
Abstract
Background
MSCs isolated from bone marrow (BM-MSCs) have well-established chondrogenic potential, but MSCs derived from the synovial membrane (SM-MSCs) and synovial fluid (SF-MSCs) are thought to possess superior chondrogenicity. This study aimed to compare the in vitro immunophenotype and trilineage and chondrogenic potential of BM-MSCs to SM-MSCs and SF-MSCs.
Methods
MSCs were isolated from bone marrow (BM-MSCs), synovial membrane (SM-MSCs), and synovial fluid (SF-MSCs) extracted from the hips (BM) and knees (SM and SF) of advanced OA patients undergoing arthroplasty. Flow cytometric analysis was used at P2 to evaluate cell stemness. The trilinear differentiation test was performed at P2. At P3, MSC-seeded collagen sponges were cultured in chondrogenic medium for 28 days. Chondrogenic gene expression was quantified by qRT-PCR. Finally, the implants were stained to assess the deposition of proteoglycans and type II collagen.
Results
Despite variability, the immunophenotyping of BM-MSCs, SM-MSCs, and SF-MSCs was quite similar. All cell types were positive for the expression of stem cell markers and negative for exclusion markers. Additionally, chondrogenic differentiation and hypertrophy were more pronounced in BM-MSCs (ACAN, SOX9, COL2B, and COL10A) than in SF-MSCs, with SM-MSCs having intermediate characteristics. Concerning matrix synthesis, the three cell types were equipotent in terms of GAG content, while BM-MSC ECM synthesis of type II collagen was superior.
Conclusions
Chondrogenic MSCs are easily collected from SM and SF in advanced human OA, but in vitro chondrogenesis that is superior to age-matched BM-MSCs should not be expected. However, due to intra-articular priming, SF-MSCs did not overexpress hypertrophic gene.
Similar content being viewed by others


Background
As hyaline cartilage lacks vasculature, neurons, and a lymphatic system, it possesses reduced self-healing potential, and as a consequence, focal or diffuse chondral defects lead to osteoarthritis (OA). Regeneration of posttraumatic injured cartilage remains an essential objective in orthopedics. In 1994, autologous chondrocyte implantation (ACI) was introduced in the clinic as a cell therapy for cartilage defects [1, 2]. During the last decade, other cell therapy and tissue engineering techniques have emerged, but most used chondrocytes, which are unique, specialized resident cartilage cells [3, 4]. However, ACI has several disadvantages: (i) development of cicatricial fibrotic cartilage, (ii) cartilage biopsy is invasive with possible donor site morbidity, (iii) the number of resident chondrocytes is low, and (iv) there is rapid dedifferentiation during monolayer cell expansion [5].
Since the discovery of mesenchymal stem cells in bone marrow [6], stem cell therapy has become a very promising and advanced scientific research topic. Stem cells originate from two primary sources: adult body tissues and embryos. Scientists are also working on ways to develop stem cells from other cells using genetic reprogramming techniques. Multipotent stromal cells are heterogeneous and contain several populations, including stem cells. The term MSCs should be used for multipotent mesenchymal stromal cells rather than mesenchymal stem cells, since the cells isolated do not contain a pure population of stem cells. The isolation of MSCs according to current criteria produces heterogeneous, non-clonal cultures of stromal cells containing stem cells with different multipotential properties, committed progenitors, and differentiated cells [7,8,9]. Concerning cartilage engineering, some adult MSCs have been promoted, such as cartilage, bone marrow (BM), fat, or periosteal mesenchymal stem cells.
The existence of chondroprogenitor cells in synovial tissue was discovered in the 2000s while characterizing synovial chondromatosis, a rheumatological disease characterized by cartilaginous nodule formation inside the synovial cavity [10], and their stemness was confirmed in 2006 [11]. In addition, human multipotent MSCs were isolated from the synovial membrane (SM-MSCs) of knee joints in 2001 [12]. MSCs from synovial fluid (SF-MSCs) were isolated later in 2004. Currently, these cells are of great interest in cartilage engineering due to their easy accessibility with arthrocentesis and their stemness trilineage differentiation, self-renewal capacity, and immunosuppressive properties [13]. In terms of chondrogenesis, both SM-MSCs and SF-MSCs should have the advantage of sharing a common marker with chondrocytes, CD44, which is involved in hyaluronan synthesis [14]. Nevertheless, to the best of our knowledge, a direct comparison of these three cell types (BM-, SM-, and SF-MSCs) is lacking in humans. Various scaffolds, environmental growth factors (especially the TGF-β superfamily) promote SM- and SF-MSC chondrogenic differentiation [15], participating in the heterogeneity of the data.
In this study, we compared the respective stemness and chondrogenicity of 2 intra-articular components of advanced OA stem cells, SM- and SF-MSCs, versus BM-MSCs, which are considered the gold standard. To this end, we first performed flow cytometry analysis and trilineage differentiation. We then investigated their respective chondrogenic differentiation (PCR, histology, and immunohistochemistry) into 3D collagen sponges for 28 days under the influence of TGF-β1 and normoxia.
Materials and methods
Isolation and expansion of MSCs derived from human bone marrow, synovial membrane, and synovial fluid
MSCs were isolated from human bone marrow following total hip (BM-MSCs) or knee arthroplasty (SM- and SF-MSCs), both for advanced osteoarthritis (OA), after informed consent and with the approval of our local ethics committee (file DC 2014—2148, authorized July 10, 2014). Each heparinized bone marrow sample was diluted in PBS (phosphate-buffered saline) solution and then centrifuged at 1500 rpm for 5 min. The pellet was resuspended in complete culture medium and then seeded in Petri dishes at 4 × 106 cells/dish at 37 °C in a humidified atmosphere containing 5% (v/v) CO2. SM samples were aseptically isolated, finely minced, washed in PBS, digested overnight with collagenase B at 2 mg/ml and 37 °C, and centrifuged at 1500 rpm for 5 min. The pellets were resuspended in complete culture medium and seeded in Petri dishes at 106 cells/dish. Human SF samples were diluted 1:6 in expansion culture medium and plated in 55 cm2 Petri dishes.
MSCs derived from various tissues (all unmatched) were expanded in monolayers separately in proliferation medium containing Dulbecco’s modified Eagle’s medium with low glucose (DMEM-LG, Gibco) supplemented with 10% fetal bovine serum (FBS, Sigma), 1 ng/ml basic fibroblast growth factor (bFGF, Miltenyi Biotec), 1% glutamine (Gibco), and 1% penicillin-streptomycin (Gibco). The dishes were cultured at 37 °C with 5% humidified CO2. The medium was unchanged for the initial 3 days and then changed twice per week until confluence was reached. The non-adherent cells were discarded through subsequent changes of the medium. When the adherent cells reached approximately 80% confluence, the MSCs were trypsinized (trypsin-EDTA 0.05%, Gibco) and plated at a density of 0.5 × 106 cells/dish. The medium was changed the following day and then every 2–3 days.
Predifferentiation medium was used at the final passage (P3) before seeding the MSCs into collagen sponges. This medium was composed of Dulbecco’s modified Eagle’s medium with high glucose (DMEM-HG, Gibco) supplemented with 10% fetal bovine serum (FBS, Sigma), sodium pyruvate (110 μg/ml, Gibco), bFGF (1 ng/ml, Miltenyi Biotech), 1% penicillin-streptomycin (Gibco), PAD chondrogenic supplements (PAD; proline (40 μg/ml, Sigma), L-ascorbic acid-2-phosphate (50 μg/ml, Sigma)), and dexamethasone (10–7 M, Sigma) [16, 17].
Trilineage differentiation potential of BM-, SM-, and SF-MSCs
Chondrogenesis
MSCs were trypsinized at P2 and centrifuged at 300×g for 10 min to form pellets (0.5 × 106 cells/pellet). Chondrogenic differentiation was induced by 3D culture for 28 days in a dedicated medium [18]. At D28, the pellets were fixed in 4% paraformaldehyde (PFA, Sigma) for 24 h at 4 °C. Samples were subsequently dehydrated and embedded in paraffin. Five-micron-thick sections were obtained using a microtome (Leica) and stained with Alcian blue to observe the proteoglycan content in the pellets.
Adipogenesis
MSCs were trypsinized at P2 and seeded in 24-well plates at 5000 cells/well. Adipogenic MSC differentiation was induced for 21 days with a dedicated medium [18]. On D21, monolayer cells were fixed with 4% PFA for 30 min and were stained with oil red (Sigma) to detect lipid vacuoles.
Osteogenesis
At P2, MSCs were seeded in 24-well plates at 10,000 cells/well and cultured in commercialized osteogenic medium (Miltenyi Biotech) for 14 days [18]. At D14, monolayer cell cultures were fixed with 4% paraformaldehyde for 30 min and stained with alizarin red (Sigma) to visualize calcium deposits.
Flow cytometry analysis of BM-, SM-, and SF-MSCs
At P2, MSCs were rinsed with a blocking solution (0.5% BSA (A-9667 Sigma Aldrich, France) in 1× PBS) and distributed at 500,000 cells/tube. The cells were centrifuged (300×g, 5 min), and the pellets were resuspended in an immunoblotting solution containing either different pairs of antibodies [anti-CD45 (BD Pharmingen), anti-CD34 (BD Pharmingen), anti-CD73 (BD Pharmingen), anti-CD90 (Beckman Coulter), or anti-CD105 (Beckman Coulter)] or 100 μL of blocking solution as a negative control. After an incubation for 45 min (4 °C, in the dark), the cells were washed with PBS (Gibco) and centrifuged (300×g, 5 min). The pellets were resuspended in 300 μL of PBS and the tubes were analyzed by using a flow cytometer (Gallios, Beckman Coulter).
Respective chondrogenic potential of BM-, SM-, and SF-MSCs in collagen sponges
The 3D culture was performed by using collagen sponges manufactured by Symatèse Biomatériaux (Chaponost, France) that were composed of 95% type I collagen and 5% type III collagen. The size of each collagen sponge was 5 mm in diameter and 2 mm in thickness. MSCs derived from the three tissue types (BM, SM, and SF) were seeded at P4 into sponges at a density of 0.5 million cells/sponge. The sponges were cultured under normoxia conditions (20% O2, v/v) at 37 °C in a humidified atmosphere containing 5% CO2 (v/v). Two conditions were tested: (i) control medium without chondrogenic growth factor composed of DMEM-HG supplemented with 1% ITS + premix (BD Biosciences), 1% glutamine (Gibco), sodium pyruvate (110 μg/ml, Gibco), 1% penicillin-streptomycin (Gibco), and PAD chondrogenic supplements (Sigma) as described previously and (ii) chondrogenic medium composed of control medium supplemented with TGF-β1 (10 ng/ml, Miltenyi Biotech). At D28, gene expression, and biochemical, histological, and immunohistochemical analyses were assessed.
Real-time RT-PCR analysis of MSC-seeded sponges at D28
At D28, the total cell RNA was extracted using a RNeasy mini kit (Qiagen), according to the manufacturer’s instructions. The RNA was quantified spectrophotometrically and reverse transcribed with an iScriptcDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s instructions [18]. Gene expression was analyzed by quantitative real-time polymerase chain reaction using the SYBERgreen master mix system according to the manufacturer’s protocol. As a control of the amplification specificity, a melting curve analysis was performed for each PCR experiment/each primers pairs. Relative quantification was determined using a standard curve made from a purified PCR product for each gene tested, with concentrations ranging from 10−3 to 10−6 ng/μl. For standardization of gene expression levels, results were expressed as the ratio of the mRNA level of each gene of interest versus the RPS29 gene. This gene (RPS29), referred to as a housekeeping gene, is typically a constitutive gene that is expressed at relatively constant levels in all cells independent of experimental conditions [17, 19], here in TGF-β1-treatment MSCs, compared to control ITS culture conditions. Each measurement was performed at least three times in three different patients.
The chondrogenic genes were type II collagen (COL2A1; NM_001844) and its isoform COL2B (NM_033150.2), aggrecan (ACAN; NM_001135), COMP (NM_000095), SRY (sex-determining region Y)-box 9 (SOX9; NM_000346), and type X collagen (COL10A1; NM_000493). The fibrotic genes were type I collagen (COL1A1; NM_000088.3) and versican (VCAN; NM_001164098). The osteogenic gene was runt-related transcription factor 2 (RUNX2; NM_001278478).
Biochemical analysis of GAG in MSC-seeded sponges at D28
Sponge lysates after digestion for RT-PCR were used to determine GAG content. This colorimetric assay uses dimethyl methylene blue (DMB, Sigma) dye, according to Goldberg’s method [20]. The absorbance was assessed at 525 nm with a spectrophotometer (Dynatech) and compared to the standard curve generated from chondroitin sulfate isolated from shark cartilage.
Histological analysis of MSC-seeded sponges at D28
After fixation with 4% PFA for 24 h at 4 °C, the sponges were dehydrated with increasing solutions of alcohol and embedded in paraffin. Sections at a thickness of 5 μm were stained with hematoxylin-erythrosin-saffron to visualize cell morphology and with Alcian blue to visualize proteoglycan content.
Immunohistochemical analysis of MSC-seeded sponges at D28
The detection of type II collagen in cartilaginous TE substitutes was performed using a primary monoclonal antibody (6B3, Labvision) at a dilution of 1/100 and the LSAB+ kit (HRP, Dako) based on avidin-biotin techniques as previously described [21]. The sections were counterstained with hematoxylin at 1/5 for 1 min (RAL, France).
Densitometric analysis of glycosaminoglycans and type II collagen using ImageJ
Histological sections stained with Alcian blue used to visualize proteoglycans or treated by immunohistochemistry to identify collagen II were digitized using a light microscope (DMD 108, Leica). The staining area percentages of Alcian blue and IHC markers for collagen type II were determined using the image analysis software ImageJ as previously described [18].
Statistical analysis
Each experiment was performed in triplicate for three patients. For gene expression, the ratio of mRNA levels of each gene to RPS29 gene expression is expressed for each condition and MSC origin at D28. The results are presented as the mean ± standard error of the mean. Statistical analysis was performed with t tests to evaluate the effect of TGF-β1 versus ITS separately for each cell group. In a second step, a significant interaction was assessed between groups by two-way ANOVA with Bonferroni’s post hoc test. Asterisks represent significant differences versus control condition (ITS 1%) (*p < 0.05; p < 0.01; and ***p < 0.001). Hash signs represent a significant difference between cellular groups (#p < 0.05; p < 0.01; and ###p < 0.001). Statistical analysis was performed with GraphPad Prism® V8.
Results
Trilineage differentiation potential of BM-, SM-, and SF-MSCs
At P2, BM-, SM-, and SF-MSCs cultured for 28 days in pellets in chondrogenic medium showed proteoglycan synthesis that was visualized by Alcian blue staining (Fig. 1a). Grossly, the BM-MSC pellets were larger and rounder than SM and SF pellets. SM-MSC pellets exhibited more intense Alcian blue staining than BM-and SF pellets. Likewise, the three lineages of MSCs were cultured in monolayers for 14 days with osteogenic medium, the presence of calcium deposits was visualized by using alizarin red staining, and the most intense staining was found in BM-MSCs. Furthermore, after 21 days of culture in the adipogenic medium, we detected lipid droplets, which are characteristic of adipogenic differentiation, by using red oil staining and found decreased intensity in SF pellets (PCR and macroscopic assessments available in Supplementary data 1 and 2).

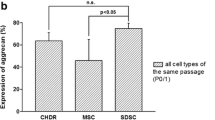
Trilineage differentiation and immunophenotyping of human SF-, SM-, and BM-MSCs. a Chondrogenic differentiation of MSCs was visualized by using Alcian blue staining of pellets cultured for 28 days in chondrogenic medium (TGF-β1). Osteogenic differentiation was induced in monolayer MSCs cultured for 14 days with osteogenic medium, and calcium deposits were highlighted using alizarin red staining. For adipogenic differentiation, MSCs were cultured in monolayers in specific medium for 21 days, and lipid droplets were observed using oil red staining. Scale bars = 200 μm. b Immunophenotyping of MSCs was assessed at the end of the monolayer second passage (P2) in the expansion medium. Data are presented as the mean of positive cells ± SEM. Please note that the percentage scale is not similar on both plots
Immunophenotyping of BM-, SM-, and SF-MSCs
All three cellular groups exhibited phenotypic characteristics that were consistent with stemness at P2. Both of the hematopoietic markers CD45 (0.1%, 1.2%, and 9.4%, respectively, NS) and CD34 (0.02%, 1.6% and 3.9%, respectively, NS) were poorly expressed, despite a nonstatistical trend showing a slight increase in SF-MSCs. HLA-DR expression was quite similar between the three kinds of MSCs, despite a nonsignificant trend showing increased expression in BM-MSCs. CD73, CD90, and CD105 are mesenchymal markers. All MSCs expressed CD73 and CD105 (> 90%, NS) very strongly. CD90 expression was variable depending on the cell type; SM-MSCs had significantly stronger CD90 expression compared to that of BM-MSCs (Fig. 1b).
Gene profiles at D28 of collagen sponges seeded with BM-, SM-, and SF-MSCs
Chondrogenic genes
As depicted in Fig. 2, TGF-β1 strongly enhanced the expression of chondrogenic genes compared to that of ITS, especially in BM-MSCs and SM-MSCs. COMP expression was significantly increased in TGF-β1-treated SM-MSCs, but ACAN, SOX9, COL2A1, and the COL2B isoform expression was more pronounced in BM-MSCs compared with that of SM-MSCs. For all genes, SF-MSC expression was significantly low.

Gene expression at D28 in collagen sponges seeded with human BM-, SM-, and SF-MSCs under ITS and TGF-β1 conditions. Relative mRNA expression was measured by real-time polymerase chain reaction of chondrogenic (COMP, ACAN, SOX9, COL2A1, and COL2B), fibrotic (COL1A1 and VCAN), and hypertrophic (COL10A1 and RUNX2) markers. All results were normalized to RPS29 mRNA expression. Data are presented as the mean ± standard error of the mean. Statistical analysis was performed with a t test to evaluate the effect of TGF-β1 versus ITS for each cellular group separately and was followed by two-way ANOVA with Bonferroni’s post hoc test on all values to evaluate differences in gene expression between all cellular groups. Asterisks represent a significant difference versus control condition (ITS 1%); *p < 0.05, p < 0.01, ***p < 0.001. Hash signs represent a significant difference between cellular groups; #p < 0.05, ##p < 0.01, ###p < 0.001
Fibrotic genes
Similarly, COL1A1 overexpression was significantly increased by TGF-β1 equally in BM-MSCs and SM-MSCs. In addition, VCAN expression was increased in BM-MSCs. Again, TGF-β1-driven COL1A1 and VCAN overexpression was significantly reduced in SF-MSCs.
Hypertrophic and osteogenic genes
COL10A1, a marker of hypertrophic differentiation, was strongly increased by TGF-β1, especially in BM-MSCs. SF-MSCs were less responsive than the other cells. RUNX2 is a transcription factor that is involved in osteogenic differentiation, and its expression was very low in the three lineages, even under the influence of TGF-β1.
GAG content in cartilage substitutes
The GAG content is shown in Fig. 3. For the three cell types, the 1% ITS control condition did not induce the production of GAG, while TGF-β1 (10 ng/mL) significantly induced synthesis in the three lineages. The average amount was 190 μg/sponge for BM-MSCs, 160 μg/mL for SM-MSCs, and 118 μg/mL for SF-MSCs, which was significantly lower than that of BM-MSCs.

GAG content at D28 in collagen sponges seeded with advanced OA human BM-, SM-, and SF-MSCs under ITS and TGF-β1. The concentration of GAG in micrograms per sponge was measured with a dimethyl methylene blue colorimetric assay. Statistical analysis was performed with a t test to evaluate the effect of TGF-β1 versus ITS for each cellular group separately and followed by two-way ANOVA with Bonferroni’s post hoc test on all values to evaluate the difference in GAG production between all cellular groups. Asterisks represent a significant difference versus control condition (ITS 1%); ***p < 0.001. Hash signs represent a significant difference between cellular group; ###p < 0.001
Histological and immunohistochemical analysis of cartilage substitutes
HES revealed homogeneous distribution and no mortality in the cells within the sponge under both ITS and TGF-β1 conditions. As depicted in Fig. 4a, Alcian blue staining revealed no GAG synthesis at D28 under ITS conditions. In contrast, TGF-β1 significantly increased proteoglycan content in the three MSC-seeded substitutes without significant differences between the three cell lineages. Type II collagen labeling revealed that ITS failed to induce any matrix deposition, while TGF-β1 provoked significant type II collagen synthesis. The densitometry measurement (Fig. 4b) demonstrated significant differences between the three cell types; type II collagen content was significantly higher in BM-MSCs than in SM- and SF-MSCs. Under normoxic conditions, no calcium deposition was observed in the sponge of each cellular group (Supplementary data 3).

Histological and immunohistochemical analyses at D28 of collagen sponges seeded with advanced OA human BM-, SM-, and SF-MSCs under ITS and TGF-β1. a The proteoglycan content was visualized by Alcian blue staining, and type II collagen was highlighted by immunohistochemistry. The scale bars represent 400 μm. All observations were carried out on three different samples for each culture condition and for each patient. b For densitometry measurement of Alcian blue staining and type II collagen, statistical analysis was performed with a t test to evaluate the effect of TGF-β1 versus ITS for each cellular group separately and was followed by two-way ANOVA with Bonferroni’s post hoc test on all values to evaluate the difference in GAG production between all cellular groups. Asterisks represent a significant difference versus control condition (ITS 1%); ***p < 0.001. Hash signs represent a significant difference between cellular group; ###p < 0.001
Discussion
The main goal of this study was to directly compare the stemness, immunophenotype, and chondrogenic potential of human advanced OA BM-MSCs, SM-MSCs, and SF-MSCs differentiated in collagen sponges. To date, such a direct comparison has not been reported in humans. First, we confirmed that these three lineages were able to differentiate into chondrocytes, osteocytes, and adipocytes with dedicated media [22,23,24]. We also observed that MSCs derived from bone marrow, synovial tissue, and synovial fluid exerted a similar immunophenotype as demonstrated by cell marker expression [25]. Despite some variability, CD105 (endoglin), a stemness marker, was very highly expressed by the three MSC lineages. This transmembrane protein is involved in the TGF-β signaling pathways. Although debated [26], it is also an excellent predictor of MSC chondrogenic differentiation. For example, it is used to select SM-MSCs [27]. In addition, our results showed strong expression of CD73 in all three cell types. This enzyme that metabolizes AMP to adenosine is described as a marker that is associated with chondrogenic differentiation, and its expression decreases significantly with chondrogenic differentiation [28].
Interestingly, the stemness marker CD90 was less positive in BM-MSCs (approximately 50%) than in SM-MSCs (approximately 75%). With regard to the CD90/Thy-1 marker, it has been shown that this marker is expressed on the MSC surface, particularly on undifferentiated MSCs [28]. Moreover, its expression decreases once the MSCs engage in chondrogenic or osteogenic differentiation [29]. In physiological conditions, it has been shown that chondrocytes do not express CD90, and this expression increases during dedifferentiation in monolayers [30], thus confirming the association of marker expression with the undifferentiated status of our MSCs. Additionally, during the expansion stages, our medium contained 1 ng/mL of bFGF, which is known to induce cell proliferation. This supplementation of bFGF can modulate the expression of cell surface markers such as CD90 in SM-MSCs [31].
The negative markers CD34 and CD45 were poorly expressed. CD34 is expressed on the surface of hematopoietic stem cells (HSCs), and CD45 is present on all nucleated hematopoietic cells (except erythrocytes and platelets). Our results confirmed that HSC markers are not expressed on BM-, SM-, and SF-MSCs at P2. Interestingly, HLA-DR, a negative marker, was moderately expressed without a significant difference between the three MSC types. These results were surprising because HLA-DR is a surface receptor of the major histocompatibility complex that is generally associated with GvHD (graft versus host disease). It has been shown previously that BM-MSCs from healthy bone marrow express HLA-DR only after stimulation by IFN-γ [32]. In another study, the heterogeneity of HLA-DR expression in human BM-MSCs was reported [33]. Again, the use of bFGF in cellular amplification may also induce the expression of HLA-DR [34]. Finally, it has been shown that the immunophenotype of BM-MSCs can be altered by in vitro proliferation [35].
Inter-individual variability is likely to contribute to the intergroup variability of immunophenotypes [36]. All of the cells used here came from elderly patients with advanced osteoarthritis of the hip or knee. Thus, the age of the donor, drug treatments, and the pathological microenvironment may interfere. Anatomical differences may be of importance. BM-MSCs are not in contact with the joint cavity, while SM-MSCs are. In addition, SF-MSCs are immersed in hypoxic and acidic joint fluid [18] and are influenced by the inflammatory environment [37,38,39], and the resident senescent cells develop a detrimental paracrine senescence-associated secretory phenotype [40]. In contrast, in our experience, this immunophenotypic heterogeneity was not found in healthy neonatal MSC populations, such as MSCs derived from human umbilical cord blood or Wharton jelly [21].
To compare the respective BM-, SM-, and SF-MSC chondrogenic potential, we chose to use a porous support biomaterial based on type I collagen sponges that allows the synthesis of extracellular matrix within this biomaterial, which is unlike the pellet model, a 3D structure based on cell-cell interactions. Collagen sponges are useful for their compatibility with clinical practice and cartilage engineering strategies [41, 42]. This biomaterial allows attachment of the cells and the expression of chondrogenic markers when the cells are seeded in the presence of TGF-β1. This collagen scaffold is biodegradable and is made with type I atelocollagen that is not immunogenic. This collagen isotype is chemically crosslinked to obtain a 3D scaffold with excellent mechanical and thermal stability [43]. With this in mind, we confirmed that TGF-β1 strongly induced chondrogenesis in BM-MSCs [44], SM-MSCs [45, 46], and SF-MSCs [18, 36, 47]. These results were consistent with our previous work showing that the use of TGF-β1 induced strong overexpression of the genes of interest, such as type 2 collagen and aggrecan, after 28 days of BM-MSC differentiation in collagen sponges [48]. Additionally, our preliminary data confirmed that BMP-2 alone, a promoter rather than an inducer [9], did not promote chondrogenesis in these three MSCs lineages embedded in collagen sponges. Besides, we observed that TGF-β1, TGF-β3, TGF-β1+BMP-2 and TGF-β3+BMP2 were quite equipotent in terms of chondrogenesis at both genic and protein levels (unpublished internal data).
We also evaluated the expression of chondrogenic genes in MSC-seeded sponges by quantitative RT-PCR. As expected, ITS control conditions did not induce the expression of any genes of interest. In contrast, as previously reported [12], TGF-β1 induced, to varying degrees, the chondrogenic differentiation of MSCs from bone marrow, synovial membrane, and synovial fluid. These results are consistent with our previous work showing that the use of TGF-β1 induced strong overexpression of chondrogenic genes such as ACAN, COL2A1, and its isoform COL2B at D28 in MSC-seeded collagen sponges [48]. Interestingly, overexpression of these genes was higher in BM-MSCs and SM-MSCs than in SF-MSCs.
We also studied the expression of the transcription factors SOX9 (the “chondromaster” gene) and RUNX2 (the “osteomaster” gene). For all three cell types, SOX9 expression was significantly increased in a similar manner, with BM-MSCs exerting the highest ratio and SF-MSCs the lowest. Conversely, RUNX2 expression remained very low, suggesting that chondrogenic differentiation did not skew towards terminal ossification. Nevertheless, COL10A1 was highly expressed in BM-MSCs, mildly in SM-MSCs, and poorly in SF-MSCs. This result is consistent with the literature because, under normoxia, chondrogenic differentiation is often associated with an increase in markers associated with hypertrophy [49,50,51]. These results fit with biochemical, histological, and immunohistochemical analyses. These invasive techniques provide qualitative information on cell distribution and the amount of neo-synthesized extracellular matrix (ECM). GAG content is less important for SF-MSCs. Interestingly, type II collagen (specific immunohistochemistry) was more abundant in BM-MSCs. It is important to note that the different cartilage substitutes did not show any calcification after 28 days, whereas calcification is classically observed with MSC-seeded alginate 3D culture under normoxia [52].
Several studies have compared the differentiation capabilities and chondrogenic potential of different sources of synovial stem cells from humans [53,54,55,56], horses [36, 57,58,59], dogs [60], and rats [61] versus bone marrow MSCs. Their results often showed superior chondrogenicity of cells derived from synovium (SM and/or SF) compared to that of BM-MSCs [62]. However, these studies were conducted in different species with various anatomical origins (hip, knee, and femoral diaphysis) and pathological conditions (temporomandibular joint dysfunction and anterior cruciate ligament rupture). Intraindividual variation in SF-MSC-related chondrogenicity between the hip and knee in patients who underwent both hip and knee arthroscopy on the same day has recently been demonstrated [63]. Furthermore, the three-dimensional differentiation system and growth factors were variable within the literature. Our study was conducted in patients with old age who were suffering from advanced OA, which may promote age-related alterations affecting the chondrogenicity of synovial MSCs, as recently demonstrated in an equine model [64] and in the clinic [65]. Additionally, changes in gene expression do not always correlate with protein synthesis, which may contribute to the differences observed in the 3 MSC lineages [66]. Apart from this in vivo senescence, whose influence on chondrogenicity remains debated [67], an in vitro replicative senescence (aging) might influence the results: environmental factors like enzyme-dependent cell detaching methods [68] or numbers of passages [69, 70] during expansion. These results are consistent with our internal data (Supplementary data 4). Interestingly, a robust work performed with BM-MSCs demonstrates that chondrogenicity is altered only after P6, osteogenic commitment after P5, while adipogenicity persists at P11. Furthermore, environmental conditioning, notably with FGF, rejuvenates MSCs’ proliferation and chondrogenic potential [71]. Finally, our chondrogenic gradient BM-MSCs > SM-MSCs > SF-MSCs is consistent with a previous study performed in similar patients on P3 with the pellet’s conditions under the influence of a mix of TGF-β3 + BMP-6 [72].
Conclusion
We demonstrated that BM-, SM-, and SF-MSCs from advanced OA patients undergoing hip or knee arthroplasty are valuable candidates for cartilage engineering, although they show different immunophenotyping and chondrogenic properties. No studies have been conducted in the clinic comparing these three MSC lineages for cartilage engineering. All cell types were able to grow in monolayers and differentiate into adipogenic, osteogenic, and chondrogenic linages. The cells also responded to various levels of TGF-β1-driven 3D chondrogenic differentiation in collagen sponges. However, there was considerable variability within and between the lineages. Most likely, these differences were due to the detrimental priming of the OA-related intra-articular inflammatory environment on SM- and SF-MSCs; bone marrow MSCs probably remain the best candidate for hyaline cartilage engineering. In contrast, SF-MSCs did not overexpress hypertrophic genes under our currently described culture conditions.
Availability of data and materials
The datasets taken during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ACAN :
-
Aggrecan (gene)
- ACI:
-
Autologous chondrocyte implantation
- ANOVA:
-
Analysis of variance
- bFGF:
-
Basic fibroblast growth factor
- BGLAP :
-
Osteocalcin (gene)
- BM:
-
Bone marrow
- BMP-2:
-
Bone morphogenetic protein 2
- CO2 :
-
Carbon dioxide
- COL10A1 :
-
Collagen type X, alpha 1 chain (gene)
- COL2A :
-
Collagen type II alpha 1 chain (gene)
- Col2b :
-
Collagen type II isoform (gene)
- COMP :
-
Cartilage oligomeric matrix protein (gene)
- DMEM:
-
Dulbecco’s modified Eagle medium
- DNA:
-
Deoxyribonucleic acid
- ECM:
-
Extracellular matrix
- EDTA:
-
Ethylene diamine tetra acetic acid
- FBS:
-
Fetal bovine serum
- GAG:
-
Glycosaminoglycan
- GvHD:
-
Graft versus host disease
- HES:
-
Hematoxylin erythrosine saffron
- HSC:
-
Hematopoietic stem cells
- IHC:
-
Immunohistochemistry
- ITS:
-
Insulin-transferrin-selenium
- MSCs:
-
Mesenchymal stromal cells
- MTT:
-
3 (4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- OA:
-
Osteoarthritis
- OSX :
-
Osterix (gene)
- P:
-
Passage 1, 2, 3
- PAD:
-
Proline, L-ascorbic acid-2-phosphate and dexamethasone
- PBS:
-
Phosphate-buffered saline
- PFA:
-
Paraformaldehyde
- qRT-PCR:
-
Quantitative reverse transcription-polymerase chain reaction
- RNA:
-
Ribonucleic acid
- RPS29 :
-
Ribosomal Protein S29 (gene)
- SF:
-
Synovial fluid
- SM:
-
Synovial membrane
- SOX9 :
-
Sex-determining region-related HMG-box 9 (gene)
- TGF-β1:
-
Transforming growth factor beta 1
- VCAN :
-
Versican (gene)
References
Welch T, Mandelbaum B, Tom M. Autologous chondrocyte implantation: past, present, and future. Sports Med Arthrosc Rev. 2016;24(2):85–91.
Ohara T, Muneta T, Nakagawa Y, et al. Hypoxia enhances proliferation through increase of colony formation rate with chondrogenic potential in primary synovial mesenchymal stem cells. J Med Dent Sci. 2016;63(4):61–70.
Lim HC, Bae JH, Song SH, Park YE, Kim SJ. Current treatments of isolated articular cartilage lesions of the knee achieve similar outcomes. Clin Orthop Relat Res. 2012;470(8):2261–7.
Dewan AK, Gibson MA, Elisseeff JH, Trice ME. Evolution of autologous chondrocyte repair and comparison to other cartilage repair techniques. Biomed Res Int. 2014;2014:272481.
Phull AR, Eo SH, Abbas Q, Ahmed M, Kim SJ. Applications of chondrocyte-based cartilage engineering: an overview. Biomed Res Int. 2016;2016:1879837.
Zakrzewski W, Dobrzynski M, Szymonowicz M, Rybak Z. Stem cells: past, present, and future. Stem Cell Res Ther. 2019;10(1):68.
Squillaro T, Peluso G, Galderisi U. Clinical trials with mesenchymal stem cells: an update. Cell Transplant. 2016;25(5):829–48.
Galderisi U, Giordano A. The gap between the physiological and therapeutic roles of mesenchymal stem cells. Med Res Rev. 2014;34(5):1100–26.
Boeuf S, Richter W. Chondrogenesis of mesenchymal stem cells: role of tissue source and inducing factors. Stem Cell Res Ther. 2010;1(4):31.
Nishimura K, Solchaga LA, Caplan AI, Yoo JU, Goldberg VM, Johnstone B. Chondroprogenitor cells of synovial tissue. Arthritis Rheum. 1999;42(12):2631–7.
Crawford A, Frazer A, Lippitt JM, Buttle DJ, Smith T. A case of chondromatosis indicates a synovial stem cell aetiology. Rheumatology (Oxford). 2006;45(12):1529–33.
De Bari C, Dell'Accio F, Tylzanowski P, Luyten FP. Multipotent mesenchymal stem cells from adult human synovial membrane. Arthritis Rheum. 2001;44(8):1928–42.
Huang YZ, Xie HQ, Silini A, et al. Mesenchymal stem/progenitor cells derived from articular cartilage, synovial membrane and synovial fluid for cartilage regeneration: current status and future perspectives. Stem Cell Rev Rep. 2017;13(5):575–86.
Archer CW, Dowthwaite GP, Francis-West P. Development of synovial joints. Birth Defects Res C Embryo Today. 2003;69(2):144–55.
Correa D, Lietman SA. Articular cartilage repair: current needs, methods and research directions. Semin Cell Dev Biol. 2017;62:67–77.
Derfoul A, Perkins GL, Hall DJ, Tuan RS. Glucocorticoids promote chondrogenic differentiation of adult human mesenchymal stem cells by enhancing expression of cartilage extracellular matrix genes. Stem Cells. 2006;24(6):1487–95.
Henrionnet C, Roeder E, Gillet R, et al. Expression of chondrogenic genes by undifferentiated vs. differentiated human mesenchymal stem cells using array technology. Biomed Mater Eng. 2010;20(3):175–81.
Neybecker P, Henrionnet C, Pape E, et al. In vitro and in vivo potentialities for cartilage repair from human advanced knee osteoarthritis synovial fluid-derived mesenchymal stem cells. Stem Cell Res Ther. 2018;9(1):329.
Cailotto F, Reboul P, Sebillaud S, Netter P, Jouzeau JY, Bianchi A. Calcium input potentiates the transforming growth factor (TGF)-beta1-dependent signaling to promote the export of inorganic pyrophosphate by articular chondrocyte. J Biol Chem. 2011;286(22):19215–28.
Goldberg RL, Kolibas LM. An improved method for determining proteoglycans synthesized by chondrocytes in culture. Connect Tissue Res. 1990;24(3–4):265–75.
Reppel L, Schiavi J, Charif N, et al. Chondrogenic induction of mesenchymal stromal/stem cells from Wharton’s jelly embedded in alginate hydrogel and without added growth factor: an alternative stem cell source for cartilage tissue engineering. Stem Cell Res Ther. 2015;6:260.
Lee WJ, Hah YS, Ock SA, et al. Cell source-dependent in vivo immunosuppressive properties of mesenchymal stem cells derived from the bone marrow and synovial fluid of minipigs. Exp Cell Res. 2015;333(2):273–88.
Koyama N, Okubo Y, Nakao K, Osawa K, Fujimura K, Bessho K. Pluripotency of mesenchymal cells derived from synovial fluid in patients with temporomandibular joint disorder. Life Sci. 2011;89(19–20):741–7.
Ando W, Kutcher JJ, Krawetz R, et al. Clonal analysis of synovial fluid stem cells to characterize and identify stable mesenchymal stromal cell/mesenchymal progenitor cell phenotypes in a porcine model: a cell source with enhanced commitment to the chondrogenic lineage. Cytotherapy. 2014;16(6):776–88.
Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–7.
Lu Z, Yan L, Pei M. Commentary on ‘Surface markers associated with chondrogenic potential of human mesenchymal stromal/stem cells’. F1000Res. 2020;9:F1000 Faculty Rev-37.
Harvanova D, Tothova T, Sarissky M, Amrichova J, Rosocha J. Isolation and characterization of synovial mesenchymal stem cells. Folia Biol (Praha). 2011;57(3):119–24.
Krampera M, Pasini A, Rigo A, et al. HB-EGF/HER-1 signaling in bone marrow mesenchymal stem cells: inducing cell expansion and reversibly preventing multilineage differentiation. Blood. 2005;106(1):59–66.
Wiesmann A, Buhring HJ, Mentrup C, Wiesmann HP. Decreased CD90 expression in human mesenchymal stem cells by applying mechanical stimulation. Head Face Med. 2006;2:8.
Giovannini S, Diaz-Romero J, Aigner T, Mainil-Varlet P, Nesic D. Population doublings and percentage of S100-positive cells as predictors of in vitro chondrogenicity of expanded human articular chondrocytes. J Cell Physiol. 2010;222(2):411–20.
Hagmann S, Moradi B, Frank S, et al. FGF-2 addition during expansion of human bone marrow-derived stromal cells alters MSC surface marker distribution and chondrogenic differentiation potential. Cell Prolif. 2013;46(4):396–407.
Le Blanc K, Tammik C, Rosendahl K, Zetterberg E, Ringden O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol. 2003;31(10):890–6.
Jones EA, Kinsey SE, English A, et al. Isolation and characterization of bone marrow multipotential mesenchymal progenitor cells. Arthritis Rheum. 2002;46(12):3349–60.
Sotiropoulou PA, Perez SA, Salagianni M, Baxevanis CN, Papamichail M. Characterization of the optimal culture conditions for clinical scale production of human mesenchymal stem cells. Stem Cells. 2006;24(2):462–71.
Bara JJ, Richards RG, Alini M, Stoddart MJ. Concise review: bone marrow-derived mesenchymal stem cells change phenotype following in vitro culture: implications for basic research and the clinic. Stem Cells. 2014;32(7):1713–23.
Zayed M, Caniglia C, Misk N, Dhar MS. Donor-matched comparison of chondrogenic potential of equine bone marrow- and synovial fluid-derived mesenchymal stem cells: implications for cartilage tissue regeneration. Front Vet Sci. 2016;3:121.
de Sousa EB, Dos Santos Junior GC, Aguiar RP, et al. Osteoarthritic synovial fluid modulates cell phenotype and metabolic behavior in vitro. Stem Cells Int. 2019;2019:8169172.
Zayed MN, Schumacher J, Misk N, Dhar MS. Effects of pro-inflammatory cytokines on chondrogenesis of equine mesenchymal stromal cells derived from bone marrow or synovial fluid. Vet J. 2016;217:26–32.
Bertram KL, Krawetz RJ. Osmolarity regulates chondrogenic differentiation potential of synovial fluid derived mesenchymal progenitor cells. Biochem Biophys Res Commun. 2012;422(3):455–61.
Jeon OH, David N, Campisi J, Elisseeff JH. Senescent cells and osteoarthritis: a painful connection. J Clin Invest. 2018;128(4):1229–37.
Branly T, Bertoni L, Contentin R, et al. Characterization and use of equine bone marrow mesenchymal stem cells in equine cartilage engineering. Study of their hyaline cartilage forming potential when cultured under hypoxia within a biomaterial in the presence of BMP-2 and TGF-ss1. Stem Cell Rev Rep. 2017;13(5):611–30.
Contentin R, Demoor M, Concari M, et al. Comparison of the Chondrogenic potential of mesenchymal stem cells derived from bone marrow and umbilical cord blood intended for cartilage tissue engineering. Stem Cell Rev Rep. 2020;16(1):126–43.
Gomez-Leduc T, Hervieu M, Legendre F, et al. Chondrogenic commitment of human umbilical cord blood-derived mesenchymal stem cells in collagen matrices for cartilage engineering. Sci Rep. 2016;6:32786.
Freyria AM, Mallein-Gerin F. Chondrocytes or adult stem cells for cartilage repair: the indisputable role of growth factors. Injury. 2012;43(3):259–65.
Yasui Y, Chijimatsu R, Hart DA, et al. Preparation of scaffold-free tissue-engineered constructs derived from human synovial mesenchymal stem cells under low oxygen tension enhances their chondrogenic differentiation capacity. Tissue Eng Part A. 2016;22(5–6):490–500.
Sugita N, Moriguchi Y, Sakaue M, et al. Optimization of human mesenchymal stem cell isolation from synovial membrane: implications for subsequent tissue engineering effectiveness. Regen Ther. 2016;5:79–85.
Liang Y, Idrees E, Szojka ARA, et al. Chondrogenic differentiation of synovial fluid mesenchymal stem cells on human meniscus-derived decellularized matrix requires exogenous growth factors. Acta Biomater. 2018;80:131–43.
Roeder E, Henrionnet C, Goebel JC, et al. Dose-response of superparamagnetic iron oxide labeling on mesenchymal stem cells chondrogenic differentiation: a multi-scale in vitro study. PLoS One. 2014;9(5):e98451.
Vinatier C, Guicheux J. Cartilage tissue engineering: from biomaterials and stem cells to osteoarthritis treatments. Ann Phys Rehabil Med. 2016;59(3):139–44.
Pelttari K, Winter A, Steck E, et al. Premature induction of hypertrophy during in vitro chondrogenesis of human mesenchymal stem cells correlates with calcification and vascular invasion after ectopic transplantation in SCID mice. Arthritis Rheum. 2006;54(10):3254–66.
Pievani A, Scagliotti V, Russo FM, et al. Comparative analysis of multilineage properties of mesenchymal stromal cells derived from fetal sources shows an advantage of mesenchymal stromal cells isolated from cord blood in chondrogenic differentiation potential. Cytotherapy. 2014;16(7):893–905.
Henrionnet C, Liang G, Roeder E, et al. Hypoxia for mesenchymal stem cell expansion and differentiation: the best way for enhancing TGFss-induced chondrogenesis and preventing calcifications in alginate beads. Tissue Eng Part A. 2017;23(17–18):913–22.
Sakaguchi Y, Sekiya I, Yagishita K, Muneta T. Comparison of human stem cells derived from various mesenchymal tissues: superiority of synovium as a cell source. Arthritis Rheum. 2005;52(8):2521–9.
Isobe Y, Koyama N, Nakao K, et al. Comparison of human mesenchymal stem cells derived from bone marrow, synovial fluid, adult dental pulp, and exfoliated deciduous tooth pulp. Int J Oral Maxillofac Surg. 2016;45(1):124–31.
Li J, Huang Y, Song J, et al. Cartilage regeneration using arthroscopic flushing fluid-derived mesenchymal stem cells encapsulated in a one-step rapid cross-linked hydrogel. Acta Biomater. 2018;79:202–15.
Danisovic L, Bohac M, Zamborsky R, et al. Comparative analysis of mesenchymal stromal cells from different tissue sources in respect to articular cartilage tissue engineering. Gen Physiol Biophys. 2016;35(2):207–14.
Zayed M, Newby S, Misk N, Donnell R, Dhar M. Xenogenic implantation of equine synovial fluid-derived mesenchymal stem cells leads to articular cartilage regeneration. Stem Cells Int. 2018;2018:1073705.
Arevalo-Turrubiarte M, Olmeo C, Accornero P, Baratta M, Martignani E. Analysis of mesenchymal cells (MSCs) from bone marrow, synovial fluid and mesenteric, neck and tail adipose tissue sources from equines. Stem Cell Res. 2019;37:101442.
Gale AL, Mammone RM, Dodson ME, Linardi RL, Ortved KF. The effect of hypoxia on chondrogenesis of equine synovial membrane-derived and bone marrow-derived mesenchymal stem cells. BMC Vet Res. 2019;15(1):201.
Sasaki A, Mizuno M, Ozeki N, et al. Canine mesenchymal stem cells from synovium have a higher chondrogenic potential than those from infrapatellar fat pad, adipose tissue, and bone marrow. PLoS One. 2018;13(8):e0202922.
Yoshimura H, Muneta T, Nimura A, Yokoyama A, Koga H, Sekiya I. Comparison of rat mesenchymal stem cells derived from bone marrow, synovium, periosteum, adipose tissue, and muscle. Cell Tissue Res. 2007;327(3):449–62.
Beane OS, Darling EM. Isolation, characterization, and differentiation of stem cells for cartilage regeneration. Ann Biomed Eng. 2012;40(10):2079–97.
Hatakeyama A, Uchida S, Utsunomiya H, et al. Isolation and characterization of synovial mesenchymal stem cell derived from hip joints: a comparative analysis with a matched control knee group. Stem Cells Int. 2017;2017:9312329.
Mazzotti E, Teti G, Falconi M, et al. Age-related alterations affecting the chondrogenic differentiation of synovial fluid mesenchymal stromal cells in an equine model. Cells. 2019;8(10):1116.
Krawetz RJ, Wu YE, Martin L, Rattner JB, Matyas JR, Hart DA. Synovial fluid progenitors expressing CD90+ from normal but not osteoarthritic joints undergo chondrogenic differentiation without micro-mass culture. PLoS One. 2012;7(8):e43616.
Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13(4):227–32.
Charif N, Li YY, Targa L, et al. Aging of bone marrow mesenchymal stromal/stem cells: implications on autologous regenerative medicine. Biomed Mater Eng. 2017;28(s1):S57–63.
Tsuji K, Ojima M, Otabe K, et al. Effects of different cell-detaching methods on the viability and cell surface antigen expression of synovial mesenchymal stem cells. Cell Transplant. 2017;26(6):1089–102.
Yang YK, Ogando CR, Wang See C, Chang TY, Barabino GA. Changes in phenotype and differentiation potential of human mesenchymal stem cells aging in vitro. Stem Cell Res Ther. 2018;9(1):131.
Turinetto V, Vitale E, Giachino C. Senescence in human mesenchymal stem cells: functional changes and implications in stem cell-based therapy. Int J Mol Sci. 2016;17(7):1164.
Pei M. Environmental preconditioning rejuvenates adult stem cells’ proliferation and chondrogenic potential. Biomaterials. 2017;117:10–23.
Alegre-Aguaron E, Desportes P, Garcia-Alvarez F, Castiella T, Larrad L, Martinez-Lorenzo MJ. Differences in surface marker expression and chondrogenic potential among various tissue-derived mesenchymal cells from elderly patients with osteoarthritis. Cells Tissues Organs. 2012;196(3):231–40.
Acknowledgements
We thank the nursing staff of Chirurgie Orthopédique & Traumatologique and Centre Emile Gallé, CHRU Nancy, for their assistance in the provision of clinical material.
Funding
Dedicated funding from “Société Française de Rhumatologie” supported this work.
Paul Neybecker was supported by a Ph.D. scholarship granted by the French Ministry of National Education, Higher Education and Research (MENESR).
Author information
Authors and Affiliations
Contributions
PN and EP contributed to experimental design, cell and tissue culture, biochemical analyses, histological analyses, statistical analyses, and manuscript preparation. DL, LG, LGa, and DM contributed to MSCs collection, study conception, results analysis, and manuscript preparation. AP, CH, and PG supervised study conception, experimental design, cell and tissue culture, biochemical analyses, animal model processing, histological analyses, statistical analyses, and manuscript revision and editing. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The clinical protocol was approved by the Ethical Committee of our Universitary Hospital (Comité de Protection des Personnes Est-III, file DC 2014—2148, authorized 2014, July 10).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
RT-PCR analysis of MSCs differentiation
Additional file 2.
Osteogenic differentiation of MSCs issued from human bone marrow (BM-MSCs), from the human synovial membrane (SM-MSCs) and fluid (SF-MSCs) at D14 without (OSTEO-) or with osteogenic medium (OSTEO+). Adipogenic differentiation of MSCs issued from human bone marrow (BM-MSCs), synovial membrane (SM-MSCs) and synovial fluid (SF-MSCs) at D21 without (ADIPO-) or with adipogenic medium (ADIPO+)
Additional file 3.
Von Kossa staining at D28 of collagen sponges seeded with advanced OA human BM, SM- and SF-MSCs under ITS and TGF-β1
Additional file 4.
The senescence was evaluated using a kit "senescence β galactosidase staining kit" according to the manufacturer's recommendations (Cell Signaling Technology)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Neybecker, P., Henrionnet, C., Pape, E. et al. Respective stemness and chondrogenic potential of mesenchymal stem cells isolated from human bone marrow, synovial membrane, and synovial fluid. Stem Cell Res Ther 11, 316 (2020). https://doi.org/10.1186/s13287-020-01786-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-020-01786-5