Similar content being viewed by others


Introduction
The normal function of the immune system is to protect organisms against invading pathogens. When such a response is directed against self-tissues, autoimmunity may ensue. However, healthy individuals can have signatures of autoimmune response as evidenced by the detection of low levels of antibodies and T cells against autoantigens that may reflect formation of natural antibodies or idiotypic networks [1,2,3,4]. Autoimmune diseases (AIDs) are clinically manifested when autoimmunity leads to tissue damage disrupting the functions of affected organs [5, 6].
AIDs are generally noted to be the leading causes of deaths in young to middle-aged women in the USA [7]. Estimates indicate a large variation in both the incidence (less than 1 per 100,000 persons to more than 20 per 100,000) and prevalence (less than 5 per 100,000 to more than 500 per 100,000) of these diseases [8]. Approximately 50 million Americans may have some form of an autoimmune disease and of these, more than 75% are women [7]. The chronic nature of many of these diseases such as multiple sclerosis (MS) can significantly impact medical costs and quality of life [8].
MS is a chronic inflammatory and demyelinating disease of the central nervous system (CNS), and it affects approximately 2.5 million people worldwide showing a female preponderance (2 to 3:1). Within the USA alone, MS affects approximately 400,000 people with 10,000 new cases diagnosed annually [9,10,11] resulting in the loss of ~ 2.5 billion to the economy [12, 13]. While, the disease can be seen in people of any age, it is commonly diagnosed in the age group of third to fifth decades. Although, no known causes are identified, it is commonly believed that a combination of genetic susceptibility and environmental factors trigger the disease-onset [9, 11]. Traditionally, four types of MS have been identified. These include relapsing-remitting MS (RRMS), secondary progressive MS, primary progressive MS, and progressive-relapsing MS (PRMS) [14], with RRMS being the most common (~ 85%) and PRMS the rarest of all (~ 5%) [11]. A recent classification emphasizes combination of active or inactive, and/or stable or progressive nature of the disease course [15]. The pathological diversity of lesions in the white and grey matter with differential mechanistic signatures provides an additional layer to the variable clinical phenotypes [16, 17]. Given this complex nature, it is a challenge to study the pathogenetic events in humans, and therefore, various animal models of experimental autoimmune encephalomyelitis (EAE) are routinely used in MS research.
EAE can be induced in a wide-range of species (rodents: rabbits, rats, and mice; and non-rodents: monkeys and pigs) [14, 18,19,20,21,22]. The two hallmarks of EAE are inflammation and demyelination, and the disease is typically mediated by autoreactive T cells [23, 24]. While EAE-induction by active immunization involves the use of myelin antigens or their immunogenic peptides in complete Freund’s adjuvant (CFA), the disease can be transferred to naïve animals by adoptively transferring myelin-reactive T cells. Three main myelin antigens have been identified to induce EAE, namely myelin basic protein (MBP), proteolipid protein (PLP), and myelin oligodendrocyte glycoprotein (MOG), and their disease-inducing peptides are also identified. These include MBP 1-11 that induces EAE in B10.PL or PL/J mice (H-2u); PLP 139-151-induced EAE in SJL mice (H-2s) and MOG 35-55-induced EAE in C57BL/6 mice (H-2b) [14, 25]. Of these models, sex differences have been well noted with the PLP 139-151-induced EAE in SJL mice. In this model, while females show chronic relapsing-remitting paralysis, the disease-course is restricted to the monophasic form in male mice [26]. These phenotypes resemble some of the clinical features of MS making the SJL model of EAE to be helpful for studying sex differences in the CNS autoimmunity [26]. Here, we review the salient features of sexual dimorphism of AIDs with an emphasis on the role of T cells in the pathogenesis of EAE.
Sexual dimorphism in the occurrence of infectious diseases vs. AIDs
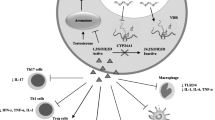
It has been known for a long time that susceptibility to various diseases differs by sex. While males are more susceptible than females to viral, bacterial, and parasitic infections, the tendency to develop autoimmune diseases is higher in females than males [27] (Fig. 1).

Sexual dimorphism with the disease occurrence, and its underlying potential immune mechanisms. It is generally believed that males are more prone to infectious diseases than females, but the latter group have a preponderance to develop autoimmune diseases. These phenotypes are shown with elbow arrows (favorable), and arrows with inhibitory lines (unfavorable). The hormonal environments in females (estrogens) and males (androgens) have been shown to influence both innate and adaptive immune cell functions. Additionally, hormonal actions on immune cells in the respective sexes can potentially be influenced by transcriptome profiles in the sex chromosomes and epigenetic modifications. Nonetheless, genetic susceptibility and exposure to environmental microbes, including alterations in the gut microbiota, if any are still the key players to trigger AIDs, but their outcomes can be modulated by sex hormones
Infectious diseases
Females are generally more resistant than males to viral infections due to the higher antibody production [28], especially during the period between puberty and menopause [27], but the conflicting reports may question this notion. While males appear to contract certain viral infections at a higher rate—such as human immunodeficiency virus, west Nile virus, hepatitis B virus, influenza virus, and Hantavirus [28, 29]—females with the same viral load as males can be at a higher risk of developing acquired immune deficiency syndrome [30]. Similarly, during the 2009 H1N1 avian influenza pandemic in Canada, women were found to be at two- to six-fold higher risk of dying than men [31]. Conversely, emerging evidence suggests that mortalities are more common in males than female individuals affected with coronavirus disease-19 that can be ascribed to other confounding factors such as smoking and behavioral changes [32,33,34]. Generally, women are known to mount higher anti-viral immune responses than men which may be beneficial to clear the virus, but prolongation of such a response can lead to increased disease-severity [31, 35]. For bacterial infections however, males were found more susceptible than females to Mycobacterium tuberculosis (M.tb), Helicobacter pylori, Coxiella burnetii, Pseudomonas aeruginosa, and Salmonella typhimurium infections [36,37,38,39,40]. Additionally, the proportion of adult males found to have symptomatic M.tb infections was two-fold higher than in females [36]. Conversely, women are more likely than men to survive from sepsis [41]. Females have a lower incidence of malaria than males [42] and experimentally, female mice also were found to be more resistant than males to Plasmodium chabaudi infection [43]. These data suggest that sex differences may vary from disease to disease of infectious origin.
AIDs
It is well conceived that most autoimmune diseases are more prevalent in females than males [44, 45]. This phenomenon has been well documented especially with AIDs mediated by autoantibodies such as Sjögren’s syndrome (female to male ratio of 16:1), systemic lupus erythematosus (SLE) (7:1), Hashimoto’s thyroiditis (19:1), and Grave’s disease (7:1), in which, about 80% of the patient population was female [46]. In the middle tier of diseases, which includes rheumatoid arthritis (RA) (3:1) and MS (2:1), the sex distribution has been 60–75% in women relative to men [46]. In fact, a study involving Danish cohorts revealed the risk for developing MS was increased more than two-fold in females, whereas in males, the disease remained unchanged over a period of 25 years [47]. Likewise, neuromyelitis optica spectrum disorder (NMOSD) is also characterized by a high female predominance and the disease-outcomes can also be influenced by the sex [48]. Interestingly, this difference is much higher in NMOSD associated with AQP4-antibodies, and less in seronegative NMOSD without pathogenic autoantibodies [49, 50]. However, for other diseases such as inflammatory bowel disease and type 1 diabetes (TID), the prevalence rates are similar for both sexes [51]. Conversely, Guillain-Barre syndrome appears to be occurring at equal or higher rates in males than females [51], whereas myasthenia gravis shows a female predominance in the early-onset as opposed to a male predominance in the late onset of the disease [52]. Likewise, myocarditis is more frequently reported in young men than their female counterparts [53]. Of note, male patients with later onset MS have a higher risk for faster disability progression suggesting that sex-differences may also be seen in the disease course [54].
Furthermore, occurrence of AIDs appears to be influenced by the reproductive cycles in affected individuals. For example, pre-pubertal cases of MS are extremely rare, with only 3–5% cases reported in individuals younger than 18 years of age. The finding that sexual dimorphism is seen mostly in post-pubertal women suggests that puberty is a critical risk factor [55]. For example, the female-to-male ratio for SLE is found to be 2–6:1 prior to puberty (9–14 years for boys and 8–13 years for girls), as opposed to 9:1 after puberty (≥ 15 years for boys and ≥ 14 years for girls) [56]. Additionally, disease severity can be influenced by pregnancy, as shown with MS, where the clinical signs of the disease are suppressed during pregnancy, especially during the third trimester. However, the risk of MS relapse is increased in the first 3 months of post-partum and returns to the pre-pregnancy level by 6 months after delivery [57, 58]. In the case of RA however, symptoms can be low or completely suppressed during gestation, whereas women with SLE often have exacerbated symptoms during pregnancy [56]. While, these observations point to a possibility that the sex hormones may determine the clinical outcomes of AIDs, primary triggers of these diseases remain largely unknown.
Factors that influence the development of AIDs
Two major factors have been implicated in the induction of AIDs. These include genetic susceptibility and exposure to environmental factors and the readers may find excellent reviews on these topics elsewhere [59, 60]. Furthermore, transcriptome profiles of sex chromosomes, specifically X, and epigenetic variations also appear to influence the occurrence of autoimmunity (Fig. 1). One such transcript is KDM6a where the animals deficient for this gene were found resistant for the development of EAE [61]. Other potential candidates include Forkhead box P3 (FoxP3) and Toll like receptor (TLR) 7 [62]. Likewise, epigenetic modifications (DNA methylation, histone modifications, chromatin remodeling, and non-coding RNAs) at major histocompatibility complex (MHC) loci may influence sex differences in MS [51, 63] (Fig. 1). Additionally, polymorphisms in the interferon (IFN)-γ and interleukin (IL)-12 receptor β genes were noted with sex differences in susceptibility to MS [64, 65]. Deficiency of the Fas/CD95 death receptor was associated with decreased apoptosis of inflammatory cells in the CNS with enhanced EAE severity. Such an association was also seen in women with MS [66], suggesting that the cellular responses might be different between sexes.
Additionally, it has been recently shown that the sex differences in autoimmunity can be influenced by the gut microbiota (Fig. 1). For example, specific pathogen-free non-obese diabetic (NOD) mice show a female preponderance to develop TID, but the germ-free mice lose such a bias [67]. Furthermore, gut flora differ between sexes, a trend reversed by male castration suggesting that androgens can influence the gut microbiota [67]. Likewise, colonization by commensal microbes led to elevated serum testosterone levels and protection of male NOD mice from developing TID [68]. Importantly, transfer of gut microbes from adult males to immature females altered the microbiota in females leading to reduced islet inflammation and autoantibody production and protection from TID occurring in conjunction with increased testosterone levels [68]. These data suggest that the gut microbiota can be an important determinant of the outcomes of sexual dimorphic nature of autoimmune diseases in those affected. In support of this preposition, microbiota composition revealed diverse microbial populations in association with chronic-progressive and chronic relapsing-remitting type of paralysis as evaluated in two mouse strains namely, C57Bl/6 and SJL mice [69]. However, existence of sex-specific altered microbiota, if any that can potentially contribute to the sex bias in EAE phenotypes, needs further investigations. Taken together, the data indicate that the immune microenvironments in males and females might be uniquely influenced by sex hormones.
Immune mechanisms of sex hormones
Expression of sex steroid receptors in immune cells
Physiologically, estrogens are responsible for female sexual characteristics, similar to androgens in males [70]. Estrogens include estrogen (E1), estradiol (E2), and estriol (E3), of which E3 is produced only during pregnancy [71]. Their effects are mediated through estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ) through the formation of homodimers or heterodimers. ERα has been detected in dendritic cells (DCs), monocytes, macrophages, natural killer (NK) cells, mast cells, B cells, and T cells [72,73,74,75,76,77]. Even though CD4 T cells express more ERα than ERβ, CD8 T cells and monocytes express low amounts of both ERs. On the contrary, B cells express higher amounts of ERβ than ERα [78].
Androgens mediate their effects predominantly by binding to androgen receptors (AR) located intracellularly [79], but they also can be expressed in a non-classic form on the cell surface [80]. Several immune cells like neutrophils, macrophages, B cells, and T cells have been shown to express AR [79, 81]. In thymic T cells, only classic AR has been detected, whereas both forms have been noted in the splenic T cells [82]. Likewise, while both macrophages and B cells can express classic AR, non-classic AR is expressed only in macrophages [83]. Since most terminally differentiated immune cells express sex hormone receptors, their functionalities can be potentially modulated by sex hormones.
Effect of sex hormones on innate immune cells
Several reports indicate significant differences in the innate immune responses between sexes (Fig. 1). For example, healthy female macaques have increased counts of most leukocyte subpopulations in their peripheral blood than their male counterparts [84]. Similarly, healthy female mice have higher numbers of leukocytes in the pleural and peritoneal cavities than do male mice [85]. Circulating NK T cells can also be more numerous in healthy women than men [86]. Male healthy mice, however, appear to have more neutrophils than do females [87]. Such variations also have been noted in the ability to respond to microbial products. For example, in the airway inflammation model of asthma, greater numbers of macrophages and DCs were found to migrate from lungs to the draining lymph nodes in females as compared to males [88]. Human monocytes from males after lipopolysaccharide (LPS) stimulation can produce more of IL-1β, tumor necrosis factor (TNF)-α, and IL-12 than those from females [89]. Similarly, compared to female neutrophils, male neutrophils release greater amounts of TNF-α in response to LPS stimulation. This hyper-responsiveness of male neutrophils to LPS has been suggested as a potential mechanism in making males more susceptible than females to sepsis [90]. Furthermore, higher levels of TLR 7 detected in females compared to males can have implications in their ability to respond to virus infections, because TLR-7 is involved in the recognition of single-stranded viral RNA molecules [91].
Effects of sex hormones on antigen-presenting cells
Most antigen-presenting cells express both ERα and ERβ [74, 92]. Estrogens can regulate the functions of monocytes/macrophages and DCs in various ways (Fig. 1). For example, E2 inhibits expression of IL-1, IL-6, and TNF-α in activated macrophages [93]. DCs pretreated with E2 can suppress antigen-presenting functions by enhancing their ability to produce the anti-inflammatory cytokines IL-4 and IL-10 [94]. However, it also has been reported that E2, acting via ERα, can promote differentiation of DCs [92]; the E2-treated DCs have superior antigen-presenting function with increased major histocompatibility complex (MHC) class II expression [95]. Similar effects also were noted with testosterone-treated macrophages [96]. Although male mice appear to have lower numbers of Langerhans cells (LC) than female mice, androgens can influence DC development [97]. Topical application of testosterone or its metabolite dihydrotestosterone (DHT) can result in a significant decrease in the density of LCs in both normal females and orchiectomized males [98]. However, DHT appears not to promote granulocyte macrophage colony-stimulating factor-driven DC differentiation [92]. Furthermore, estrogen or progesterone can activate macrophages and promote wound healing through angiogenesis and tissue remodeling [99]. Androgens also can modulate inflammatory responses during acute wound healing, as evidenced by the observation that castration or blockage of androgens can result in suppressed recruitment of macrophages [100, 101], as well as the experimental observation that AR-deficient mice show accelerated wound healing [79] (Fig. 1). These observations suggest that the innate immune functions can be modulated by estrogens or androgens similarly.
Effect of sex hormones on adaptive immune cells
Adaptive immune responses are mediated by B cells and T cells. While some of the common lymphoid progenitors originated in the bone marrow can be educated within bone marrow to become B cells, some progenitors go to thymus and mature to become CD4 or CD8 T cells. T cells and B cells recognize self-antigens in the corresponding primary lymphoid organs. While strong recognition of self-antigens leads to the death of immature lymphocytes by negative selection, weak recognition favors positive selection of developing lymphocytes, indicating that the lymphocytes present in the peripheral repertoires must have seen the self-antigens. Conversely, if the self-antigens are not expressed in the generative lymphoid organs, then the developing lymphocytes can escape central tolerance. This has been clearly demonstrated in the case of PLP 139-151 as the naïve repertoire of SJL mice contain a significant proportion of PLP 139-151-reactive T cells [102]. Mechanistically, this phenomenon has been ascribed to the thymic expression of truncated form of PLP, called DM-20 isoform that contains a deletion in the coding region, representing the motif, PLP 139-151 [102,103,104]. Furthermore, in addition to repressive effects on lymphopoiesis, estrogens and testosterone can directly modulate the expression of autoimmune regulator (AIRE) protein that has a pivotal role in the thymic expression of self-antigens [105]. While estrogen suppresses AIRE via epigenetic changes [106, 107], androgens promote AIRE’s expression, an effect that can be abolished by castration [106, 108]. Whether enhanced expression of AIRE in the male thymus can be directly related to their low susceptibility to autoimmune diseases needs further clarifications.
Additionally, sex hormones have been shown to modulate lymphocyte development (Fig. 1). AR can inhibit T cell development in the thymus, as castrated animals exhibit thymic enlargement and increased numbers of lymphocytes that can be reversed by androgen-replacement therapy [83, 109, 110]. E2 has been shown to decrease B cell lymphopoiesis, since pregnancy levels of estrogens have been correlated with both a significant reduction in B cell numbers and activity of B lymphocyte precursors in the bone marrow [111]. Experimentally, formation of B cells was reduced in the bone marrow of mice treated with E2, while castration or ovariectomy led to increase in B lymphopoiesis ER-dependently [112, 113]. In addition, E2 can dampen B cell receptor (BCR) signals and favor the generation of marginal zone B cells and survival of autoreactive B cells [114, 115]. Similar suppressive effects were noted with androgen on B cell development. Assessment of B cell progenitors in the bone marrow of castrated mice revealed a dramatic increase in late pro-B cell levels, leading to an increase in the numbers of peripheral B cells, but to a lesser degree in pre-B and immature B cell populations [116, 117]. Estrogens can block T cell development and cause thymic atrophy in an ERα-dependent manner [118].
As to the peripheral repertoires, both human and macaque females appear to possess a higher number of circulating CD4 T cells, including CD4/CD8 ratios, than males [89, 119]. Likewise, human peripheral blood CD4 T cells from females produce relatively higher levels of the T-helper (Th) 1 cytokine, IFN-γ, than from males [120]. As to MS, although autoantibodies contribute to the disease pathogenesis, no sex-specific variations have been noted with antibodies in affected individuals. However, the peripheral repertoires of female humans and non-human primates can contain a relatively high proportion of activated B cells [84, 121], suggesting that lymphocyte responses can be potentially dictated by the inherent production of hormones specific to each sex.
Effects of sex hormones on the effector lymphocyte responses
Sex hormones have been shown to exert anti-inflammatory effects (Fig. 1), and therapeutically, estrogens and DHT and their derivatives have been used in various diseases (Table 1). Specifically, as to MS, reduced brain lesions and relapse rates were noted with estrogen therapy accompanied with reduced inflammatory cytokines (Th1 and TNF-α) [122, 123]. Likewise, DHT treatment was associated with decreased fatigue and increased gray matter volume with a corresponding decrease in CD4 T cell infiltrates and IL-2 production, and increase in TGF-β1 secretion [124, 125]. Experimentally, low doses of estrogens have been shown to stimulate Th1 responses, whereas high doses equivalent to pregnancy levels can promote Th2 response in primary cultures [126, 127]. Estrogens also can stimulate the production of regulatory T cells (Tregs) by upregulating the expression of FoxP3 [128, 129], and other non-FoxP3-expressing Treg subsets such as B regulatory cells (Bregs), CD8+CD122+ Treg cells, and CD11b+ CD206+ ARG-1+ M2 like macrophages, among others [130]. EAE mice treated with E2 or E3 show reduced disease severity through inhibition of Th1 and Th17 cytokine production with a corresponding increase in Th2 cytokines [126, 131]. Similarly, testosterone also ameliorates EAE severity with a Th2 bias, as androgen-treated T cell lines, as opposed to untreated cultures, secrete a lower amount of IFN-γ compared to IL-10 [132,133,134]. Although testosterone appears not to promote differentiation of murine Treg cells, high testosterone and low estrogen conditions may promote skewing of Th1/Th17 responses toward Treg cells [135]. Recent reports suggest that males possess high frequencies of innate lymphoid cells (ILC) 2, and IL-33 produced from mast cells facilitate induction of non-pathogenic Th2 rather than encephalitogenic Th17 cytokines in the females [136]. But determination of antigen-specificity of these Th subsets has remained a major challenge in the field.
In our research, we made efforts to understand the cellular basis for sex bias in the occurrence of EAE in SJL mice by testing the hypothesis that the EAE-phenotypic differences between sexes are due to defects in antigen-specific, CD4 T cell responses. To this end, we created MHC class II (IAs) tetramers and dextramers for PLP 139-151 that can detect antigen-specific T cells with a high degree of specificity and sensitivity [137]. By enumerating the precursor frequencies of PLP-specific CD4 T cells flow cytometrically, we noted that the lymph node cells derived from male and female SJL mice responded equally to PLP 139-151, suggesting no defect in their ability to respond to self-antigens. We have also verified this phenomenon for an environmental microbe-derived epitope that cross-reacts with PLP 139-151 [138]. Furthermore, dextramer staining analysis of CNS infiltrates also did not reveal any significant variations between sexes with PLP-specific T cells as evaluated by flow cytometry (Fig. 2, top panel). Next, we established a novel in situ dextramer staining method to localize PLP-specific CD4 T cells in the brains of EAE mice by laser scanning confocal microscopy (LSCM) [139]. By evaluating brains obtained from male and female mice affected with EAE, we found the PLP dextramer+ cells to be scattered all through the tissues with equal proportions in both male and female mice, ruling out defects in the migration of antigen-specific T cells into the CNS (Fig. 2, bottom panel). Finally, T cells harvested from the brains of EAE mice and the T cell cultures stimulated with PLP 139-151 in vitro showed comparable expression of most of the positive and negative regulators of T cell activation in both male and female mice (unpublished observations). Based on these findings, we envision a scenario in which equal numbers of PLP-reactive, pathogenic T cells infiltrate into the brains in both male and female SJL mice, but their survivability may differ between sexes raising a question whether differences exist in the biochemical pathways between DHT and estrogen.

Enumeration of PLP 139-151-specific CD4 T cells in the CNS infiltrates from EAE mice. Male and female SJL mice were immunized with PLP 139-151, and brains and spinal cords were harvested from EAE-mice that showed paralytic signs. Mononuclear cells isolated from these tissues were stained with PLP 139-151 (specific) or control (Theiler’s murine encephalomyelitis virus [TMEV] 70-86) dextramers and the dextramer+ CD4+ cells were then analyzed. Representative flow cytometric plots are shown (top panel). By establishing in situ dextramer staining technique using LSCM, PLP 139-151-specific, CD4 T cells were analyzed in the brains harvested from male and female mice (bottom panel). CD4 T cells, green; dextramers, red; merged (circles, dext+ CD4+ T cells; insets represent enlarged views of dext+ CD4+ T cells). Original magnification × 1000; bar = 20 μm. Mean ± SEM values are shown (n = 3)
Biochemical mechanisms of sex hormones
Sex hormones mediate their cellular functions through both the genomic/nuclear and nongenomic/membrane signaling pathways, with the expected end result being transcriptional regulation [140, 141] that may affect cell proliferation or cell death [142,143,144]. For example, in breast cancer cells, E2 stimulates cell growth by augmenting transition from G1 to S phase, leading to activation of cyclin-dependent kinase and retinoblastoma protein phosphorylation [145, 146]. Whereas other groups have also demonstrated that E2 is capable of inducing apoptosis in breast and prostate cancer cells, thymocytes, monocytes, macrophages, neuronal cells, and T cells [147,148,149,150]. Similarly, androgens also can regulate apoptosis in breast and prostate cancer cells, human renal tubular leukemic and primary cells, including monocytes and macrophages and T cells [151,152,153]. Recently, autophagy-associated cell death has been described that involves the upregulation of autophagy flux, its machinery and the accumulation of autophagosomes [154]. A relationship has been shown recently between sex hormones, apoptosis, and autophagy. For example, pregnancy levels of E2 and progesterone exert stimulatory effects on autophagy in mammary epithelial cells by suppressing mammalian target of rapamycin (mTOR) activation that occurs in association with apoptotic cell death [155]. Additionally, E2 may regulate transcription factors targeted by autophagy, miRNAs, and histone modifications [156]. Likewise, E2 was shown to inhibit osteoblast apoptosis by promoting autophagy via the mTOR pathway [157]. But, less is known about androgens, and they were shown to promote prostate cancer cell growth through the induction of autophagy, in part through the production of reactive oxygen species [158]. Because both autophagy and apoptosis are well-controlled biological processes that play important roles in tissue homeostasis and disease, dissecting the cross-talk between the two, if any in the context of sex hormones, may lead to identification of molecules that affect both processes [159, 160].
To address the above theme, we established an in vitro system to determine the mechanistic basis for DHT-mediated effects in autoreactive T cells, since DHT has been successfully used to treat EAE. Unexpectedly, we noted that DHT reduced the proliferative responses to PLP 139-151, but the effects were not selective, since both proliferating and non-proliferating cells were equally affected [161]. Likewise, using MHC class II dextramers, we failed to note any immune deviation toward Th2 phenotype in antigen-specific T cells; rather, cells capable of producing all major inflammatory cytokines (Th1 and Th17), including Th2 cytokines, were reduced in DHT-treated cells. We also showed that DHT-mediated effects involved the induction of cell death, which also was associated with autophagy in autoreactive T cells [161]. Although our data did not support the notion that DHT-mediated effects accompany the appearance of IL-10-producing cells [132,133,134], production of IL-10 by non-T cell sources in vivo or in mixed T cell cultures in response to DHT-treatment cannot be discounted. Previous reports indicate that DHT can ameliorate EAE when administered either during induction or in the effector phase of the disease process [132, 134]. Our observation that DHT induces cell death of both proliferating and non-proliferating T cells may mean that the DHT-mediated effects might have occurred due to cell death. Importantly, we have also demonstrated that cell death can occur in conjunction with autophagy in DHT-treated cells [161], suggesting that common signaling cascades, or crosstalk, may exist between the two processes. Although dissecting this complexity is a challenge, using model systems that are deficient for apoptosis and autophagy machineries, such as caspase-3- and ATG-deficient mice, may be helpful. These studies may then provide avenues to identify molecules responsive to DHT that can affect both apoptosis and autophagy processes.
Perspectives and significance
As discussed above, autoimmune diseases are more prevalent in females than males and such a discrepancy also exists in the animal models, as shown with PLP 139-151-induced EAE in SJL mice [60, 138]. Essentially, PLP-reactive T cells generated in males can induce EAE in males comparable to the EAE-phenotype in females induced by cells generated in the female SJL mice [138]. Conversely, cells from males can induce only mild disease in females [138], suggesting that the microenvironment of recipients may determine the EAE-outcomes. By investigating the underlying mechanisms, we had previously noted that the EAE-resistant, male B10.S mice possess higher frequencies of Treg cells specific to PLP 139-151 than SJL mice, and depletion of Treg cells enabled B10.S mice to develop severe EAE [162, 163]. While these observations provide a cellular basis for EAE-susceptibility and EAE-resistance phenotypes, male hormones appear to play a critical role in the suppression of EAE. In support of this notion, a number of studies [124, 125, 132, 136, 164,165,166] indicate therapeutic benefits of testosterone by ameliorating the EAE-severity or clinical remissions in MS patients that are accompanied with increased gray matter volume, reduced Th1/Th17 inflammatory cytokines (IFN-γ, IL-2, and IL-17A), skewness of Th1/Th17:Treg ratio toward Tregs, shift of immune response toward Th2 type (IL-10), increased NK cell populations, and significant reductions in CNS infiltrations containing CD4 T cells [124, 125, 132, 135, 164, 165, 167]. Based on our observations with DHT [161], we did not recognize the phenomenon of immune deviation from pro- to anti-inflammatory cytokine switch; rather, DHT was found to suppress T cell responses regardless of their antigen-specificity that involve apoptosis and/or autophagy as the possible underlying mechanisms [161]. Additionally, we performed a few pilot experiments and determined that estrogens mediate effects similar to DHT (data not shown). Whether all sex hormones mediate their functions through common pathways such as apoptosis and autophagy is currently unknown. Proving this concept to be true may then widen the applications of sex hormone-dependent molecules as drug targets for a range of diseases, including metabolic syndromes, aging, and osteoporosis. Such discoveries also may potentially reduce the need to use small molecules like selective androgen receptor modulators. As a result, it may be possible to minimize side effects observed with sex hormones.
Availability of data and materials
Not applicable.
Abbreviations
- AIDs:
-
Autoimmune diseases
- AIRE:
-
Autoimmune regulator
- AR:
-
Androgen receptors
- BCR:
-
B cell receptor
- Bregs:
-
B regulatory cells
- CFA:
-
Complete Freund’s adjuvant
- CNS:
-
Central nervous system
- DCs:
-
Dendritic cells
- DHT:
-
Dihydrotestosterone
- E1:
-
Estrogen
- E2:
-
Estradiol
- E3:
-
Estriol
- EAE:
-
Experimental autoimmune encephalomyelitis
- ERα:
-
Estrogen receptor alpha
- ERβ:
-
Estrogen receptor beta
- FoxP3:
-
Forkhead box P3
- IFN:
-
Interferon
- IL:
-
Interleukin
- ILCs:
-
Innate lymphoid cells
- LC:
-
Langerhans cells
- LCSM:
-
Laser scanning confocal microscopy
- LPS:
-
Lipopolysaccharide
- M.tb:
-
Mycobacterium tuberculosis
- MBP:
-
Myelin basic protein
- MHC:
-
Major histocompatibility complex
- MOG:
-
Myelin oligodendrocyte glycoprotein
- MS:
-
Multiple sclerosis
- mTOR:
-
Mammalian target of rapamycin
- NK:
-
Natural killer
- NMOSD:
-
Neuromyelitis optica spectrum disorder
- NOD:
-
Non-obese diabetic
- PLP:
-
Proteolipid protein
- PRMS:
-
Progressive-relapsing multiple sclerosis
- RA:
-
Rheumatoid arthritis
- RRMS:
-
Relapsing-remitting multiple sclerosis
- SLE:
-
Systemic lupus erythematosus
- Th:
-
T helper
- TID:
-
Type I diabetes
- TLR:
-
Toll like receptor
- TMEV:
-
Theiler’s murine encephalomyelitis virus
- TNF:
-
Tumor necrosis factor
- Treg:
-
T regulatory cells
References
Elkon K, Casali P. Nature and functions of autoantibodies. Nat Clin Pract Rheumatol. 2008;4(9):491–8.
Palma J, Tokarz-Deptula B, Deptula J, Deptula W. Natural antibodies—facts known and unknown. Cent Eur J Immunol. 2018;43(4):466–75.
Zhang J. T-cell vaccination for autoimmune diseases: immunologic lessons and clinical experience in multiple sclerosis. Expert Rev Vaccines. 2002;1(3):285–92.
Coutinho A. Will the idiotypic network help to solve natural tolerance? Trends Immunol. 2003;24(2):53–4.
Lasrado N, Yalaka B, Reddy J. Triggers of inflammatory heart disease. Front Cell Dev Biol. 2020;8(192).
Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest. 2015;125(6):2228–33.
Walsh SJ, Rau LM. Autoimmune diseases: a leading cause of death among young and middle-aged women in the United States. Am J Public Health. 2000;90(9):1463.
Cooper GS, Stroehla BC. The epidemiology of autoimmune diseases. Autoimmun Rev. 2003;2(3):119–25.
McAlpine D, Compston A. McAlpine’s multiple sclerosis: Elsevier Health Sciences; 2005.
Bishop M, Rumrill PD. Multiple sclerosis: Etiology, symptoms, incidence and prevalence, and implications for community living and employment. Work. 2015.
Goldenberg MM. Multiple sclerosis review. Pharm Therapeutics. 2012;37(3):175.
Hartung DM. Economics and cost-effectiveness of multiple sclerosis Therapies in the USA. Neurotherapeutics. 2017;14(4):1018–26.
Owens GM. Economic burden of multiple sclerosis and the role of managed sare organizations in multiple sclerosis management. Am J Manag Care. 2016;22(6 Suppl):s151–8.
Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. 2011;164(4):1079–106.
Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sorensen PS, Thompson AJ, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–86.
Lassmann H. Multiple sclerosis pathology. Cold Spring Harb Perspect Med. 2018;8(3).
Elkjaer ML, Frisch T, Reynolds R, Kacprowski T, Burton M, Kruse TA, et al. Molecular signature of different lesion types in the brain white matter of patients with progressive multiple sclerosis. Acta Neuropathol Commun. 2019;7(1):205.
Lebar R, Boutry JM, Vincent C, Robineaux R, Voisin GA. Studies on autoimmune encephalomyelitis in the guinea pig. II. An in vitro investigation on the nature, properties, and specificity of the serum-demyelinating factor. J Immunol. 1976;116(5):1439–46.
Rivers TM, Sprunt DH, Berry GP. Observations on attempts to produce acute disseminated encephalomyelitis in monkeys. J Exp Med. 1933;58(1):39–53.
Genain CP, Nguyen MH, Letvin NL, Pearl R, Davis RL, Adelman M, et al. Antibody facilitation of multiple sclerosis-like lesions in a nonhuman primate. J Clin Invest. 1995;96(6):2966–74.
Adelmann M, Wood J, Benzel I, Fiori P, Lassmann H, Matthieu JM, et al. The N-terminal domain of the myelin oligodendrocyte glycoprotein (MOG) induces acute demyelinating experimental autoimmune encephalomyelitis in the Lewis rat. J Neuroimmunol. 1995;63(1):17–27.
Morris-Downes MM, Smith PA, Rundle JL, Piddlesden SJ, Baker D, Pham-Dinh D, et al. Pathological and regulatory effects of anti-myelin antibodies in experimental allergic encephalomyelitis in mice. J Neuroimmunol. 2002;125(1-2):114–24.
Hedegaard CJ, Krakauer M, Bendtzen K, Lund H, Sellebjerg F, Nielsen CH. T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology. 2008;125(2):161–9.
Crane IJ, Forrester JV. Th1 and Th2 lymphocytes in autoimmune disease. Crit Rev Immunol. 2005;25(2).
Miller SD, Karpus WJ. Experimental autoimmune encephalomyelitis in the mouse. Current protocols in immunology / edited by John E Coligan [et al]. 2007;Chapter 15:Unit 15.1.
Bebo BF Jr, Vandenbark AA, Offner H. Male SJL mice do not relapse after induction of EAE with PLP 139-151. J Neurosci Res. 1996;45(6):680–9.
Klein SL, Roberts CW. Sex and gender differences in infection and treatments for infectious diseases: Springer; 2015.
Klein SL. Sex influences immune responses to viruses, and efficacy of prophylaxis and treatments for viral diseases. Bioessays. 2012;34(12):1050–9.
Armien B, Pascale JM, Bayard V, Munoz C, Mosca I, Guerrero G, et al. High seroprevalence of hantavirus infection on the Azuero peninsula of Panama. Am J Trop Med Hyg. 2004;70(6):682–7.
Farzadegan H, Hoover DR, Astemborski J, Lyles CM, Margolick JB, Markham RB, et al. Sex differences in HIV-1 viral load and progression to AIDS. Lancet. 1998;352(9139):1510–4.
Klein SL, Passaretti C, Anker M, Olukoya P, Pekosz A. The impact of sex, gender and pregnancy on 2009 H1N1 disease. Biol Sex Differ. 2010;1(1):5.
Cai H. Sex difference and smoking predisposition in patients with COVID-19. Lancet Respir Med. 2020.
Liu X, Luo W-T, Li Y, Li C-N, Hong Z-S, Chen H-L, et al. Psychological status and behavior changes of the public during the COVID-19 epidemic in China. Infect Dis Poverty. 2020;9(1):58.
Jin JM, Bai P, He W, Wu F, Liu XF, Han DM, et al. Gender differences in patients with COVID-19: focus on severity and mortality. Front Public Health. 2020;8:152.
Klein SL, Huber S. Sex differences in susceptibility to viral infection. Sex hormones and immunity to infection. Berlin Heidelberg: Springer-Verlag; 2010. p. 93–122.
Neyrolles O, Quintana-Murci L. Sexual inequality in tuberculosis. PLoS Med. 2009;6(12):e1000199.
Valliani A, Khan F, Chagani B, Khuwaja AK, Majid S, Hashmi S, et al. Factors associated with Helicobacter pylori infection, results from a developing country - Pakistan. Asian Pac J Cancer Prev. 2013;14(1):53–6.
Leone M, Honstettre A, Lepidi H, Capo C, Bayard F, Raoult D, et al. Effect of sex on Coxiella burnetii infection: protective role of 17beta-estradiol. J Infect Dis. 2004;189(2):339–45.
Sivanmaliappan TS, Sevanan M. Antimicrobial susceptibility patterns of Pseudomonas aeruginosa from diabetes patients with foot ulcers. Int J Microbiol. 2011;2011.
Afroz R, Hanaki K, Tudin R. Factors affecting waste generation: a study in a waste management program in Dhaka City, Bangladesh. Environ Monit Assess. 2011;179(1-4):509–19.
Schroder J, Kahlke V, Book M, Stuber F. Gender differences in sepsis: genetically determined? Shock. 2000;14(3):307–10 discussion 10-3.
Pathak S, Rege M, Gogtay NJ, Aigal U, Sharma SK, Valecha N, et al. Age-dependent sex bias in clinical malarial disease in hypoendemic regions. PLoS One. 2012;7(4):e35592.
Wunderlich F, Benten WPM, Lieberherr M, Guo Z, Stamm O, Wrehlke C, et al. Testosterone signaling in T cells and macrophages. Steroids. 2002;67(6):535–8.
Beeson PB. Age and sex associations of 40 autoimmune diseases. Am J Med. 1994;96(5):457–62.
Gleicher N, Barad DH. Gender as risk factor for autoimmune diseases. J Autoimmun. 2007;28(1):1–6.
Brandt JE, Priori R, Valesini G, Fairweather D. Sex differences in Sjogren's syndrome: a comprehensive review of immune mechanisms. Biol Sex Differ. 2015;6:19.
Magyari M. Gender differences in multiple sclerosis epidemiology and treatment response. Dan Med J. 2016;63(3).
Kim SM, Waters P, Woodhall M, Kim YJ, Kim JA, Cheon SY, et al. Gender effect on neuromyelitis optica spectrum disorder with aquaporin4-immunoglobulin G. Mult Scler. 2016.
Papp V, Iljicsov A, Rajda C, Magyari M, Koch-Henriksen N, Petersen T, et al. A population-based epidemiological study of neuromyelitis optica spectrum disorder in Hungary. Eur J Neurol. 2020;27(2):308–17.
Papp V, Illes Z, Magyari M, Koch-Henriksen N, Kant M, Pfleger CC, et al. Nationwide prevalence and incidence study of neuromyelitis optica spectrum disorder in Denmark. Neurology. 2018;91(24):e2265–e75.
Ngo ST, Steyn FJ, McCombe PA. Gender differences in autoimmune disease. Front Neuroendocrinol. 2014;35(3):347–69.
Pal J, Rozsa C, Komoly S, Illes Z. Clinical and biological heterogeneity of autoimmune myasthenia gravis. J Neuroimmunol. 2011;231(1-2):43–54.
Fairweather D, Cooper LT Jr, Blauwet LA. Sex and gender differences in myocarditis and dilated cardiomyopathy. Curr Probl Cardiol. 2013;38(1):7–46.
D'Amico E, Patti F, Zanghi A, Chisari CG, Lo Fermo S, Zappia M. Late-onset and young-onset relapsing-remitting multiple sclerosis: evidence from a retrospective long-term follow-up study. Eur J Neurol. 2018;25(12):1425–31.
Chitnis T. Role of puberty in multiple sclerosis risk and course. Clin Immunol. 2013;149(2):192–200.
Tedeschi SK, Bermas B, Costenbader KH. Sexual disparities in the incidence and course of SLE and RA. Clin Immunol. 2013;149(2):211–8.
Confavreux C, Hutchinson M, Hours MM, Cortinovis-Tourniaire P, Moreau T. Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group. N Engl J Med. 1998;339(5):285–91.
Hughes SE, Spelman T, Gray OM, Boz C, Trojano M, Lugaresi A, et al. Predictors and dynamics of postpartum relapses in women with multiple sclerosis. Mult Scler. 2014;20(6):739–46.
Jorg S, Grohme DA, Erzler M, Binsfeld M, Haghikia A, Muller DN, et al. Environmental factors in autoimmune diseases and their role in multiple sclerosis. Cell Mol Life Sci. 2016;73(24):4611–22.
Voskuhl RR, Gold SM. Sex-related factors in multiple sclerosis susceptibility and progression. Nat Rev Neurol. 2012;8(5):255–63.
Itoh Y, Golden LC, Itoh N, Matsukawa MA, Ren E, Tse V, et al. The X-linked histone demethylase Kdm6a in CD4+ T lymphocytes modulates autoimmunity. J Clin Invest. 2019;130:3852–63.
Voskuhl RR, Sawalha AH, Itoh Y. Sex chromosome contributions to sex differences in multiple sclerosis susceptibility and progression. Mult Scler. 2018;24(1):22–31.
Qureshi IA, Mehler MF. Genetic and epigenetic underpinnings of sex differences in the brain and in neurological and psychiatric disease susceptibility. Prog Brain Res. 2010;186:77–95.
Kantarci OH, Goris A, Hebrink DD, Heggarty S, Cunningham S, Alloza I, et al. IFNG polymorphisms are associated with gender differences in susceptibility to multiple sclerosis. Genes Immun. 2005;6(2):153–61.
Miteva L, Trenova A, Slavov G, Stanilova S. IL12B gene polymorphisms have sex-specific effects in relapsing-remitting multiple sclerosis. Acta Neurol Belg. 2019;119(1):83–93.
Kantarci OH, Hebrink DD, Achenbach SJ, Atkinson EJ, de Andrade M, McMurray CT, et al. CD95 polymorphisms are associated with susceptibility to MS in women. A population-based study of CD95 and CD95L in MS. J Neuroimmunol. 2004;146(1-2):162–70.
Yurkovetskiy L, Burrows M, Khan AA, Graham L, Volchkov P, Becker L, et al. Gender bias in autoimmunity is influenced by microbiota. Immunity. 2013;39(2):400–12.
Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science. 2013;339(6123):1084–8.
Gandy KAO, Zhang J, Nagarkatti P, Nagarkatti M. The role of gut microbiota in shaping the relapse-remitting and chronic-progressive forms of multiple sclerosis in mouse models. Sci Rep. 2019;9(1):6923.
Bhatia A, Sekhon HK, Kaur G. Sex hormones and immune dimorphism. Sci World J. 2014;2014.
Distler W, Gabbe SG, Freeman RK, Mestman JH, Goebelsmann U. Estriol in pregnancy. Am J Obstet Gynecol. 1978;130(4):424–31.
Grimaldi CM, Cleary J, Dagtas AS, Moussai D, Diamond B. Estrogen alters thresholds for B cell apoptosis and activation. J Clin Invest. 2002;109(12):1625–33.
Komi J, Lassila O. Nonsteroidal anti-estrogens inhibit the functional differentiation of human monocyte-derived dendritic cells. Blood. 2000;95(9):2875–82.
Harkonen PL, Vaananen HK. Monocyte-macrophage system as a target for estrogen and selective estrogen receptor modulators. Ann N Y Acad Sci. 2006;1089:218–27.
Curran EM, Berghaus LJ, Vernetti NJ, Saporita AJ, Lubahn DB, Estes DM. Natural killer cells express estrogen receptor-alpha and estrogen receptor-beta and can respond to estrogen via a non-estrogen receptor-alpha-mediated pathway. Cell Immunol. 2001;214(1):12–20.
Lelu K, Laffont S, Delpy L, Paulet PE, Perinat T, Tschanz SA, et al. Estrogen receptor alpha signaling in T lymphocytes is required for estradiol-mediated inhibition of Th1 and Th17 cell differentiation and protection against experimental autoimmune encephalomyelitis. J Immunol. 2011;187(5):2386–93.
Zaitsu M, Narita S, Lambert KC, Grady JJ, Estes DM, Curran EM, et al. Estradiol activates mast cells via a non-genomic estrogen receptor-alpha and calcium influx. Mol Immunol. 2007;44(8):1977–85.
Phiel KL, Henderson RA, Adelman SJ, Elloso MM. Differential estrogen receptor gene expression in human peripheral blood mononuclear cell populations. Immunol Lett. 2005;97(1):107–13.
Lai J-J, Lai K-P, Zeng W, Chuang K-H, Altuwaijri S, Chang C. Androgen receptor influences on body defense system via modulation of innate and adaptive immune systems: lessons from conditional AR knockout mice. Am J Pathol. 2012;181(5):1504–12.
Walker WH. Non-classical actions of testosterone and spermatogenesis. Philos Trans R Soc Lond Ser B Biol Sci. 2010;365(1546):1557–69.
Mantalaris A, Panoskaltsis N, Sakai Y, Bourne P, Chang C, Messing EM, et al. Localization of androgen receptor expression in human bone marrow. J Pathol. 2001;193(3):361–6.
Benten WP, Lieberherr M, Giese G, Wrehlke C, Stamm O, Sekeris CE, et al. Functional testosterone receptors in plasma membranes of T cells. FASEB J. 1999;13(1):123–33.
Kovats S, Carreras E, Agrawal H. Sex steroid receptors in immune cells. Sex hormones and immunity to infection: Springer; 2010. p. 53–91.
Xia HJ, Zhang GH, Wang RR, Zheng YT. The influence of age and sex on the cell counts of peripheral blood leukocyte subpopulations in Chinese rhesus macaques. Cell Mol Immunol. 2009;6(6):433–40.
Scotland RS, Stables MJ, Madalli S, Watson P, Gilroy DW. Sex differences in resident immune cell phenotype underlie more efficient acute inflammatory responses in female mice. Blood. 2011;118(22):5918–27.
Kee SJ, Park YW, Cho YN, Jin HM, Kim MJ, Lee SJ, et al. Age- and gender-related differences in circulating natural killer T cells and their subset levels in healthy Korean adults. Hum Immunol. 2012;73(10):1011–6.
Doeing DC, Borowicz JL, Crockett ET. Gender dimorphism in differential peripheral blood leukocyte counts in mice using cardiac, tail, foot, and saphenous vein puncture methods. BMC Clin Pathol. 2003;3(1):3.
Melgert BN, Oriss TB, Qi Z, Dixon-McCarthy B, Geerlings M, Hylkema MN, et al. Macrophages: regulators of sex differences in asthma? Am J Respir Cell Mol Biol. 2010;42(5):595–603.
Bouman A, Schipper M, Heineman MJ, Faas MM. Gender difference in the non-specific and specific immune response in humans. Am J Reprod Immunol. 2004;52(1):19–26.
Aomatsu M, Kato T, Kasahara E, Kitagawa S. Gender difference in tumor necrosis factor-alpha production in human neutrophils stimulated by lipopolysaccharide and interferon-gamma. Biochem Biophys Res Commun. 2013;441(1):220–5.
Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312(5780):1669–72.
Paharkova-Vatchkova V, Maldonado R, Kovats S. Estrogen preferentially promotes the differentiation of CD11c+ CD11b(intermediate) dendritic cells from bone marrow precursors. J Immunol. 2004;172(3):1426–36.
Murphy AJ, Guyre PM, Wira CR, Pioli PA. Estradiol regulates expression of estrogen receptor ERalpha46 in human macrophages. PLoS One. 2009;4(5):e5539.
Liu HY, Buenafe AC, Matejuk A, Ito A, Zamora A, Dwyer J, et al. Estrogen inhibition of EAE involves effects on dendritic cell function. J Neurosci Res. 2002;70(2):238–48.
Nalbandian G, Kovats S. Understanding sex biases in immunity: effects of estrogen on the differentiation and function of antigen-presenting cells. Immunol Res. 2005;31(2):91–106.
D'Agostino P, Milano S, Barbera C, Di Bella G, La Rosa M, Ferlazzo V, et al. Sex hormones modulate inflammatory mediators produced by macrophages. Ann N Y Acad Sci. 1999;876:426–9.
Koyama Y, Nagao S, Ohashi K, Takahashi H, Marunouchi T. Sex differences in the densities of epidermal Langerhans cells of the mouse. J Invest Dermatol. 1987;88(5):541–4.
Koyama Y, Nagao S, Ohashi K, Takahashi H, Marunouchi T. Effect of systemic and topical application of testosterone propionate on the density of epidermal Langerhans cells in the mouse. J Invest Dermatol. 1989;92(1):86–90.
Chiche L, Jourde N, Ulmann C, Mancini J, Darque A, Bardin N, et al. Seasonal variations of systemic lupus erythematosus flares in southern France. Eur J Internal Med. 2012;23(3):250–4.
Ashcroft GS, Mills SJ. Androgen receptor-mediated inhibition of cutaneous wound healing. J Clin Invest. 2002;110(5):615–24.
Gilliver SC, Ashworth JJ, Mills SJ, Hardman MJ, Ashcroft GS. Androgens modulate the inflammatory response during acute wound healing. J Cell Sci. 2006;119(Pt 4):722–32.
Anderson AC, Nicholson LB, Legge KL, Turchin V, Zaghouani H, Kuchroo VK. High frequency of autoreactive myelin proteolipid protein–specific T cells in the periphery of naive mice mechanisms of selection of the self-reactive repertoire. J Exp Med. 2000;191(5):761–70.
Scabeni S, Lapilla M, Musio S, Gallo B, Ciusani E, Steinman L, et al. CD4+ CD25+ regulatory T cells specific for a thymus-expressed antigen prevent the development of anaphylaxis to self. J Immunol. 2008;180(7):4433–40.
Klein L, Klugmann M, Nave KA, Tuohy VK, Kyewski B. Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat Med. 2000;6(1):56–61.
Mathis D, Benoist C. A decade of AIRE. Nat Rev Immunol. 2007;7(8):645–50.
Dragin N, Bismuth J, Cizeron-Clairac G, Biferi MG, Berthault C, Serraf A, et al. Estrogen-mediated downregulation of AIRE influences sexual dimorphism in autoimmune diseases. J Clin Invest. 2016;126(4):1525–37.
Berrih-Aknin S, Panse RL, Dragin N. AIRE: a missing link to explain female susceptibility to autoimmune diseases. Ann N Y Acad Sci. 2018;1412(1):21–32.
Brown MA, Su MA. An inconvenient variable: sex hormones and their impact on T cell responses. J Immunol. 2019;202(7):1927–33.
Olsen NJ, Olson G, Viselli SM, Gu X, Kovacs WJ. Androgen receptors in thymic epithelium modulate thymus size and thymocyte development. Endocrinology. 2001;142(3):1278–83.
Fitzpatrick F, Lepault F, Homo-Delarche F, Bach JF, Dardenne M. Influence of castration, alone or combined with thymectomy, on the development of diabetes in the nonobese diabetic mouse. Endocrinology. 1991;129(3):1382–90.
Medina KL, Smithson G, Kincade PW. Suppression of B lymphopoiesis during normal pregnancy. J Exp Med. 1993;178(5):1507–15.
Thurmond TS, Murante FG, Staples JE, Silverstone AE, Korach KS, Gasiewicz TA. Role of estrogen receptor alpha in hematopoietic stem cell development and B lymphocyte maturation in the male mouse. Endocrinology. 2000;141(7):2309–18.
Erlandsson MC, Jonsson CA, Islander U, Ohlsson C, Carlsten H. Oestrogen receptor specificity in oestradiol-mediated effects on B lymphopoiesis and immunoglobulin production in male mice. Immunology. 2003;108(3):346–51.
Hill L, Jeganathan V, Chinnasamy P, Grimaldi C, Diamond B. Differential roles of estrogen receptors alpha and beta in control of B-cell maturation and selection. Mol Med. 2011;17(3-4):211–20.
Grimaldi CM, Michael DJ, Diamond B. Cutting edge: expansion and activation of a population of autoreactive marginal zone B cells in a model of estrogen-induced lupus. J Immunol. 2001;167(4):1886–90.
Ellis TM, Moser MT, Le PT, Flanigan RC, Kwon ED. Alterations in peripheral B cells and B cell progenitors following androgen ablation in mice. Int Immunol. 2001;13(4):553–8.
Smithson G, Couse JF, Lubahn DB, Korach KS, Kincade PW. The role of estrogen receptors and androgen receptors in sex steroid regulation of B lymphopoiesis. J Immunol. 1998;161(1):27–34.
Rijhsinghani AG, Thompson K, Bhatia SK, Waldschmidt TJ. Estrogen blocks early T cell development in the thymus. Am J Reprod Immunol. 1996;36(5):269–77.
Das BR, Bhanushali AA, Khadapkar R, Jeswani KD, Bhavsar M, Dasgupta A. Reference ranges for lymphocyte subsets in adults from western India: influence of sex, age and method of enumeration. Indian J Med Sci. 2008;62(10):397–406.
Zhang MA, Rego D, Moshkova M, Kebir H, Chruscinski A, Nguyen H, et al. Peroxisome proliferator-activated receptor (PPAR)alpha and -gamma regulate IFNgamma and IL-17A production by human T cells in a sex-specific way. Proc Natl Acad Sci U S A. 2012;109(24):9505–10.
Rovati B, Mariucci S, Poma R, Tinelli C, Delfanti S, Pedrazzoli P. An eight-colour flow cytometric method for the detection of reference values of lymphocyte subsets in selected healthy donors. Clin Exp Med. 2014;14(3):249–59.
Soldan SS, Alvarez Retuerto AI, Sicotte NL, Voskuhl RR. Immune modulation in multiple sclerosis patients treated with the pregnancy hormone estriol. J Immunol. 2003;171(11):6267–74.
Sicotte NL, Liva SM, Klutch R, Pfeiffer P, Bouvier S, Odesa S, et al. Treatment of multiple sclerosis with the pregnancy hormone estriol. Ann Neurol. 2002;52(4):421–8.
Gold SM, Chalifoux S, Giesser BS, Voskuhl RR. Immune modulation and increased neurotrophic factor production in multiple sclerosis patients treated with testosterone. J Neuroinflammation. 2008;5:32.
Kurth F, Luders E, Sicotte NL, Gaser C, Giesser BS, Swerdloff RS, et al. Neuroprotective effects of testosterone treatment in men with multiple sclerosis. Neuroimage Clin. 2014;4:454–60.
Maret A, Coudert JD, Garidou L, Foucras G, Gourdy P, Krust A, et al. Estradiol enhances primary antigen-specific CD4 T cell responses and Th1 development in vivo. Essential role of estrogen receptor alpha expression in hematopoietic cells. Eur J Immunol. 2003;33(2):512–21.
Bebo BF Jr, Fyfe-Johnson A, Adlard K, Beam AG, Vandenbark AA, Offner H. Low-dose estrogen therapy ameliorates experimental autoimmune encephalomyelitis in two different inbred mouse strains. J Immunol. 2001;166(3):2080–9.
Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1). Int Immunol. 2007;19(3):337–43.
Tai P, Wang J, Jin H, Song X, Yan J, Kang Y, et al. Induction of regulatory T cells by physiological level estrogen. J Cell Physiol. 2008;214(2):456–64.
Seifert HA, Benedek G, Nguyen H, Kent G, Vandenbark AA, Offner H. Estrogen protects both sexes against EAE by promoting common regulatory cell subtypes independent of endogenous estrogen. Metab Brain Dis. 2017;32(5):1747–54.
Haghmorad D, Amini AA, Mahmoudi MB, Rastin M, Hosseini M, Mahmoudi M. Pregnancy level of estrogen attenuates experimental autoimmune encephalomyelitis in both ovariectomized and pregnant C57BL/6 mice through expansion of Treg and Th2 cells. J Neuroimmunol. 2014;277(1-2):85–95.
Dalal M, Kim S, Voskuhl RR. Testosterone therapy ameliorates experimental autoimmune encephalomyelitis and induces a T helper 2 bias in the autoantigen-specific T lymphocyte response. J Immunol. 1997;159(1):3–6.
Liva SM, Voskuhl RR. Testosterone acts directly on CD4+ T lymphocytes to increase IL-10 production. J Immunol. 2001;167(4):2060–7.
Bebo BF Jr, Schuster JC, Vandenbark AA, Offner H. Androgens alter the cytokine profile and reduce encephalitogenicity of myelin-reactive T cells. J Immunol. 1999;162(1):35–40.
Massa MG, David C, Jorg S, Berg J, Gisevius B, Hirschberg S, et al. Testosterone differentially affects T cells and neurons in murine and human models of neuroinflammation and neurodegeneration. Am J Pathol. 2017;187(7):1613–22.
Russi AE, Ebel ME, Yang Y, Brown MA. Male-specific IL-33 expression regulates sex-dimorphic EAE susceptibility. Proc Natl Acad Sci U S A. 2018;115(7):E1520–E9.
Massilamany C, Upadhyaya B, Gangaplara A, Kuszynski C, Reddy J. Detection of autoreactive CD4 T cells using major histocompatibility complex class II dextramers. BMC Immunol. 2011;12(1):40.
Massilamany C, Thulasingam S, Steffen D, Reddy J. Gender differences in CNS autoimmunity induced by mimicry epitope for PLP 139-151 in SJL mice. J Neuroimmunol. 2011;230(1-2):95–104.
Massilamany C, Gangaplara A, Jia T, Elowsky C, Li Q, Zhou Y, et al. In situ detection of autoreactive CD4 T cells in brain and heart using major histocompatibility complex class II dextramers. J Vis Exp. 2014;90:e51679.
Cui J, Shen Y, Li R. Estrogen synthesis and signaling pathways during aging: from periphery to brain. Trends Mol Med. 2013;19(3):197–209.
Bennett NC, Gardiner RA, Hooper JD, Johnson DW, Gobe GC. Molecular cell biology of androgen receptor signalling. Int J Biochem Cell Biol. 2010;42(6):813–27.
Monteiro R, Teixeira D, Calhau C. Estrogen signaling in metabolic inflammation. Mediat Inflamm. 2014;2014:615917.
Foradori C, Weiser M, Handa R. Non-genomic actions of androgens. Front Neuroendocrinol. 2008;29(2):169–81.
Lewis-Wambi JS, Jordan VC. Estrogen regulation of apoptosis: how can one hormone stimulate and inhibit? Breast Cancer Res. 2009;11(3).
Foster JS, Wimalasena J. Estrogen regulates activity of cyclin-dependent kinases and retinoblastoma protein phosphorylation in breast cancer cells. Mol Endocrinol. 1996;10(5):488–98.
Altucci L, Addeo R, Cicatiello L, Dauvois S, Parker MG, Truss M, et al. 17beta-estradiol induces cyclin D1 gene transcription, p36D1-p34cdk4 complex activation and p105Rb phosphorylation during mitogenic stimulation of G (1)-arrested human breast cancer cells. Oncogene. 1996;12(11):2315–24.
Jordan VC. The 38th David A. Karnofsky lecture: the paradoxical actions of estrogen in breast cancer--survival or death? J Clin Oncol. 2008;26(18):3073–82.
Robertson CN, Roberson KM, Padilla GM, O'Brien ET, Cook JM, Kim CS, et al. Induction of apoptosis by diethylstilbestrol in hormone-insensitive prostate cancer cells. J Natl Cancer Inst. 1996;88(13):908–17.
Mor G, Sapi E, Abrahams VM, Rutherford T, Song J, Hao XY, et al. Interaction of the estrogen receptors with the Fas ligand promoter in human monocytes. J Immunol. 2003;170(1):114–22.
McMurray RW, Suwannaroj S, Ndebele K, Jenkins JK. Differential effects of sex steroids on T and B cells: modulation of cell cycle phase distribution, apoptosis and bcl-2 protein levels. Pathobiology. 2001;69(1):44–58.
Verzola D, Gandolfo MT, Salvatore F, Villaggio B, Gianiorio F, Traverso P, et al. Testosterone promotes apoptotic damage in human renal tubular cells. Kidney Int. 2004;65(4):1252–61.
Cutolo M, Capellino S, Montagna P, Ghiorzo P, Sulli A, Villaggio B. Sex hormone modulation of cell growth and apoptosis of the human monocytic/macrophage cell line. Arthritis Res Ther. 2005;7(5):R1124–32.
Thomas P, Pang Y, Dong J, Berg AH. Identification and characterization of membrane androgen receptors in the ZIP9 zinc transporter subfamily: II. Role of human ZIP9 in testosterone-induced prostate and breast cancer cell apoptosis. Endocrinology. 2014;155(11):4250–65.
Tsujimoto Y, Shimizu S. Another way to die: autophagic programmed cell death. Cell Death Differ. 2005;12(Suppl 2):1528–34.
Sobolewska A, Gajewska M, Zarzyńska J, Gajkowska B, Motyl T. IGF-I, EGF, and sex steroids regulate autophagy in bovine mammary epithelial cells via the mTOR pathway. Eur J Cell Biol. 2009;88(2):117–30.
Xiang J, Liu X, Ren J, Chen K, Wang HL, Miao YY, et al. How does estrogen work on autophagy? Autophagy. 2019;15(2):197–211.
Yang YH, Chen K, Li B, Chen JW, Zheng XF, Wang YR, et al. Estradiol inhibits osteoblast apoptosis via promotion of autophagy through the ER-ERK-mTOR pathway. Apoptosis. 2013;18(11):1363–75.
Shi Y, Han JJ, Tennakoon JB, Mehta FF, Merchant FA, Burns AR, et al. Androgens promote prostate cancer cell growth through induction of autophagy. Mol Endocrinol. 2013;27(2):280–95.
Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833(12):3448–59.
Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16(7):966–75.
Jia T, Anandhan A, Massilamany C, Rajasekaran RA, Franco R, Reddy J. Association of autophagy in the cell death mediated by dihydrotestosterone in autoreactive T cells independent of antigenic stimulation. J NeuroImmune Pharmacol. 2015;10(4):620–34.
Reddy J, Illes Z, Zhang X, Encinas J, Pyrdol J, Nicholson L, et al. Myelin proteolipid protein-specific CD4+CD25+ regulatory cells mediate genetic resistance to experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2004;101(43):15434–9.
Reddy J, Waldner H, Zhang X, Illes Z, Wucherpfennig KW, Sobel RA, et al. Cutting edge: CD4+CD25+ regulatory T cells contribute to gender differences in susceptibility to experimental autoimmune encephalomyelitis. J Immunol. 2005;175(9):5591–5.
Gold SM, Voskuhl RR. Estrogen and testosterone therapies in multiple sclerosis. Prog Brain Res. 2009;175:239–51.
Ziehn MO, Avedisian AA, Dervin SM, Umeda EA, O'Dell TJ, Voskuhl RR. Therapeutic testosterone administration preserves excitatory synaptic transmission in the hippocampus during autoimmune demyelinating disease. J Neurosci. 2012;32(36):12312–24.
Giatti S, Rigolio R, Romano S, Mitro N, Viviani B, Cavaletti G, et al. Dihydrotestosterone as a Protective Agent in Chronic Experimental Autoimmune Encephalomyelitis. Neuroendocrinology. 2015;101(4):296–308.
Palaszynski KM, Loo KK, Ashouri JF, Liu HB, Voskuhl RR. Androgens are protective in experimental autoimmune encephalomyelitis: implications for multiple sclerosis. J Neuroimmunol. 2004;146(1-2):144–52.
Voskuhl RR, Wang H, Wu TC, Sicotte NL, Nakamura K, Kurth F, et al. Estriol combined with glatiramer acetate for women with relapsing-remitting multiple sclerosis: randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(1):35–46.
Palaszynski KM, Liu H, Loo KK, Voskuhl RR. Estriol treatment ameliorates disease in males with experimental autoimmune encephalomyelitis: implications for multiple sclerosis. J Neuroimmunol. 2004;149(1-2):84–9.
Kim S, Liva SM, Dalal MA, Verity MA, Voskuhl RR. Estriol ameliorates autoimmune demyelinating disease: implications for multiple sclerosis. Neurology. 1999;52(6):1230–8.
Hall GM, Daniels M, Huskisson EC, Spector TD. A randomised controlled trial of the effect of hormone replacement therapy on disease activity in postmenopausal rheumatoid arthritis. Ann Rheum Dis. 1994;53(2):112–6.
Ganesan K, Balachandran C, Manohar BM, Puvanakrishnan R. Comparative studies on the interplay of testosterone, estrogen and progesterone in collagen induced arthritis in rats. Bone. 2008;43(4):758–65.
Holmdahl R, Jansson L, Andersson M. Female sex hormones suppress development of collagen-induced arthritis in mice. Arthritis Rheum. 1986;29(12):1501–9.
Holmdahl R, Jansson L, Meyerson B, Klareskog L. Oestrogen induced suppression of collagen arthritis: I. Long term oestradiol treatment of DBA/1 mice reduces severity and incidence of arthritis and decreases the anti type II collagen immune response. Clin Exp Immunol. 1987;70(2):372–8.
Booji A, Biewenga-Booji CM, Huber-Bruning O, Cornelis C, Jacobs JW, Bijlsma JW. Androgens as adjuvant treatment in postmenopausal female patients with rheumatoid arthritis. Ann Rheum Dis. 1996;55(11):811–5.
Cutolo M, Balleari E, Giusti M, Intra E, Accardo S. Androgen replacement therapy in male patients with rheumatoid arthritis. Arthritis Rheum. 1991;34(1):1–5.
Keith RC, Sokolove J, Edelman BL, Lahey L, Redente EF, Holers VM, et al. Testosterone is protective in the sexually dimorphic development of arthritis and lung disease in SKG mice. Arthritis Rheum. 2013;65(6):1487–93.
Ganesan K, Balachandran C, Manohar BM, Puvanakrishnan R. Effects of testosterone, estrogen and progesterone on TNF-alpha mediated cellular damage in rat arthritic synovial fibroblasts. Rheumatol Int. 2012;32(10):3181–8.
van Vollenhoven RF, Morabito LM, Engleman EG, McGuire JL. Treatment of systemic lupus erythematosus with dehydroepiandrosterone: 50 patients treated up to 12 months. J Rheumatol. 1998;25(2):285–9.
van Vollenhoven RF, Engleman EG, McGuire JL. An open study of dehydroepiandrosterone in systemic lupus erythematosus. Arthritis Rheum. 1994;37(9):1305–10.
Olsen NJ, Kovacs WJ. Case report: testosterone treatment of systemic lupus erythematosus in a patient with Klinefelter's syndrome. Am J Med Sci. 1995;310(4):158–60.
Lucas JA, Ahmed SA, Casey ML, MacDonald PC. Prevention of autoantibody formation and prolonged survival in New Zealand black/New Zealand white F1 mice fed dehydroisoandrosterone. J Clin Invest. 1985;75(6):2091–3.
Virkki LM, Porola P, Forsblad-d’Elia H, Valtysdottir S, Solovieva SA, Konttinen YT. Dehydroepiandrosterone (DHEA) substitution treatment for severe fatigue in DHEA-deficient patients with primary Sjogren’s syndrome. Arthritis Care Res. 2010;62(1):118–24.
Forsblad-d'Elia H, Carlsten H, Labrie F, Konttinen YT, Ohlsson C. Low serum levels of sex steroids are associated with disease characteristics in primary Sjogren's syndrome; supplementation with dehydroepiandrosterone restores the concentrations. J Clin Endocrinol Metab. 2009;94(6):2044–51.
Vendramini AC, Soo C, Sullivan DA. Testosterone-induced suppression of autoimmune disease in lacrimal tissue of a mouse model (NZB/NZW F1) of Sjogren's syndrome. Invest Ophthalmol Vis Sci. 1991;32(11):3002–6.
Ariga H, Edwards J, Sullivan DA. Androgen control of autoimmune expression in lacrimal glands of MRL/Mp-lpr/lpr mice. Clin Immunol Immunopathol. 1989;53(3):499–508.
Sato EH, Sullivan DA. Comparative influence of steroid hormones and immunosuppressive agents on autoimmune expression in lacrimal glands of a female mouse model of Sjogren's syndrome. Invest Ophthalmol Vis Sci. 1994;35(5):2632–42.
Krysiak R, Kowalcze K, Okopien B. The effect of testosterone on thyroid autoimmunity in euthyroid men with Hashimoto's thyroiditis and low testosterone levels. J Clin Pharm Ther. 2019;44(5):742–9.
Ahmed SA, Penhale WJ. The influence of testosterone on the development of autoimmune thyroiditis in thymectomized and irradiated rats. Clin Exp Immunol. 1982;48(2):367–74.
Nasser M, Haider A, Saad F, Kurtz W, Doros G, Fijak M, et al. Testosterone therapy in men with Crohn's disease improves the clinical course of the disease: data from long-term observational registry study. Horm Mol Biol Clin Invest. 2015;22(3):111–7.
Haider A, Kurtz W, Giltay EJ, Gooren LJ, Saad F. Administration of testosterone to elderly hypogonadal men with Crohn's disease improves their Crohn’s Disease Activity Index: a pilot study. Horm Mol Biol Clin Invest. 2010;2(3):287–92.
Saad F, Haider A, Gooren L. Hypogonadal men with psoriasis benefit from long-term testosterone replacement therapy - a series of 15 case reports. Andrologia. 2016;48(3):341–6.
Saad F, Yassin A, Almehmadi Y, Doros G, Gooren L. Effects of long-term testosterone replacement therapy, with a temporary intermission, on glycemic control of nine hypogonadal men with type 1 diabetes mellitus - a series of case reports. Aging Male. 2015;18(3):164–8.
Liu L, Wu L, Gao A, Zhang Q, Lv H, Xu L, et al. The influence of dihydrotestosterone on the development of Graves' disease in female BALB/c Mice. Thyroid. 2016;26(3):449–57.
Zhao F, Wu L, Wang Y, Liu L, Yang F, Sun Y, et al. Dihydrotestosterone regulates oxidative stress and immunosuppressive cytokines in a female BALB/c mouse model of Graves' disease. Autoimmunity. 2019;52(3):117–25.
Schwinge D, Carambia A, Quaas A, Krech T, Wegscheid C, Tiegs G, et al. Testosterone suppresses hepatic inflammation by the downregulation of IL-17, CXCL-9, and CXCL-10 in a mouse model of experimental acute cholangitis. J Immunol. 2015;194(6):2522–30.
Fijak M, Schneider E, Klug J, Bhushan S, Hackstein H, Schuler G, et al. Testosterone replacement effectively inhibits the development of experimental autoimmune orchitis in rats: evidence for a direct role of testosterone on regulatory T cell expansion. J Immunol. 2011;186(9):5162–72.
Acknowledgements
Not applicable.
Funding
This work was partially supported by the Transformational grant from the American Heart Association [18TPA34170206].
Author information
Authors and Affiliations
Contributions
All authors contributed to the synthesis of literature and writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lasrado, N., Jia, T., Massilamany, C. et al. Mechanisms of sex hormones in autoimmunity: focus on EAE. Biol Sex Differ 11, 50 (2020). https://doi.org/10.1186/s13293-020-00325-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13293-020-00325-4