
Abstract
Background
Retinal pigment epithelium (RPE) replacement has been proposed as an efficacious treatment for age-related macular degeneration (AMD), which is the primary cause of vision loss in the elderly worldwide. The embryonic stem cell (ESC) microenvironment has been demonstrated to enable mature cells to gain a powerful proliferative ability and even enhance the stem/progenitor phenotype via activation of the phosphoinositide 3-kinase (PI3K) signaling pathway. As the PI3K signaling pathway plays a pivotal role in proliferation and homeostasis of RPE, we hypothesize that the stemness and proliferative capability of RPE can be enhanced by the ESC microenvironment via activation of the PI3K signaling pathway.
Methods
To investigate whether the ESC microenvironment improves the stem cell phenotype and proliferation properties of human RPE (hRPE) cells by regulating the PI3K signaling pathway, primary hRPE cells were cocultured with either ESCs or human corneal epithelial cells (CECs) for 72 h, after which their proliferation, apoptosis, cell cycle progression, and colony formation were assayed to evaluate changes in their biological characteristics. Gene expression was detected by real-time PCR and protein levels were determined by western blotting or immunofluorescence. LY294002, an antagonist of the PI3K signaling pathway, was used to further confirm the mechanism involved.
Results
In comparison to hRPE cells cultured alone, hRPE cells cocultured with ESCs had an increased proliferative capacity, reduced apoptotic rate, and higher colony-forming efficiency. The expression of the stem cell-associated marker KLF4 and the differentiation marker CRALBP increased and decreased, respectively, in hRPE cells isolated from the ESC coculture. Furthermore, PI3K pathway-related genes were significantly upregulated in hRPE cells after exposure to ESCs. LY294002 reversed the pro-proliferative effect of ESCs on hRPE cells. In contrast, CECs did not share the ability of ESCs to influence the biological behavior and gene expression of hRPE cells.
Conclusions
Our findings indicate that the ESC microenvironment enhances stemness and proliferation of hRPE cells, partially via activation of the PI3K signaling pathway. This study may have a significant impact and clinical implication on cell therapy in regenerative medicine, specifically for age-related macular degeneration.
Similar content being viewed by others


Background
Age-related macular degeneration (AMD) is one of the leading irreversible causes of vision loss in the elderly worldwide. With the increasing longevity of the population, the prevalence of AMD is rising annually [1,2,3,4,5]. Although the exact cause of AMD remains unclear, it is universally acknowledged that aged or dysfunctional retinal pigment epithelium (RPE) cells play a vital role in the initial pathogenesis of AMD [6]. Consequently, RPE replacement has been proposed as a treatment for AMD.
In 1991, an autologous pedicle RPE graft by Peyman et al. in a patient with end-stage AMD resulted in improved visual acuity, suggesting that RPE replacement could be an efficacious treatment [7]. Unfortunately, patients who require cell transplantation usually do not have enough viable RPE cells to repopulate the entire macula adequately [8]. In addition, lipofuscin and melanosomes undergoing significant age-related changes will influence phototoxicity and detract from the normal functioning of the RPE and hinder the benefits of RPE transplantation [9]. Researchers have successfully attempted to promote RPE cell proliferation and functional recovery by mesenchymal stem cells [10,11,12], such as umbilical cord Wharton’s jelly-derived mesenchymal stem cells (WJ-MSCs) [13]. But the inevitable immune rejection by allogeneic cells cannot be ignored. Recent studies in regenerative medicine have revealed that human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) can be induced to differentiate into functional RPE cells, providing hope of using pluripotent stem cells to rescue visual function. However, certain problems with these approaches limited their clinical application, such as being costly and time-consuming, and having low efficiency and the potential for tumorigenesis [10, 14,15,16]. Consequently, the development of a convenient, efficient, and safe method to obtain sufficient viable autologous RPE cells is urgently required in AMD therapy.
Salero et al. discovered that RPE stem cells (RPESCs) are present in the human RPE layer, thus providing new insight for RPE replacement therapy [17]. RPESCs have a multipotency and self-renewal potential and can differentiate into RPE cells that produce neural and mesenchymal progeny. It was demonstrated by Blenkinsop et al. that RPESC-derived RPEs preserve the native RPE morphology, electrophysiology, and gene and protein expression patterns [18]. When grown on transwell polyester membranes, the RPESC-derived RPEs can be transplanted into rabbit subretinally and survive with retained characteristics in the subretinal space for at least 4 weeks [19]. Research has shown that proper culture conditions could turn off suppressive factors to activate RPESC proliferation [20]. Therefore, activating the surviving RPE within a patient’s eye is a strategy for replenishing the RPE layer and may also benefit the neural retina by producing favorable growth factors or improving RPE support of neural retinal cell function. Moreover, surgical injury and immunosuppression could potentially be avoided by this strategy. Therefore, it is crucial to explore ways to promote the proliferation and differentiation potential of RPESCs both in vitro and in vivo.
The embryonic stem cell (ESC) microenvironment can ameliorate or even reverse the aging process and enable mature cells to gain a powerful proliferative ability [21,22,23,24,25]. Yousef et al. have confirmed that the proliferation of mouse and human muscle progenitors can be enhanced by hESC-conditioned medium [24]. Similarly, our previous work demonstrated that the ESC microenvironment markedly improved the stem cell phenotype and proliferation properties of corneal epithelial cells and even human corneal endothelial cells that are considered unable to proliferate and that the PI3K signaling pathway is indispensable for these pro-regenerative effects [26,27,28,29,30]. Therefore, we hypothesize that the stemness and proliferative capability of RPE can be enhanced by the ESC microenvironment via activation of the PI3K signaling pathway. To test this hypothesis, the present study examined the effects of coculturing primary human RPE (hRPE) cells with ESCs on the proliferation, apoptosis, cell cycle progression, and colony formation of hRPE cells. Our findings may open a novel therapeutic avenue for AMD and other diseases of RPE dysfunction.
Methods
Cell culture
hRPE cells were extracted from eyecups of bulbus oculi subsequent to the application of corneal transplantation, performed at the State Key Laboratory of Ophthalmology (Guangzhou, China). The cell culture, storage, and subculture were performed following previously described guidelines [17]. hRPE cells were cultured in DMEM/F12 (Corning) with 10% FBS and 1% penicillin-streptomycin. Subsequent experiments were performed on the fourth or fifth generation of hRPE cells, when the cells had reached the optimum level of cell viability and morphology, in which the cell confluence rate reached between 70 and 90%. Cell identification was conducted using immunofluorescent staining of CRALBP protein, RPE65, and S-100 antibodies as described below.
Mouse ESCs were gifts from Professor Andy Peng Xiang. ESCs were cultured in knockout Dulbecco’s modified Eagle’s medium (DMEM; Gibco) with 10% FBS (Gibco), 0.1 mM non-essential amino acid (Gibco), 1% GlutaMAX media (Gibco), 0.055 mM 2-mercaptoethanol (Gibco), 5 × 105 units leukemia inhibitory factor (Millipore, USA), and 1% penicillin-streptomycin. Green fluorescent protein-labeled ESCs were constructed as described previously [31] and grown in ESC culture medium.
The CEC cell line, established in our laboratory previously [32], used as a kind of differentiated cell, without indifferentiability, to coculture with RPE cells was cultured in DMEM with 10% FBS, 10 ng/ml human epidermal growth factor (hEGF, Pepro Tech, USA), 5 mg/ml insulin (Sigma, USA), 5 mg/ml human transferrin (Sigma), 0.4 mg/ml hydrocortisone (MB-Chem, India), 2 mM L-glutamine (Gibco), and 1% penicillin-streptomycin.
hRPE cells and CECs were stained with cell-labeling solution (CM- DiI or DiD, Invitrogen) according to the manufacturer’s protocol. 6 × 105 DiD-labeled hRPE cells were plated in a 75-cm2 culture flask with 6 × 105 green fluorescent protein-labeled ESCs or CECs in hRPE culture medium. hRPE cells were collected after 72 h using fluorescence-activated cell sorting (BD FACSAria Fusion, USA). For control groups, hRPE cells were cultured alone in hRPE culture medium.
Cell cycle analysis
Cells were resuspended with 75% alcohol at − 20 °C overnight. After that, cells were incubated with 1 mg/ml RNase A stock solution for 30 min at 37 °C, stained with 100 μg/ml propidium iodide (BD) for 5 min at room temperature, and assessed on an LSRFortessa flow cytometer (BD). Data analysis was conducted using Modfit software.
Apoptosis assay
We followed a previous method to evaluate the apoptosis assay [30]. Staining cells were assessed by BD LSRFortessa flow cytometer using Annexin V-APC/7-aminoactinomycin D (Invitrogen) according to the manufacturer’s protocol.
Clone formation assay
The clone formation assay was performed as previously described [30]. Cells were seeded into 6-well plates (1000 hRPE cells/well) and cultured for 10 days. Clones were visualized by crystal violet staining and counted.
CCK-8 cell proliferation assay
As previously described [30], hRPE cells (700 cells/well) were seeded in a 96-well plate. Twenty-four hours after seeding, CCK-8 reagent (Dojindo Molecular Technologies, Japan) was added to the cell culture media for 1 h at 37 °C. The cell proliferation curve was generated according to the optical density measured at 450 nm (BioTek, USA).
RT-qPCR
Total RNA was isolated from cell cultures and tissues using a RNeasy Plus Mini kit (Qiagen, Germany) according to the manufacturer’s instructions and then quantified by absorption at 260 nm as previously described [31]. cDNA was synthesized with a PrimeScript™ RT Master Mix (Takara, Japan) and used for qPCR with SYBR® Premix Ex Taq™ (Takara) in a StepOnePlus thermal cycler (ABI, USA). The GAPDH gene was used as an internal control. The PCR primer sequences are listed in Table 1.
Western blot analysis
Protein expression in hRPE cells from four groups was assessed using western blotting as previously described [30]. The primary antibodies used are described in Table 2. The secondary antibody used was HRP-conjugated goat anti-rabbit IgG (1:2000, Sigma). Localization of antibodies was detected by an enhanced chemiluminescence kit (Amersham, Piscataway, NJ).
Immunofluorescence assay
The immunofluorescence assay was performed according to the previously described methods [30]. The primary antibodies used were shown in Table 2. The secondary antibodies used were Alexa 488 goat-anti mouse lgG (1:1000, Invitrogen) and Alexa Fluor 594 donkey anti-rabbit IgG (1:1000, Invitrogen). The cells were analyzed under an LSM780 or LSM800 confocal microscope (Zeiss, Germany).
Statistical analyses
All values were presented as the means ± standard deviation (SD). The statistical analyses were performed with GraphPad Prism software. A 2-tailed unpaired Student’s t test was used for analyses comparing 2 groups. P values < 0.05 were considered significant.
Results
Phenotype of hRPE cells and mouse ESCs
The primary adherent pigmented hRPE cells at first plating (P0) reached 90% confluence after 7 days of cultivation (Fig. 1a). They grew as cobblestone cultures and contained a great quantity of pigmentation. During culture, the pigment was diluted upon cell division and the cells gradually acquired a fusiform, largely depigmented morphology. The results of western blotting indicated that the differentiation marker proteins CRALBP, S-100, and RPE65 were expressed in the hRPE cells at P4 (Fig. 1b). Immunofluorescence staining also showed CRALBP expression in the hRPE cells (Fig. 1c).

Characteristics of hRPE cells and ESCs. a Representative images of hRPE cells by phase microscopy. b Western blotting of CRALBP, S-100, and RPE65 in hRPE cells. β-actin served as the internal control. c Immunofluorescence assays of CRALBP in hRPE cells. Scale bar, 50 μm. d Representative images of ESCs by phase microscopy. e Immunofluorescence assays of OCT4 and KLF4 in ESCs. Scale bar, 50 μm
The mouse ESCs exhibited a clonal or islet appearance. Under a light microscope, the clone was bright and round with a clear, sharp boundary (Fig. 1d). Immunofluorescence assays showed that the stem cell markers OCT4 and KLF4 were expressed in the ESCs (Fig. 1e).
Effects of coculture with ESCs on morphological changes in hRPE cells
The role of ESCs in regulating the morphology of hRPE cells was investigated. The hRPE cells at P5 in the control group presented a fusiform pattern (Fig. 2a). On the other hand, the P5 hRPE cells in the hRPE+ESC group showed an epithelioid shape with a homogeneous morphology that is more similar to the primary cultured cells originating from the eyecups, which maintained a normal cell property of contact inhibition. Immunofluorescent staining indicated reduced OCT4 expression in ESCs cocultured with hRPE cells (Fig. 2b). After culturing for 72 h, hRPE cells from all groups were collected for experiments below.

Effects of coculture with ESCs on morphological changes in hRPE cells. a Representative images of morphology by phase microscopy. b The expression of OCT4 in ESC before and after coculture as determined by immunofluorescent staining. Scale bar, 50 μm
Coculture with ESCs enhances the proliferative capacity of hRPE cells
We next investigated the potential effects of ESCs on the proliferation of hRPE cells. The CCK-8 test (a common assay for detecting cell proliferation) was used to obtain the growth curve of hRPE cells from each group. During the slow-growing latent phase in days 1 and 2, no marked differences of optical density (OD) values were detected among the three groups (Fig. 3a). However, on the third day, the hRPE+ESC group showed significantly higher OD values than the control group. All three groups entered the logarithmic growth phase on the fourth day but the slope of the growth curve for the hRPE+ESC group was higher than the other two groups. These observations indicate that the hRPE cells treated with ESCs possessed a relatively stronger growth capacity. In contrast, there were no significant differences in the growth curves between the hRPE group and the hRPE+CEC group.

Coculture with ESCs increases the proliferation rate and lowers the apoptosis rate. a Proliferation of hRPE cells sorted from the control (hRPE), hRPE+CEC, hRPE+ESC, and hRPE+ESC+LY294002 groups, as assessed by a CCK8 proliferation assay. b Percentages of apoptotic hRPE cells, as assessed by flow cytometer. Data are means ± SDs. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001
To study the effects of ESCs on cell apoptosis, we employed Annexin V-APC/7-AAD staining and flow cytometry analysis. We found that the percentages of apoptotic hRPE cells from the Ctrl, hRPE+CEC, and hRPE+ESC groups were 13.73 ± 0.9912%, 13.7 ± 1.512%, and 9.473 ± 1.835%, respectively (Fig. 3b). There were fewer apoptotic cells in the hRPE+ESC group than in the hRPE group (P = 0.0065), while the percentages of apoptotic cells were almost the same in the hRPE and hRPE+CEC groups. These results suggest that ESCs inhibit apoptosis in hRPE cells.
Cell cycle progression was further evaluated by flow cytometry. As shown in Fig. 4a, the percentage of hRPE cells entering the cell cycle was significantly higher in the hRPE+ESC group than the other groups (P < 0.05). In the hRPE+ESC group, 33.94% ± 2.191% of cells were entering S phase, whereas in the hRPE group, 14.71% ± 2.468% of cells were in S phase. Consistent with the flow cytometry results, hRPE cells cocultured with ESCs showed higher expression levels of the cell cycle promoters cyclin A2, cyclin B1, and cyclin D1, and lower expression levels of the cell cycle negative regulators p21 and p27, both transcriptionally and translationally (Fig. 4b–d). In contrast, neither the cell cycle distribution nor the cell cycle-related protein expression levels of hRPE cells were significantly changed by coculture with CECs.

ESC-treated hRPE cells had a higher proliferation rate with a faster cell cycle turnaround time. a Proportion of cell cycle distribution in C918 cells, as assessed by flow cytometry. b Expression of the cell cycle related factors in hRPE cells, as assessed by RT-qPCR. c Western blotting of cyclin proteins and p21, p27 in hRPE cells. β-actin served as the internal control. d The expression of cyclin B1, cyclin D1, and p21 in hRPE cells as determined by immunofluorescent staining. Scale bar, 50 μm. Data are means ± SDs. *P < 0.05; **P < 0.01; ***P < 0.001
Coculture with ESCs enhances the stem cell phenotype of hRPE cells
As shown in Fig. 2a, the hRPE cells cocultured with ESCs had smaller cell sizes with bigger nucleus to cytoplasm (N:C) ratios than cells in the control group (Fig. 5a). No obvious differences in morphology and N:C ratio was observed between the control and hRPE+CEC groups. To evaluate the clonal growth capacity, the hRPE cells from the three groups were seeded without a feeder layer to assess colony-forming efficiency (CFE). At day 7, the CFE of the hRPEs in the ESC-treated group reached 10.65 ± 0.6856%, whereas the CFE of the control and hRPE+CEC groups were 3 ± 0.4082% and 2.925 ± 0.3775%, respectively (Fig. 5b).

Coculture with ESCs enhances the stem cell phenotype of hRPE cells. a The N:C ratio analysis of hRPE cells. b Colony-forming efficacy and representative images of clones formed by hRPE cells. c Expression of the RPE differentiation markers (CRALBP and PEDF) and stem cell-associated marker (KLF4) in hRPE cells, as assessed by RT-qPCR. c Western blotting of CRALBP and KLF4 in hRPE cells. β-actin served as the internal control. Data are means ± SDs. *P < 0.05; ***P < 0.001; ****P < 0.0001
RT-qPCR analysis revealed that the mRNA expression of the RPE differentiation markers CRALBP and PEDF by hRPE cells in the hRPE+ESC group was significantly decreased by 0.264- and 0.315-fold, respectively, in comparison to the control group (Fig. 5c). However, the expression of KLF4, a marker associated with early stem cells and reprogramming, was markedly increased by 167-fold in hRPE cells after coculturing with ESCs (Fig. 5c). Consistent with the mRNA expression profiles, western blot analysis revealed that hRPE cells from the control group strongly expressed CRALBP, while hRPE cells in the hRPE+ESC group showed weaker CRALBP expression (Fig. 5d). KLF4 was barely detectable in the hRPE cells in the control group but markedly upregulated in ESC-treated hRPE cells (Fig. 5d). In contrast, the expression of RPE-specific and stem cell-associated markers in hRPEs from the hRPE+CEC group was not significantly different from that of the control group. These observations indicate that ESCs can enhance the stem cell phenotype of hRPE cells.
ESCs enhance the proliferative capacity of hRPE cells by activating the PI3K pathway
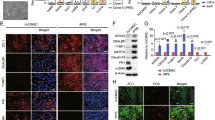
Our previous finding [27] demonstrated that the ESC microenvironment promoted the proliferation of corneal epithelial cells via activation of the PI3K/Akt signaling pathway. Therefore, we further examined whether coculturing with ESCs enhances the proliferative capacity of hRPE cells by upregulating the PI3K pathway. We performed RT-PCR validation of key PI3K pathway genes, including PAR2, FAK, PI3K, PDK1/2, and AKT, and found that they were significantly upregulated in hRPE cells after exposure to ESCs (Fig. 6a–c). In contrast, the expressions of these genes were not enhanced in hRPE cells from the CEC group compared with the hRPE group.

ESCs enhance the proliferative capacity of hRPE cells by activating the PI3K pathway. a Expression of PI3K pathway genes in hRPE cells, as assessed by RT-qPCR. b Western blotting of PI3K pathway genes in hRPE cells. β-actin served as the internal control. c The expression of FAK and P3K in hRPE cells as determined by immunofluorescent staining. Scale bar, 50 μm. d Immunofluorescence assays of pAKT in hRPE cells. Scale bar, 50 μm. Data are means ± SDs. **P < 0.01; ***P < 0.001
To determine whether PI3K pathway activity is necessary for the pro-proliferative effect of the ESC microenvironment, we added the PI3K antagonist, LY294002 to disrupt PI3K signaling (Fig. 6d). LY294002 abolished the pro-proliferative effect of the ESCs on hRPE cells in the ESC co-culture system; the growth of hRPE cells was reduced and ESC-inhibited apoptosis was blocked (Figs. 3 and 4). Meanwhile, expression of the cell cycle promoters cyclin A2, cyclin B1, and cyclin D1 decreased, while the expression of p21 and p27 increased significantly (Fig. 4b–d). These findings indicate that the PI3K pathway is indeed activated by ESCs in hRPE cells, which in turn boosts proliferation. Interestingly, although LY294002 decreased the N:C ratio and clonal growth capacity of hRPEs (Fig. 5a, b), it did not affect their expression of RPE-specific and stem cell-associated markers (Fig. 5c, d).
Discussion
A variety of signaling pathways, such as the MAPK, PI3K, and Notch pathways, become deficient or inactive in senescent cells, including senescent adult stem cells, leading to anti-proliferation of the organ’s stem cells, which then prevents their regeneration [33,34,35,36]. With the rapid development in bioengineering, in vivo applications of biopolymers that release signaling pathway agonists could evoke efficient regenerative responses from senescent cells [37,38,39]. However, achieving “young” levels of signaling not only promotes cell growth, but also cause undesired changes, including but not limited to oncogenic transformation [40,41,42]. Additionally, even young cells are doomed to perish when introduced into an old organ without a youthful niche [43, 44]. Though the recent study demonstrated that sub-tenon WJ-MSCs administration was effective on reactivating the degenerated photoreceptors in dormant phase [13], the long-term effects may be weakened with the gradual aging of WJ-MSCs in vivo, and the senescence-associated secretory phenotype secreted by senescent WJ-MSCs may even produce adverse effects. Therefore, the idea of providing endogenous or transplanted stem and progenitor cells with better microenvironments is being pursued by many researchers in the field of regenerative medicine.
We and others have demonstrated that the ESC microenvironment manifests a pro-regenerative activity that enhances and, more importantly, rejuvenates the regenerative capacity and promotes the proliferation of differentiated cells. Differentiated ESCs and tissue-specific adult stem cells yield much weaker results than ESCs [21, 24, 43]. Administration of sub-tenon WJ-MSCs could stop the progression of retinal degeneration and rescue photoreceptors in the dormant phase with the paracrine effects of WJ-MSCs [13]. Interactions between cells involve multi-directional signaling. In the coculture system, the stem cell microenvironment serves as a niche by releasing cytokines via paracrine and autocrine pathways as well as direct signal transmission via cell-to-cell contact. We have previously shown that the enhancement of the stem cell phenotype and proliferation of CECs by ESCs was much more remarkable in the cell-to-cell contact coculture system than in the non-contact system which only exists the paracrine effect. Furthermore, CECs with strong proliferation after being cocultured with ESCs were not reprogrammed to pluripotency and were not tumorigenic, suggesting that the ESC microenvironment could safely and effectively improve the proliferative potential of adult cells [26]. Accordingly, we cultured hRPE cells alone or with ESCs in a cell-to-cell contact coculture system. Compared with the control group, ESC-treated hRPE cells had a higher proliferation rate with a faster cell cycle turnaround time, a decreased apoptotic rate, an increased clone formation rate, and an increased stemness. However, CECs do not share the ability of ESCs to influence the regenerative behavior of hRPE cells, which support the notion that non-embryonic cells cannot promote the proliferative ability of terminal cells.
With increased aging, RPE cells show a reduced activation of the PI3K/Akt pathway and are less tolerant to tissue damage [45]. Studies have shown that epidermal growth factor and thrombin can stimulate RPE cell survival and proliferation through activating the PI3K pathway [46, 47]. Our previous studies demonstrated that the proliferation of corneal epithelial cells was significantly promoted by the ESC microenvironment via activation of the PI3K/Akt signaling pathway [26, 29]. Consistent with these findings, the present study showed that ESCs upregulate the PI3K pathway of hRPE cells, which accounts for their pro-proliferative effect.
An accumulation of evidence supports a key role for the PI3K pathway in cell cycle progression. Transcription and translation of multiple cyclins have been shown to be dependent on the activation of the PI3K/Akt pathway [48]. Cyclin D1 plays a central role in cell cycle progression from G1 to S phase [49], and its accumulation induces RPE cell proliferation [47]. Cyclin A is involved in DNA replication in S phase and is required for G2/M phase transition [50]. Cyclin B facilitates G2/M progression; depletion of cyclin B causes a dramatic G2 arrest and reduction in mitotic cells in hTert-RPE1 cells [51]. Moreover, PI3K activation reduces the levels of cyclin-dependent kinase inhibitors (CDKIs). Both p21 and p27 are important CDKIs with a wide range of kinase inhibition activity that can effectively block the activity of cyclin/CDK complexes to prevent cells from going through the G1/S phase checkpoint and inhibit cell proliferation [52]. It was revealed that the level of p21 increases in an age-dependent manner; hence, p21 has been used as a senescence marker [53,54,55]. It has also been demonstrated that increased p21 expression in RPE cells inhibits their proliferation in vitro [56]. Consistent with these findings, our experiments showed that ESCs activate the PI3K pathway of hRPE cells, which accounts for their pro-proliferation effect via upregulating cyclins and downregulating CDKIs.
Tissue stem cells have been found in tissues with lower self-renewal demands such as the nervous system, and where a rapid rate of cellular turnover is required, such as the skin [57, 58]. Adult stem cells are an important cell source in regenerative medicine. Normally, cell differentiation starts from stem cells that progress to transiently amplifying cells (TACs) and finally to terminally differentiated cells. This process can be delayed or even reversed by certain factors or conditions [59]. It has been shown that a sub-population of multipotent, self-renewing RPESCs is present in the human RPE layer [17]. Although there are no definitive markers to identify RPESCs, RPESCs show higher expression of c-MYC and KLF4 and lower expression of CRALBP, Tyr, and PEDF compared to RPE cells [17, 60]. A combination of stem cell-associated and differentiation markers has enabled scientists to identify putative RPESCs. The present study found that the isolated hRPE cells from the coculture systems expressed a significantly higher level of the key stemness factor KLF4 at the protein and transcription levels and decreased level of the differentiation marker CRALBP. Our results suggest that ESCs, through coculture, can enhance the stem/progenitor phenotype of hRPE cells. As KLF4 is an upstream regulator of the PI3K/Akt pathway [61], the activation of KLF4 in hRPE cells by ESCs cannot be affected by PI3K inhibitors.
Conclusions
Our study provides evidence that the ESC microenvironment can enhance the undifferentiated status and proliferation properties of hRPE cells. This finding opens new avenues for the potential therapeutic application of ESCs in regenerative medicine. Further studies on the functional enhancement of hRPE cells in the coculture system may shed light on the use of such cells in regenerative medicine.
Availability of data and materials
Most data generated or analyzed during this study are included in the article. Additional data sets generated and/or analyzed during the study and other relevant information are available from the corresponding authors upon reasonable request.
Abbreviations
- AMD:
-
Age-related macular degeneration
- RPE:
-
Retinal pigment epithelium
- ESC:
-
Embryonic stem cell
- iPSCs:
-
Induced pluripotent stem cells
- RPESCs:
-
Retinal pigment epithelium stem cells
- CEC:
-
corneal epithelial cell
- CCK8:
-
Cell Counting Kit-8
- DiI:
-
1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate
- DiD:
-
1,1-Dioctadecyl-3,3,3,3-tetramethylindodicarbocyanine
- PI3K:
-
Phosphoinositide 3-kinase
- PDK2:
-
Pyruvate dehydrogenase kinase isoform 2
- mTOR:
-
Mechanistic target of rapamycin
- MSCs:
-
Mesenchymal stem cells
- RT-qPCR:
-
Reverse transcription polymerase chain reaction
- KLF4:
-
Kruppel-like factor 4
- SASP:
-
Senescence-associated secretory phenotype
- PAR2:
-
Protease-activated receptor 2
- CRALBP:
-
Cellular retinaldehyde-binding protein
- OCT4:
-
Octamer-binding transcription factor 4
- PTEN:
-
Phosphatase and tensin homologue deleted on chromosome ten
- FAK:
-
Focal adhesion kinase
References
Friedman DS, O’Colmain BJ, Munoz B, Tomany SC, McCarty C, de Jong PT, et al. Prevalence of age-related macular degeneration in the United States. Archives of ophthalmology (Chicago, Ill: 1960). 2004; 122(4): 564–72.
Klein R, Klein BE, Lee KE, Cruickshanks KJ, Gangnon RE. Changes in visual acuity in a population over a 15-year period: the Beaver Dam Eye Study. Am J Ophthalmol. 2006;142(4):539–49.
Klein R, Knudtson MD, Lee KE, Gangnon RE, Klein BE. Age-period-cohort effect on the incidence of age-related macular degeneration: the Beaver Dam Eye Study. Ophthalmology. 2008;115(9):1460–7.
Lindekleiv H, Erke MG. Projected prevalence of age-related macular degeneration in Scandinavia 2012-2040. Acta Ophthalmol. 2013;91(4):307–11.
Vingerling JR, Dielemans I, Hofman A, Grobbee DE, Hijmering M, Kramer CF, et al. The prevalence of age-related maculopathy in the Rotterdam Study. Ophthalmology. 1995;102(2):205–10.
Nowak JZ. AMD--the retinal disease with an unprecised etiopathogenesis in search of effective therapeutics. Acta Pol Pharm. 2014;71(6):16.
Peyman GA, Blinder KJ, Paris CL, Alturki. A technique for retinal pigment epithelium transplantation for age-related macular degeneration secondary to extensive subfoveal scarring. Ophthalmic Surg. 1991;22(2):6.
Dang Y, Zhang C, Zhu Y. Stem cell therapies for age-related macular degeneration: the past, present, and future. Clin Interv Aging. 2015;10:255–64.
Boulton M, Roanowska M, Wess T. Ageing of the retinal pigment epithelium: implications for transplantation. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2004; 242(1): 76–84.
Huang C, Zhang J, Ao M, Li Y, Zhang C, Xu Y, et al. Combination of retinal pigment epithelium cell-conditioned medium and photoreceptor outer segments stimulate mesenchymal stem cell differentiation toward a functional retinal pigment epithelium cell phenotype. J Cell Biochem. 2012;113(2):590–8.
Gong L, Wu Q, Song B, Lu B, Zhang Y. Differentiation of rat mesenchymal stem cells transplanted into the subretinal space of sodium iodate-injected rats. Clin Exp Ophthalmol. 2008;36(7):666–71.
Tomita M, Adachi Y, Yamada H, Takahashi K, Kiuchi K, Oyaizu H, et al. Bone marrow-derived stem cells can differentiate into retinal cells in injured rat retina. Stem cells (Dayton, Ohio). 2002;20(4):279–83.
Özmert E, Arslan U. Management of retinitis pigmentosa by Wharton’s jelly derived mesenchymal stem cells: preliminary clinical results. Stem Cell Res Ther. 2020;11(1):25.
Buchholz DE, Hikita ST, Rowland TJ, Friedrich AM, Hinman CR, Johnson LV, et al. Derivation of functional retinal pigmented epithelium from induced pluripotent stem cells. Stem cells (Dayton, Ohio). 2009; 27(10): 2427–34.
Vaajasaari H, Ilmarinen T, Juuti-Uusitalo K, Rajala K, Onnela N, Narkilahti S, et al. Toward the defined and xeno-free differentiation of functional human pluripotent stem cell-derived retinal pigment epithelial cells. Mol Vis. 2011;17:558–75.
Zhang K, Liu GH, Yi F, Montserrat N, Hishida T, Esteban CR, et al. Direct conversion of human fibroblasts into retinal pigment epithelium-like cells by defined factors. Protein & cell. 2014;5(1):48–58.
Salero E, Blenkinsop Timothy A, Corneo B, Harris A, Rabin D, Stern Jeffrey H, et al. Adult human RPE can be activated into a multipotent stem cell that produces mesenchymal derivatives. Cell Stem Cell. 2012;10(1):88–95.
Blenkinsop TA, Saini JS, Maminishkis A, Bharti K, Wan Q, Banzon T, et al. Human adult retinal pigment epithelial stem cell-derived RPE monolayers exhibit key physiological characteristics of native tissue. Invest Ophthalmol Vis Sci. 2015;56(12):7085–99.
Stanzel BV, Liu Z, Somboonthanakij S, Wongsawad W, Brinken R, Eter N, et al. Human RPE stem cells grown into polarized RPE monolayers on a polyester matrix are maintained after grafting into rabbit subretinal space. Stem cell reports. 2014;2(1):64–77.
Saini JS, Temple S, Stern JH. Human retinal pigment epithelium stem cell (RPESC). Adv Exp Med Biol. 2016;854:557–62.
Conboy IM, Yousef H, Conboy MJ. Embryonic anti-aging niche. Aging. 2011;3(5):555–63.
Singla DK, Singla RD, McDonald DE. Factors released from embryonic stem cells inhibit apoptosis in H9c2 cells through PI3K/Akt but not ERK pathway. Am J Physiol Heart Circ Physiol. 2008;295(2):H907–13.
Chou YF, Chen HH, Eijpe M, Yabuuchi A, Chenoweth JG, Tesar P, et al. The growth factor environment defines distinct pluripotent ground states in novel blastocyst-derived stem cells. Cell. 2008;135(3):449–61.
Yousef H, Conboy MJ, Mamiya H, Zeiderman M, Schlesinger C, Schaffer DV, et al. Mechanisms of action of hESC-secreted proteins that enhance human and mouse myogenesis. Aging. 2014;6(8):602–20.
Guo Y, Graham-Evans B, Broxmeyer HE. Murine embryonic stem cells secrete cytokines/growth modulators that enhance cell survival/anti-apoptosis and stimulate colony formation of murine hematopoietic progenitor cells. Stem cells (Dayton, Ohio). 2006; 24(4): 850–6.
Zhou J, Chen F, Xiao J, Li C, Liu Y, Ding Y, et al. Enhanced functional properties of corneal epithelial cells by coculture with embryonic stem cells via the integrin beta1-FAK-PI3K/Akt pathway. Int J Biochem Cell Biol. 2011;43(8):1168–77.
Lu X, Chen D, Liu Z, Li C, Liu Y, Zhou J, et al. Enhanced survival in vitro of human corneal endothelial cells using mouse embryonic stem cell conditioned medium. Mol Vis. 2010;16:611–22.
Liu Y, Ding Y, Ma P, Wu Z, Duan H, Liu Z, et al. Enhancement of long-term proliferative capacity of rabbit corneal epithelial cells by embryonic stem cell conditioned medium. Tissue engineering Part C, Methods. 2010;16(4):793–802.
Liu Z, Wan P, Duan H, Zhou J, Tan B, Liu Y, et al. ES micro-environment enhances stemness and inhibits apoptosis in human limbal stem cells via the maintenance of telomerase activity. PLoS One. 2013;8(1):e53576.
Liu J, Huang Z, Yang L, Wang X, Wang S, Li C, et al. Embryonic stem cells modulate the cancer-permissive microenvironment of human uveal melanoma. Theranostics. 2019;9(16):4764–78.
Zhou C, Huang Z, Li P, Li W, Liu Y, Li C, et al. Safety and efficacy of embryonic stem cell microenvironment in a leukemia mouse model. Stem Cells Dev. 2014;23(15):1741–54.
Liu J, Song G, Wang Z, Huang B, Gao Q, Liu B, et al. Establishment of a corneal epithelial cell line spontaneously derived from human limbal cells. Exp Eye Res. 2007;84(3):599–609.
Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007;8(9):703–13.
Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443(7110):448–52.
Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443(7110):453–7.
Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443(7110):421–6.
Huebsch N, Mooney DJ. Inspiration and application in the evolution of biomaterials. Nature. 2009;462(7272):426–32.
Park H, Cannizzaro C, Vunjak-Novakovic G, Langer R, Vacanti CA, Farokhzad OC. Nanofabrication and microfabrication of functional materials for tissue engineering. Tissue Eng. 2007;13(8):1867–77.
Sands RW, Mooney DJ. Polymers to direct cell fate by controlling the microenvironment. Curr Opin Biotechnol. 2007;18(5):448–53.
Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26(22):3227–39.
Lea IA, Jackson MA, Li X, Bailey S, Peddada SD, Dunnick JK. Genetic pathways and mutation profiles of human cancers: site- and exposure-specific patterns. Carcinogenesis. 2007;28(9):1851–8.
Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L. Rational targeting of Notch signaling in cancer. Oncogene. 2008;27(38):5124–31.
Carlson ME, Conboy IM. Loss of stem cell regenerative capacity within aged niches. Aging Cell. 2007;6(3):371–82.
Carlson BM, Faulkner JA. Muscle transplantation between young and old rats: age of host determines recovery. Am J Phys. 1989;256(6 Pt 1):C1262–6.
He Y, Leung KW, Ren Y, Pei J, Ge J, Tombran-Tink J. PEDF improves mitochondrial function in RPE cells during oxidative stress. Invest Ophthalmol Vis Sci. 2014;55(10):6742–55.
Defoe DM, Grindstaff RD. Epidermal growth factor stimulation of RPE cell survival: contribution of phosphatidylinositol 3-kinase and mitogen-activated protein kinase pathways. Exp Eye Res. 2004;79(1):51–9.
Parrales A, Lopez E, Lopez-Colome AM. Thrombin activation of PI3K/PDK1/Akt signaling promotes cyclin D1 upregulation and RPE cell proliferation. Biochim Biophys Acta. 2011;1813(10):1758–66.
Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13(3):195–203.
Coqueret O. Linking cyclins to transcriptional control. Gene. 2002;299(1–2):35–55.
Oredsson SM. Polyamine dependence of normal cell-cycle progression. Biochem Soc Trans. 2003;31(2):366–70.
Soni DV, Sramkoski RM, Lam M, Stefan T, Jacobberger JW. Cyclin B1 is rate limiting but not essential for mitotic entry and progression in mammalian somatic cells. Cell Cycle. 2008;7(9):1285–300.
Cazzalini O, Scovassi AI, Savio M, Stivala LA, Prosperi E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat Res. 2010;704(1–3):12–20.
Enomoto K, Mimura T, Harris DL, Joyce NC. Age differences in cyclin-dependent kinase inhibitor expression and rb hyperphosphorylation in human corneal endothelial cells. Invest Ophthalmol Vis Sci. 2006;47(10):4330–40.
Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodriguez-Ubreva J, Rebollo E, et al. Autophagy maintains stemness by preventing senescence. Nature. 2016;529(7584):37–42.
Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530(7589):184–9.
Wang Y, Yuan Z, You C, Han J, Li H, Zhang Z, et al. Overexpression p21WAF1/CIP1 in suppressing retinal pigment epithelial cells and progression of proliferative vitreoretinopathy via inhibition CDK2 and cyclin E. BMC Ophthalmol. 2014;14:144.
Clarke DL, Johansson CB, Wilbertz J, Veress B, Nilsson E, Karlstrom H, et al. Generalized potential of adult neural stem cells. Science (New York, NY). 2000; 288(5471): 1660–3.
Ghazizadeh S, Taichman LB. Multiple classes of stem cells in cutaneous epithelium: a lineage analysis of adult mouse skin. EMBO J. 2001;20(6):1215–22.
Qin M, Tai G, Collas P, Polak JM, Bishop AE. Cell extract-derived differentiation of embryonic stem cells. Stem cells (Dayton, Ohio). 2005; 23(6): 712–8.
Mariotti C, Lazzarini R, Nicolai M, Saitta A, Orsini E, Orciani M, et al. Comparative study between amniotic-fluid mesenchymal stem cells and retinal pigmented epithelium (RPE) stem cells ability to differentiate towards RPE cells. Cell Tissue Res. 2015;362(1):21–31.
Chang YL, Zhou PJ, Wei L, Li W, Ji Z, Fang YX, et al. MicroRNA-7 inhibits the stemness of prostate cancer stem-like cells and tumorigenesis by repressing KLF4/PI3K/Akt/p21 pathway. Oncotarget. 2015;6(27):24017–31.
Acknowledgements
The authors thank Professor Andy Peng Xiang, from the Center for Stem Cell Biology and Tissue Engineering, the Key Laboratory for Stem Cells and Tissue Engineering, Ministry of Education, Sun Yat-Sen University for providing mouse ESCs. We would like to thank Editage (www.editage.cn) for English language editing.
Funding
This work was supported by The National Key R&D program of China (2018YFC1106000) and Guangdong Basic and Applied Basic Research Foundation (2019A1515110618).
Author information
Authors and Affiliations
Contributions
ZCW conceived and designed the study. ZCW, JHL, and LY participated in designing the experiments and writing the manuscript. XRW, JHL, and LY conducted all in vitro experiments. CYL, SBW, ZQH, and YQC prepared the RNA, performed the RNA integrity analysis, and conducted western blot analyses. CXL and YL assisted with flow cytometry, and YL made the charts and performed statistical analyses. JHL and LY contributed equally to this work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance with the Declaration of Helsinki and approved by medical ethics committee of Zhongshan Ophthalmology Center, Sun Yat-sen University (Code#2020KYPJ013).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, J., Yang, L., Wang, X. et al. Embryonic stem cell microenvironment enhances proliferation of human retinal pigment epithelium cells by activating the PI3K signaling pathway. Stem Cell Res Ther 11, 411 (2020). https://doi.org/10.1186/s13287-020-01923-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-020-01923-0