
Abstract
Background
Since cartilage-derived stem/progenitor cells (CSPCs) were first identified in articular cartilage using differential adhesion to fibronectin, their self-renewal capacity and niche-specific lineage preference for chondrogenesis have propelled their application for cartilage tissue engineering. In many adult tissues, stem/progenitor cells are recognised to be involved in tissue homeostasis. However, the role of nasoseptal CSPCs has not yet been elucidated. Our aim was to isolate and characterise nasoseptal CSPCs alongside nasoseptal chondrocyte populations and determine chondrogenic capacity.
Methods
Here, we isolated nasoseptal CSPCs using differential adhesion to fibronectin and assessed their colony forming efficiency, proliferation kinetics, karyotype and trilineage potential. CSPCs were characterised alongside non-fibronectin-adherent nasoseptal chondrocytes (DNCs) and cartilage-derived cells (CDCs, a heterogenous combination of DNCs and CSPCs) by assessing differences in gene expression profiles using PCR Stem Cell Array, immunophenotype using flow cytometry and chondrogencity using RT-PCR and histology.
Results
CSPCs were clonogenic with increased gene expression of the neuroectodermal markers NCAM1 and N-Cadherin, as well as Cyclins D1 and D2, compared to DNCs. All three cell populations expressed recognised mesenchymal stem cell surface markers (CD29, CD44, CD73, CD90), yet only CSPCs and CDCs showed multilineage differentiation potential. CDC populations expressed significantly higher levels of type 2 collagen and bone morphogenetic protein 2 genes, with greater cartilage extracellular matrix secretion. When DNCs were cultured in isolation, there was reduced chondrogenicity and higher expression of type 1 collagen, stromal cell-derived factor 1 (SDF-1), CD73 and CD90, recognised markers of a fibroblast-like phenotype.
Conclusions
Fibronectin-adherent CSPCs demonstrate a unique gene expression profile compared to non-fibronectin-adherent DNCs. DNCs cultured in isolation, without CSPCs, express fibroblastic phenotype with reduced chondrogenicity. Mixed populations of stem/progenitor cells and chondrocytes were required for optimal chondrogenesis, suggesting that CSPCs may be required to retain phenotypic stability and chondrogenic potential of DNCs. Crosstalk between DNCs and CSPCs is proposed based on SDF-1 signalling.
Similar content being viewed by others

Introduction
The ability to successfully tissue engineer cartilage would have a significant impact on the ability to reconstruct cartilaginous defects and thereby restore function. Contemporary cartilage tissue engineered implants, often using unrelated adult stem cell sources, do not produce stable, physiologically relevant cartilage [1, 2]. Diseases with great health economic and physical burdens such as osteoarthritis, propagated by the low self-repair properties of cartilage, have stimulated research into articular cartilage tissue engineering [3,4,5]. There has been a wide range of alloplastic, autologous and composite attempts to produce cartilage implants, which have been associated with a variety of complications including inflammation, resorption, extrusion, infection and migration [6,7,8]. Cell integration in the form of chondrocytes has been used to reduce complication rates with mixed results [6, 9,10,11,12]. The use of stem cells in cartilage tissue engineering was a major development in the field. Although mesenchymal stem cells (MSCs) from unrelated sources including the bone marrow, adipose tissue and skeletal muscle demonstrate chondrogenic potential, the majority of studies have shown that these MSCs often lead to fibrotic and calcified cartilage with poor mechanical properties and low physiological relevance [10, 13,14,15,16].
Since they were first identified in the articular cartilage using differential adhesion to fibronectin [17,18,19,20], cartilage-derived stem/progenitor cells (CSPCs) have been added to the repertoire for cartilage tissue engineering [21,22,23,24]. CPSCs have been characterised by convention of the International Society of Cellular Therapy (ISCT) based on the demonstration of plastic adherence, multipotency and positivity for stem-cell-related surface markers [17, 19, 25, 26]. As a clonogenic, renewable cell source derived from the specific tissue that requires replacement, CSPCs are of interest due to their niche-specific lineage preference for chondrogenesis [27,28,29].
In the present study, we isolated cells from human nasoseptal cartilage due to relative ease of access for biopsy and favourable donor site morbidity versus articular cartilage [30]. This donor site offers several other advantages including the ability of nasal chondrocytes to adapt to heterotopic transplantation to articular sites and potent chondrogenic potential which is hyaline specific [31,32,33]. Although previous studies have suggested a role for isolated CSPCs for cartilage tissue engineering, none have characterised them alongside chondrocytes or in mixed populations to investigate their possible role in phenotype modulation and chondrogenesis. As seen in vivo, the tissue microenvironment and stem cell niche plays a significant role in contributing to the phenotypic stability of cells and chondrogenesis [34]. The aim of this study was therefore to fully characterise heterogenous cell populations in nasoseptal cartilage. Cartilage stem/progenitor cells (CSPCs) were isolated using differential adhesion to fibronectin and characterised alongside differentiated nasoseptal chondrocytes (DNCs) and cartilage-derived cells (CDCs, a combination of DNCs and CSPCs). We demonstrate that heterogenous cell populations have greater chondrogenic potential than the use of stem/progenitor cells alone, indicating important crosstalk that may contribute to phenotypic stability.
Materials and methods
Isolation of human nasoseptal cartilage stem/progenitor cells (CSPCs)
Adult human nasoseptal cartilage was obtained from healthy donors undergoing septorhinoplasty after obtaining informed consent at Singleton and Morriston Hospitals, Swansea, UK. All procedures were approved by the ABM University Health Board (IRAS ID 99202). Surface fibrous tissue was removed, and the remaining blood was washed away using Dulbecco’s modified Eagle medium (DMEM, Thermofisher Scientific, Waltham, MA, USA) prior to mincing the cartilage into ~ 1-mm3 pieces. Sequential tissue digestion was performed with gentle agitation using 2 mg/ml pronase (Roche, Basel, Switzerland) solution for 40 min and 2.4 mg/ml collagenase (Sigma-Aldrich, St. Louis, MO, USA) for 16–18 h, all at 37 °C [35]. The enzymatic solutions were prepared using DMEM supplemented with 1% penicillin-streptomycin solution (PS, Thermofisher Scientific), 1 mM d-glucose solution (Thermofisher Scientific) and 0.1% minimum essential medium (MEM) non-essential amino acids (NEAA, Thermofisher Scientific). Digested tissue was filtered through a 40-μm cell strainer (VWR, Radnor, PA, USA) and centrifuged for 5 min at 500g. Cells were re-suspended in chondro-medium (CM, DMEM supplemented with 10% foetal bovine serum (FBS, Thermofisher Scientific), 1% PS, 1 mM d-glucose and 0.1% MEM NEAA) and plated (1) in culture grade plastic surface (i.e. CDCs) or (2) in fibronectin-coated surface using a previously reported protocol [18, 20, 29, 36]. Briefly, 6-well plates (Sigma-Aldrich) were coated with 10 mg/ml fibronectin (Sigma-Aldrich) in Dulbecco’s phosphate-buffered saline (DPBS, pH 7.4, Thermofisher Scientific) containing 0.5 mM magnesium chloride (MgCl2) and 0.9 mM calcium chloride (CaCl2). Cells were seeded at low density (2000 cells/well) in CM and incubated for 20 min in standard culture conditions (i.e. 5% CO2 and 37 °C). Non-adherent cells were collected and seeded on new non-coated 6-well plates (i.e. DNCs). Fresh CM was added to the fibronectin-adherent CSPCs. Once confluent, all three cell populations were passaged on culture grade plastic under standard culture conditions and the same cell density, with medium changes every 2–3 days. Cells were trypsinyzed (0.05% trypsin-EDTA, Thermofisher Scientific) for 5 min at 37 °C, centrifuged at 587g for 5 min, resuspended in fresh CM and re-seeded at 6.7 × 103 cells/cm2. Cells were kept in culture under standard conditions up to passage 13 (P13).
Growth kinetics of CDCs, DNCs and CSPCs
Short-term cell proliferation was determined using the RTCA iCELLigence™ system (ACEA Biosciences, San Diego, CA, USA). P2 cells were seeded in 8-well E-plates at 10,000 cells/well and CM under standard culture conditions. Cell attachment and proliferation were monitored in real time based on cellular impedance. Wells containing CM only were used as negative controls. The cell index (CI) is a function of the cell number and ratio of cells at different time intervals; CI = 0 when there is no cell adhesion. The CI in a RTCA system is the result of the impedance induced by adherent cells to the electron flow. CI is calculated as follows: CI = (impedance at time point n-impedance in the absence of cells)/nominal impedance value. Measurements for CI were taken every minute for the first 2 h and then every hour for 24 h for all three cell populations (CDC, DNC and CSPC).
Long-term proliferative capacity in culture was determined by measuring cumulative population doublings (PD) at each cell passage [37]. Cell growth was determined between P1 and P13 by direct cell counts using trypan blue exclusion method. PDs were calculated using the formula below where N represents cells harvested/cells seeded and used to plot growth curves.
Colony forming efficiency (CFE) assay of CSPCs
CSPCs were seeded at P0 at 200 cells/cm2 and cultured in standard culture conditions with medium changes every 2–3 days. After 14 days, fibroblast-like colony forming units (CFU-F) were counted under an optical microscope. A colony was defined as > 32 cells [18]. Colony forming efficiency (CFE) was calculated as a percentage of CFU-F from the initial number of cells seeded [36, 38] and used as a predictor for the proportion of CSPCs within the original nasoseptal cartilage cell population.
Trilineage differentiation of CDCs, DNCs and CSPCs
For osteogenic and adipogenic differentiation, cells were seeded (osteogenic, 20,000 cells/well and adipogenic, 40,000 cells/well) onto 12-well plates (Sigma-Aldrich) and cultured in CM for 3 days under standard culture conditions. The medium was then changed to either StemPro™ osteogenesis differentiation medium (Thermofisher Scientific) or StemPro™ adipogenesis differentiation medium (Thermofisher Scientific). Cultures were maintained for 21 days under standard culture conditions with medium changes every 2–3 days. For chondrogenic differentiation, 500,000 cells were resuspended in 1.5 ml Eppendorf in StemPro™ chondrogenesis differentiation medium (Thermofisher Scientific) and centrifuged for 5 min at 783g. Pellets were cultured for 21 days under standard culture conditions with medium changes every 2–3 days. Negative control samples of each cell type were maintained in CM. After 21 days, cells were fixed in 4% paraformaldehyde (PFA, Alfa Aesar, Haverhill, MA, USA) for 30 min. Osteogenesis and adipogenesis were confirmed using 2% Alizarin Red S and 0.3% Oil Red O, respectively. A 1% Alcian blue solution prepared in 0.1 N hydrochloric acid (HCl) was used to confirm the presence of glycosaminoglycans (GAGs) in chondrogenic differentiation. Stains were visualised using phase-contrast AmScope MD35 microscope (AmScope, Irvine, CA, USA).
Histological staining
Cells were fixed in 4% PFA for 30 min and washed in phosphate-buffered saline (PBS, Thermofisher Scientific). Cells were stained with 1% Alcian blue (TCS Biosciences, Buckingham, UK) solution for 15 min and washed with water, while others were stained with 0.1% toluidine blue (TCS Biosciences) solution in water for 3 min following wash with water. For safranin-O stain, cells were exposed to 0.1% fast green (TCS Biosciences) solution for 10 min, immersed in 1% acetic acid for 10 s (Sigma-Aldrich), stained with 0.1% safranin-O (TCS Biosciences) for 20 min and washed with water. All stains were visualised using phase-contrast AmScope MD35 (AmScope).
Flow cytometry
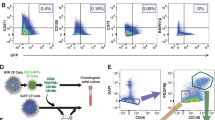
CDCs, DNCs and CSPCs from P1 and P8 were immunophenotypically characterised using flow cytometry and a panel of mouse anti-human monoclonal antibodies against CD29, CD34, CD44, CD45, CD56, CD73 and CD90 (all from Biolegend, Supplementary Table S1). P1 and P8 cells were used to assess phenotype at both early and late passages. Unstained cell populations were used as controls. A minimum of 10,000 events were collected for each sample, and data was acquired using a Novocyte® flow cytometer (ACEA Biosciences) and analysed by FlowJo® software (FlowJo, LLC, Ashland, OR, USA). The geometric mean fluorescence intensity (MFI) for each cell surface marker was used as quantitative measure of expression relative to the unstained controls to allow the level of expression to be compared between the three cell populations. Percentage of cells expressing each cell surface marker was used as a further quantitative measure to allow comparisons between the three cell populations.
RNA extraction, quantitative real-time PCR and PCR array
Total RNA was extracted using Trizol (Thermofisher Scientific) and chloroform (Sigma-Aldrich) and purified using RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. RNA concentration was determined using NanoDrop (Thermofisher Scientific) and converted into cDNA using Superscript IV reverse transcriptase (Thermofisher Scientific) following manufacturer’s protocol. Quantitative PCR (qPCR) was completed using Human Stem Cell RT2 Profiler™ PCR Array (Qiagen, Supplementary Figure S1) according to the manufacturer’s protocol and using iCycler Real-Time PCR system (Bio-rad, Hercules, CA, USA). Gene expression was quantified using GeneGlobe Data Analysis Center software (Qiagen) and normalised to RPLP0, using CDCs as a control. A volcano plot was generated to compare CSPCs and DNCs.
Cytogenetic analysis
Karyotyping was performed through a collaboration with the Institute of Medical Genetics from Cardiff and Vale University Health Board. Cells from passage 1 and passage 4 from 5 human donors were used to investigate whether prolonged expansion during in vitro culture causes any gross karyotype changes that may warrant further genotoxicology studies, with a view to autologous cartilage-derived cells being utilised for tissue engineering in the future. Passage 4 was chosen because it provides enough cells for seeding into 3D scaffolds and there are several reports in the literature that CSPCs can maintain capacity to be reprogrammed into chondrocytes even after passage 4 [39, 40]. Briefly, cells at 70% confluence were exposed overnight and under standard culture conditions to BioWhittaker® Amniochrome II medium (Lonza, Basel, Switzerland) containing 37.5 μg/ml of 5′-Bromo-2′-deoxyuridine (BrdU, Sigma-Aldrich) and 0.6 μg/ml Colcemid (Invitrogen, Carlsbad, CA, USA). Cultures were washed with PBS, trypsinised using trypsin/EDTA solution (Life Technologies, Carlsbad, CA, USA) for 2 min and resuspended in a 1:10 FBS (Thermofisher Scientific) and sterile water solution. Cell suspensions were fixed by adding 3:1 methanol-acetic acid fixative (VWR, BDH Chemicals) followed by centrifugation at 487g for 10 min (performed twice) and 2:1 methanol-acetic acid followed by another centrifugation. Pellets were resuspended in 2:1 methanol-acetic acid fixative, spread on slides and dried at a relative humidity of 50%. For Giemsa banding (GTG-banding), slides (aged 3–5 days at room temperature) were placed in trypsin solution for 5–10 s, rinsed in 3 changes of normal saline and stained in 10–20% RA Lamb Giemsa stain (Thermofisher Scientific) in phosphate buffer pH 6.8 (VWR, BDH Chemicals) for 1.5 min. After rinsing in 3 changes of phosphate buffer pH 6.8, slides were dried and mounted in Entellan mountant (Merck, Kenilworth, NJ, USA).
Statistical analysis
Statistical data are represented as means ± standard error of the mean (SEM) unless otherwise indicated. One-way ANOVA was applied to calculate p values. Statistical differences between groups for the same experimental set were determined using Tukey post hoc test. Statistical analysis was performed using Minitab® 18 (Minitab, Inc., State College, PA, USA). A p value ≤ 0.05 was considered significant.
Results
CSPCs show increased expression of CCND1, CCND2, NCAM1 and CDH2 genes compared to DNCs
CSPCs were isolated using differential adhesion to fibronectin from fifteen patient donors following routine septorhinoplasties (Fig. 1). Cells which were not adhered to fibronectin were referred to as DNCs, and the original cell population containing both populations were referred to as CDCs. Nasoseptal cartilage samples (292 ± 124 mg) yielded 11,022 cells/mg of tissue with over 90% viability.

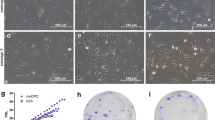
Isolation of nasoseptal cartilage-derived cells. a Gross morphology of nasoseptal cartilage taken from patients undergoing septorhinoplasty. Scale bar, 4 mm. b, c Safranin-O staining of nasoseptal cartilage indicating the fibrous tissue removed (dashed line) prior to enzymatic digestion. Scale bars, 100 μm. d Schematics of the isolation of CDC, DNC and CSPC populations. Briefly, at day 1, the tissue is subjected to enzymatic digestion for 16–18 h. CDCs are seeded in culture grade plastic while the remaining cells are separated based on fibronectin adherence: after 20 min on fibronectin, the non-adherent population (DNCs) is transferred to a separate culture plate. Once confluent all three cell populations were passaged on culture grade plastic under standard culture conditions and the same cell seeding density
Genetic and flow cytometric profiles of the fibronectin-adherent (CSPC) and non-adherent (DNC) cell populations were examined at P1. There is an increase in the expression of neural cell adhesion molecule 1 (NCAM1/CD56) by 2.2-fold (p value < 0.01) and N-cadherin (CDH2) by 1.7-fold (p value 0.001) in CSPC versus DNC populations. Proliferative genes, cyclin D1 (CCND1) and cyclin D2 (CCND2), were also shown to be significantly upregulated in CSPCs compared to DNCs (p < 0.05) (Fig. 2a). Additionally, a decrease in type 1 collagen (COL1A1) expression (p value < 0.01) was observed (Fig. 2a). A hierarchical clustering dendrogram analysis of a heat map of mean 2-ΔCt values for selected genes from PCR array revealed that the significantly upregulated genes NCAM1, CDH2, CCND1 and CCND2 in CSPCs cluster together (Fig. 2b). Both CSPC and DNC populations demonstrated cell surface expression of CD56, recognised MSC markers (CD29, CD44, CD73, CD90) and a lack of haematopoietic markers (CD34, CD45) (Fig. 2c).

Genetic and flow cytometric profiles of CSPC and DNC cell populations. a Volcano plot of PCR array depicting the average of log2 (fold change) and p value of the mRNA expression level of stem cell markers in CSPCs versus DNCs (n = 7). b Heat map and hierarchical clustering dendrogram of the average 2-ΔCt values of co-regulated genes that were significant upregulated or downregulated in at least three biological samples in the CSPC and DNC populations. c Flow cytometry characterisation of cell surface markers on CSPCs and DNCs
CSPCs are a slow proliferating clonogenic subpopulation from human nasoseptal cartilage
CSPCs were demonstrated to be clonogenic with an average CFE of 3.2 ± 0.15 (Fig. 3a), suggesting 3.2% of cells in the original isolated nasoseptal cell population are CSPCs. All three cell populations showed comparable long-term growth curves and achieved at least 20 population doublings over 13 passages (over 200 days in culture). The slope of the CDC growth curve appeared to reduce after passage 9 potentially indicating senescence (Fig. 3b). The impedance-based proliferation assay demonstrated that the DNC population had greater proliferative capacity by reaching a significantly higher maximal cell index compared to the slower-cycling CSPC population (0.80 ± 0.05 and 0.58 ± 0.04, respectively, p < 0.001, Fig. 3c). Karyotype analysis indicated no chromosomal abnormalities for all three cell populations after four passages in culture (P1 vs P4) and thereby feasibility for in vitro expansion towards cartilage tissue engineering applications (Fig. 3d). As part of the genes evaluated by PCR array, CCND2 was shown to be significantly downregulated in DNCs (8.3-fold, p < 0.001) and CSPCs (5.3-fold, p < 0.001) with respect to CDCs (Fig. 4a).

Characterisation of proliferating CDC, DNC and CSPC populations. a Colony forming efficiency (CFE; left axis—red) of CSPCs from nine donors and number of cells/colony (right axis—black) after 14 days in culture. Upper panel insert: representative crystal violet staining of CSPC colonies. Scale bars = 10 mm and 100 μm (left to right). b Population doublings (PD) of CDC, DNC and CSPC cultures along cell passages (P1–P13). Inset represents patient variability (n = 5 at passage 4). c Changes in cell index over time according to RTCA iCELLigence™ assay (n = 3) with bar graphs to demonstrate maximum cell index reached after 24 h in culture, using RTCA iCELLigence™ assay (n = 3). Results as mean ± SEM, ***p < 0.001. d G-band karyotype analysis of CDCs, DNCs and CSPCs at P4 (46XY) representative analysis from 5 human donors analysed for changes at P1 vs P4

Chondrogenic potential of CDC, DNC and CSPC populations. a Relative gene expression of chondrogenic-, proliferative- and progenitor-related genes in CDCs, DNCs and CSPCs. Expression levels were normalised to the level of RPLP0, and relative expression was calculated against CDCs. CCND2, cell cycle cyclin D2; BMP2, bone morphogenetic protein 2; COL1A1, type 1 collagen; COL2A1, type 2 collagen; ACAN, aggrecan; CXCL12, C-X-C motif chemokine 12; NCAM1, neural cell adhesion molecule 1; SOX9, sex-determining region Y box 9. Results as mean ± SEM from 6 independent experiments (n = 6) in technical triplicates, *p < 0.05, **p < 0.01 and ***p < 0.001. b–j Histology staining of b–d CDCs, e–g DNCs and h–j CSPCs at P2 using b, e and h Alcian blue; c, f and i Safranin-O; and d, g and j toluidine blue. Images are representative from 3 independent experiments (n = 3). Scale bars, 100 μm
CDCs demonstrate greater chondrogenicity
The chondrogenic potential of the three populations was investigated at the gene and protein level. PCR arrays showed CDCs expressed significantly higher levels of type 2 collagen (COL2A1) than DNC and CSPC populations (Fig. 4a). Interestingly, COL2A1 was significantly downregulated in CSPC subpopulation by 24-fold (p < 0.01) with respect to CDCs and 5.5-fold (p < 0.05) with respect to DNCs (Fig. 4a). Bone morphogenetic protein-2 (BMP2) expression has exhibited significant downregulation in DNCs and CSPCs by 2.2-fold and 2.3-fold respectively compared to CDCs (p < 0.001, Fig. 4a). However, aggrecan (ACAN) and SOX9 mRNA expression is observed to be expressed at similar levels in all cell types (Fig. 4a). In contrast, at the protein level, CDCs are observed to have enhanced glycosaminoglycan secretion as evident by the intense Alcian blue (Fig. 4b, e and h) and safranin O (Fig. 4c, f and i) staining in comparison to DNC and CSPC populations. While the toluidine blue staining is of similar intensity in all three cell populations, the distribution of the staining is sparser in the DNCs and CSPCs when compared to the CDC population (Fig. 4d,g and j).
CDCs and CSPCs demonstrate multilineage potential in vitro
The ability of chondrocyte populations to commit to trilineage differentiation was compared in vitro (Fig. 5a–i). CDC and CSPC populations demonstrated trilineage commitment whereas DNCs have limited lineage plasticity. DNC cultures possessed no osteogenic and poor adipogenic potential (Fig. 5e, h) as evident by limited staining with Alizarin red and Oil Red O respectively. All three subpopulations (CDCs, DNCs and CSPCs) stained positively for Alcian blue following chondrogenic differentiation in pellet culture (Fig. 5a–c).

Nasoseptal CSPCs are multipotent and positive for MSCs markers. a–i Multilineage differentiation ability of CDCs (a, d, g), DNCs (b, e, h) and CSPCs (c, f, i) into a–c chondrogenic, d–f osteogenic and g–i adipogenic lineages at P2. Images are representative from 3 independent experiments (n = 3). Scale bars, 100 μm. j–l Flow cytometry characterisation of CDCs, DNCs and CSPCs. j Dot blots representative of cell populations from P1. k, l Mean fluorescence intensity (MFI) calculated for P1 and P8 cells, respectively. Results as mean ± SEM from three biological repeats (n = 3), *p < 0.05, **p < 0.01 and ***p < 0.001
DNCs commit to a fibroblastic lineage in the absence of CSPCs
Early passage (P1) nasoseptal cells were further evaluated to determine if the cultured cell populations present distinct immunophenotype profiles (Fig. 5j, k and Table 1). CD29 expression was shown to be at a similar level across all cell types, showing no significant differences (> 165 MFI, Fig. 5k and > 97%, Table 1). CD44 was significantly downregulated in CSPCs in comparison to DNCs with a MFI fold decrease of 2.1 (p < 0.05, Fig. 5k). Although CSPCs present lower CD44 MFI than CDCs (6.81 and 9.92 MFI, respectively, p > 0.05, Fig. 5k), there is higher expression percentage in CSPCs (83.9% and 70.4%, respectively, p > 0.05, Table 1), indicating that this population has more cells expressing CD44, but those expressing it have fewer receptors present. The expression of CD56 is significantly higher in DNCs and CSPCs when compared to CDCs (2.1 and 1.7 MFI fold increase, respectively, p < 0.05, Fig. 5k and 51.3% and 47.9% increase, respectively, p < 0.0001, Table 1).
DNCs showed a significantly higher MFI for CD73 and CD90 when compared to the two other cell populations (2.1- and 1.5-fold increase for CD73 when compared to CDCs and CSPCs, respectively, p < 0.05, and 14.2- and 6.3-fold increase for CD90 when compared to CDCs and CSPCs, respectively, p < 0.001, Fig. 5k). CD73 showed over 99% expression in all cell populations, with DNCs showing higher expression, which was not found to be significant (p > 0.05, Table 1). On the other hand, CD90 expression was significantly increased in DNCs when compared to the other two cell populations (6.6% increase compared to CDCs, p < 0.0001, and around 0.4% increase compared to CSPCs, p < 0.05, Table 1).
Other markers indicative of fibroblastic lineage were found to be elevated in the DNC population including COL1A1 (8.8-fold with respect to CDCs p < 0.001 and 5-fold with respect to CSPCs p < 0.001, Fig. 4a) and C-X-C motif chemokine 12 (CXCL12) (6.8-fold with respect to CDCs p < 0.001 and 5.3-fold in comparison to CSPCs p < 0.01, Fig. 4a). MSC markers have been investigated at a later passage (P8) to determine the effects of culturing DNC and CSPC populations separately; however, there were no significant changes across cell populations indicating that these immunophenotypic differences are not maintained after prolonged culture (Fig. 5l).
Discussion
To date, no optimal cell source has been identified for cartilage tissue engineering purposes. The aneural and avascular nature of cartilage tissue should theoretically make it an easier tissue to replicate compared to other specialised tissue types with more heterogenous functions. However, recapitulating native, functional cartilage remains a challenge [2] and has therefore been the focus for tissue engineering research for many years [41, 42]. Chondrocytes are well documented to undergo fibroblastic differentiation after prolonged expansion in traditional monolayer culture with loss of chondrogenicity [43, 44]. MSCs tend to produce calcified or hypertrophic cartilage [10, 13, 14]. Cartilage-specific stem cells have shown diminished capacity to retain optimal chondrogenic potential when cultured in isolation [45, 46], exhibiting low chondrogenic markers and failing to form a functional matrix in vivo [47,48,49,50]. Nevertheless, it is well established that almost all connective tissues contain tissue-specific stem/progenitor cells which have a significant role in tissue homeostasis and maintenance [51, 52]. Recent studies also speculate that tissue-specific stem/progenitor cells help limit dedifferentiation and maintain phenotypic stability in other tissue types by modulating the local environment [52].
In this study, we successfully isolated CSPCs from human nasoseptal cartilage and characterised them alongside DNC and CDCs. Trilineage differentiation analysis indicated that the multilineage potential observed in CDCs was due to the presence of the CSPC subpopulation, evident from the absence of osteogenic and adipogenic staining in the DNC population. DNCs showed higher proliferation rates than CSPCs, which is in line with the literature addressing the slow proliferation rates of stem/progenitors cultured separately [53]. Interestingly, Cyclin D1 and Cyclin D2 gene expression was greater in CSPCs despite their lower proliferation rates. The differentiation fate of stem cells is tightly tied to cell cycle regulation and cyclins D1 and 2, whose activity is required for cell cycle G1/S transition, has been associated with stem/progenitor cells from a variety of tissue types and can control cell fate and differentiation through transcriptional networks and epigenetic modifiers [54, 55].
All cell populations were positive for recognised MSC markers including CD29, CD44, CD73 and CD90 while being negative for the haematopoietic markers CD34 and CD45. To our knowledge, this is the first study to report CD56/NCAM1 expression in all subpopulations of adult chondrocytes which may be attributed to the previously reported neuroectodermal developmental origin of nasoseptal cartilage in the literature [32]. NCAM1 has been shown to be expressed in embryonic chondroprogenitor cells and mediates cell-cell adhesion during the early stages of cartilage development [56, 57]. However, NCAM1 was not previously thought to be expressed in adult chondrocytes, where the primary interactions were believed to be integrin mediated cell-matrix contacts [58]. Accumulating evidence suggests that biomechanical signals originating from cell-cell adhesion are critical for stem cell lineage specification, and studies have suggested CD56/NCAM1 as a putative marker of connective tissue stem/progenitor cells [59,60,61], but further work is required to determine whether it is a marker of stemness or dedifferentiation in nasoseptal chondrocytes. It may be that in the mixed CDC populations chondrocytes remain in a more differentiated state hence lower MFI and percentage CD56/NCAM1 expression, in keeping with its role during early condensation stage of cartilage development [53].
Our findings suggest that CSPCs may act as a supporter cell to stabilise the chondrogenic phenotype of DNCs. When cultured in isolation, DNCs and CSPCs demonstrated significantly reduced gene expression of chondrogenic markers such as type 2 collagen as well as proteoglycan histological staining compared to the mixed CDC population. DNCs and CPSCs also showed significant downregulation of BMP2, which is recognised in the literature for its requirement in chondrogenic matrix synthesis [62]. Interestingly, DNCs adopt a fibroblastic phenotype in the absence of CSPCs, evidenced by increased expression of COL1A1 and cell surface expression of CD73 and CD90, which are known fibroblastic markers [63, 64] whose expression increases in a time-dependent manner [65]. The expression of the fibroblastic marker, CXCL12, which encodes stromal cell-derived factor 1 (SDF-1), was also observed to be significantly increased in the DNC population following a similar trend to COL1A1 and CD90 [66]. This is of interest as SDF-1 is a chemokine that acts on surrounding cells and has multiple roles including promoting proliferation and progenitor differentiation [67, 68].
Our study indicates that the co-existence of CSPC and DNC populations produces the optimal chondrogenic phenotype in vitro and the putative mechanism for this is proposed in Fig. 6. Collectively, the results indicate a potential regulatory role for DNCs in recruitment of CSPCs through SDF-1, which may in turn either differentiate into chondrocytes or release extracellular signalling factors to promote DNC chondrogenic phenotype stability, thereby contributing to the greater chondrogenicity observed in the CDC population [69, 70] (Fig. 6). DNC-CSPC interaction may be essential, to maintain chondrogenic gene expression and extracellular matrix synthesis. These findings are supported by a previous in vivo study, where mixed populations, i.e. CDCs, maintained a more chondrogenic phenotype when injected intramuscularly in mice when compared to an isolated population of chondroprogenitors [48]. When cultured individually, the CSPC and DNC populations have distinct CCND1, CCND2, COL1A1, COL2A1, CXCL12, NCAM1 and CDH2 gene expression profiles, indicating either progenitor status or fibroblastic lineage respectively [59, 60] but lacking optimal chondrogenicity.

Proposed mechanism of the support role of CSPCs in the nasoseptal cartilage cell niche. a CDC population comprises DNCs and CSPCs which reside together in the cartilage niche, expressing high levels of BMP2, COL2 and CCND2 and secreting normal hyaline cartilage extracellular matrix (ECM). The crosstalk between the two cell populations may be based on SDF-1: DNCs secrete SDF-1 which induces CSPC differentiation into DNCs, supporting the maintenance of a chondrogenic environment. When b DNCs and c CSPCs are separated and cultured individually, their expression profile and phenotype are altered. c CSPCs alone tend to maintain their progenitor-like phenotype, showing increased expression of CD56/NCAM. These cells are clonogenic and hold multilineage differentiation potential as demonstrated here. b When DNCs are deprived of CSPCs, these will change from a chondrogenic to a fibroblast-like phenotype, expressing high levels of COL1, SDF-1, CD90 and CD73. Consequently, DNCs alone will have decreased chondrogenicity and poor secretion of cartilage ECM. CCND2, cell cycle cyclin D2; BMP2, bone morphogenetic protein 2; COL1A1, type 1 collagen; COL2A1, type 2 collagen; SDF-1, stromal cell-derived factor 1; NCAM, neural cell adhesion molecule. Created using BioRender©
Conclusions
This study provides insight into the role of nasoseptal CSPCs in the in vitro chondrogenic niche and maintenance of phenotypic stability of nasoseptal chondrocytes through influence on dedifferentiation. Mixed populations of stem/progenitor cells and chondrocytes were required for optimal chondrogenesis. Further work will elucidate the molecular mechanisms underlying this phenomenon, enriching the translational potential of tissue engineered cartilage and associated cell-based therapies.
Availability of data and materials
The datasets generated during the current study are available in the figshare repository which can be downloaded from https://figshare.com/s/f2fb966577d8c655d171. All other datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ACAN:
-
Aggrecan gene
- BMP2:
-
Bone morphogenetic protein-2
- CCND2:
-
Cyclin D2
- CCND1:
-
Cyclin D1
- CDC:
-
Cartilage-derived cell
- CFE:
-
Colony forming efficiency
- CFU-F:
-
Fibroblast-like colony forming units
- CM:
-
Chondro-medium
- COL1A1:
-
Type 1 collagen
- COL2A1:
-
Type 2 collagen
- CSPC:
-
Cartilage-derived stem/progenitor cell
- CXCL12:
-
C-X-C motif chemokine 12 or stromal cell-derived factor 1
- DMEM:
-
Dulbecco’s modified Eagle medium
- DNC:
-
Differentiated nasoseptal chondrocyte
- FBS:
-
Foetal bovine serum
- GAG:
-
Glycosaminoglycan
- ISCT:
-
International Society of Cellular Therapy
- MEM:
-
Minimum essential medium
- MSC:
-
Mesenchymal stem cell
- NCAM1:
-
Neural cell adhesion molecule 1 or CD56
- NEAA:
-
Non-essential amino acids
- PFA:
-
Paraformaldehyde
- RPLP0:
-
Ribosomal protein lateral stalk subunit P0
- SDF-1:
-
Stromal cell-derived factor 1
- SOX9:
-
Sex-determining region Y box 9
References
Bichara DA, O'Sullivan NA, Pomerantseva I, Zhao X, Sundback CA, Vacanti JP, et al. The tissue-engineered auricle: past, present, and future. Tissue Eng Part B Rev. 2012;18(1):51–61.
Abbott RD, Kaplan DL. Strategies for improving the physiological relevance of human engineered tissues. Trends Biotechnol. 2015;33(7):401–7.
Tiku ML, Sabaawy HE. Cartilage regeneration for treatment of osteoarthritis: a paradigm for nonsurgical intervention. Ther Adv Musculoskeletal Dis. 2015;7(3):76–87.
Huey DJ, Hu JC, Athanasiou KA. Unlike bone, cartilage regeneration remains elusive. Science. 2012;338(6109):917–21.
Cruz IB, Severo AL, Azzolin VF, Garcia LF, Kuhn A, Lech O. Regenerative potential of the cartilaginous tissue in mesenchymal stem cells: update, limitations, and challenges. Rev Bras Ortop. 2017;52(1):2–10.
Lee SJ, Broda C, Atala A, Yoo JJ. Engineered cartilage covered ear implants for auricular cartilage reconstruction. Biomacromolecules. 2011;12(2):306–13.
Cao Y, Vacanti JP, Paige KT, Upton J, Vacanti CA. Transplantation of chondrocytes utilizing a polymer-cell construct to produce tissue-engineered cartilage in the shape of a human ear. Plast Reconstr Surg. 1997;100(2):297–302 discussion 3-4.
Raghavan U, Jones NS, Romo T 3rd. Immediate autogenous cartilage grafts in rhinoplasty after alloplastic implant rejection. Arch Facial Plast Surg. 2004;6(3):192–6.
Zeng L, Yao Y, Wang D-A, Chen X. Effect of microcavitary alginate hydrogel with different pore sizes on chondrocyte culture for cartilage tissue engineering. Mater Sci Eng C. 2014;34:168–75.
Mauck RL, Yuan X, Tuan RS. Chondrogenic differentiation and functional maturation of bovine mesenchymal stem cells in long-term agarose culture. Osteoarthr Cartil. 2006;14(2):179–89.
Schütz K, Despang F, Lode A, Gelinsky M. Cell-laden biphasic scaffolds with anisotropic structure for the regeneration of osteochondral tissue. J Tissue Eng Regen Med. 2016;10(5):404–17.
Yamaoka H, Asato H, Ogasawara T, Nishizawa S, Takahashi T, Nakatsuka T, et al. Cartilage tissue engineering using human auricular chondrocytes embedded in different hydrogel materials. J Biomed Mater Res A. 2006;78A(1):1–11.
Farrell MJ, Fisher MB, Huang AH, Shin JI, Farrell KM, Mauck RL. Functional properties of bone marrow-derived MSC-based engineered cartilage are unstable with very long-term in vitro culture. J Biomech. 2014;47(9):2173–82.
Herlofsen SR, Küchler AM, Melvik JE, Brinchmann JE. Chondrogenic differentiation of human bone marrow-derived mesenchymal stem cells in self-gelling alginate discs reveals novel chondrogenic signature gene clusters. Tissue Eng A. 2011;17(7–8):1003–13.
Fisher JN, Tessaro I, Bertocco T, Peretti GM, Mangiavini L. The application of stem cells from different tissues to cartilage repair. Stem Cells Int. 2017;2017:2761678.
Baugé C, Boumédiene K. Use of adult stem cells for cartilage tissue engineering: current status and future developments. Stem Cells Int. 2015;2015:438026.
Alsalameh S, Amin R, Gemba T, Lotz M. Identification of mesenchymal progenitor cells in normal and osteoarthritic human articular cartilage. Arthritis Rheum. 2004;50(5):1522–32.
Dowthwaite GP, Bishop JC, Redman SN, Khan IM, Rooney P, Evans DJ, et al. The surface of articular cartilage contains a progenitor cell population. J Cell Sci. 2004;117(Pt 6):889–97.
Fickert S, Fiedler J, Brenner RE. Identification of subpopulations with characteristics of mesenchymal progenitor cells from human osteoarthritic cartilage using triple staining for cell surface markers. Arthritis Res Ther. 2004;6(5):R422–32.
Williams R, Khan IM, Richardson K, Nelson L, McCarthy HE, Analbelsi T, et al. Identification and clonal characterisation of a progenitor cell sub-population in normal human articular cartilage. PLoS One. 2010;5(10):e13246.
Levato R, Webb WR, Otto IA, Mensinga A, Zhang Y, van Rijen M, et al. The bio in the ink: cartilage regeneration with bioprintable hydrogels and articular cartilage-derived progenitor cells. Acta Biomater. 2017;61:41–53.
Derks M, Sturm T, Haverich A, Hilfiker A. Isolation and chondrogenic differentiation of porcine perichondrial progenitor cells for the purpose of cartilage tissue engineering. Cells Tissues Organs. 2013;198(3):179–89.
Kobayashi S, Takebe T, Inui M, Iwai S, Kan H, Zheng YW, et al. Reconstruction of human elastic cartilage by a CD44+ CD90+ stem cell in the ear perichondrium. Proc Natl Acad Sci U S A. 2011;108(35):14479–84.
Anderson DE, Markway BD, Weekes KJ, McCarthy HE, Johnstone B. Physioxia promotes the articular chondrocyte-like phenotype in human chondroprogenitor-derived self-organized tissue. Tissue Eng Part A. 2018;24(3-4):264–74.
McCarthy HE, Bara JJ, Brakspear K, Singhrao SK, Archer CW. The comparison of equine articular cartilage progenitor cells and bone marrow-derived stromal cells as potential cell sources for cartilage repair in the horse. Vet J. 2012;192(3):345–51.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–7.
Seol D, McCabe DJ, Choe H, Zheng H, Yu Y, Jang K, et al. Chondrogenic progenitor cells respond to cartilage injury. Arthritis Rheum. 2012;64(11):3626–37.
Pizzute T, Lynch K, Pei M. Impact of tissue-specific stem cells on lineage-specific differentiation: a focus on the musculoskeletal system. Stem Cell Rev. 2015;11(1):119–32.
Jessop ZM, Manivannan S, Zhang Y, Thornton CA, Narayan R, Whitaker IS. Tissue specific stem/progenitor cells for cartilage tissue engineering: a systematic review of the literature. Appl Phys Rev. 2019;6(031301).
do Amaral RJ, CaS P, Kochem MC, Silva KR, Aniceto M, Claudio-da-Silva C, et al. Isolation of human nasoseptal chondrogenic cells: a promise for cartilage engineering. Stem Cell Res. 2012;8(2):292–9.
Mumme M, Barbero A, Miot S, Wixmerten A, Feliciano S, Wolf F, et al. Nasal chondrocyte-based engineered autologous cartilage tissue for repair of articular cartilage defects: an observational first-in-human trial. Lancet. 2016;388(10055):1985–94.
Pelttari K, Mumme M, Barbero A, Martin I. Nasal chondrocytes as a neural crest-derived cell source for regenerative medicine. Curr Opin Biotechnol. 2017;47:1–6.
Pelttari K, Pippenger B, Mumme M, Feliciano S, Scotti C, Mainil-Varlet P, et al. Adult human neural crest-derived cells for articular cartilage repair. Sci Transl Med. 2014;6(251):251ra119.
Dell’Accio F, Bari CD, Luyten FP. Microenvironment and phenotypic stability specify tissue formation by human articular cartilage-derived cells in vivo. Exp Cell Res. 2003;287(1):16–27.
Oseni AO, Butler PE, Seifalian AM. Optimization of chondrocyte isolation and characterization for large-scale cartilage tissue engineering. J Surg Res. 2013;181(1):41–8.
Jones PH, Watt FM. Separation of human epidermal stem cells from transit amplifying cells on the basis of differences in integrin function and expression. Cell. 1993;73(4):713–24.
Rodriguez R, Tornin J, Suarez C, Astudillo A, Rubio R, Yauk C, et al. Expression of FUS-CHOP fusion protein in immortalized/transformed human mesenchymal stem cells drives mixoid liposarcoma formation. Stem Cells. 2013;31(10):2061–72.
Mensing N, Gasse H, Hambruch N, Haeger JD, Pfarrer C, Staszyk C. Isolation and characterization of multipotent mesenchymal stromal cells from the gingiva and the periodontal ligament of the horse. BMC Vet Res. 2011;7:42.
Bernstein P, Sperling I, Corbeil D, Hempel U, Fickert S. Progenitor cells from cartilage--no osteoarthritis-grade-specific differences in stem cell marker expression. Biotechnol Prog. 2013;29(1):206–12.
Benz K, Stippich C, Freudigmann C, Mollenhauer JA, Aicher WK. Maintenance of "stem cell" features of cartilage cell sub-populations during in vitro propagation. J Transl Med. 2013;11:27.
Sterodimas A, de Faria J. Human auricular tissue engineering in an immunocompetent animal model. Aesthet Surg J. 2013;33(2):283–9.
Jessop ZM, Javed M, Otto IA, Combellack EJ, Morgan S, Breugem CC, et al. Combining regenerative medicine strategies to provide durable reconstructive options: auricular cartilage tissue engineering. Stem Cell Res Ther. 2016;7:19.
Hamada T, Sakai T, Hiraiwa H, Nakashima M, Ono Y, Mitsuyama H, et al. Surface markers and gene expression to characterize the differentiation of monolayer expanded human articular chondrocytes. Nagoya J Med Sci. 2013;75(1–2):101–11.
Darling EM, Athanasiou KA. Rapid phenotypic changes in passaged articular chondrocyte subpopulations. J Orthop Res. 2005;23(2):425–32.
Jiang Y, Cai Y, Zhang W, Yin Z, Hu C, Tong T, et al. Human cartilage-derived progenitor cells from committed chondrocytes for efficient cartilage repair and regeneration. Stem Cells Transl Med. 2016;5(6):733–44.
Tallheden T, Dennis JE, Lennon DP, Sjögren-Jansson E, Caplan AI, Lindahl A. Phenotypic plasticity of human articular chondrocytes. J Bone Joint Surg Am. 2003;85-A(Suppl 2):93–100.
Khan IM, Bishop JC, Gilbert S, Archer CW. Clonal chondroprogenitors maintain telomerase activity and Sox9 expression during extended monolayer culture and retain chondrogenic potential. Osteoarthr Cartil. 2009;17(4):518–28.
Marcus P, De Bari C, Dell’Accio F, Archer CW. Articular chondroprogenitor cells maintain chondrogenic potential but fail to form a functional matrix when implanted into muscles of SCID mice. Cartilage. 2014;5(4):231–40.
Seol D, Zhou C, Brouillette MJ, Song I, Yu Y, Choe HH, et al. Characteristics of meniscus progenitor cells migrated from injured meniscus. J Orthop Res. 2017;35(9):1966–72.
Zhou C, Zheng H, Seol D, Yu Y, Martin JA. Gene expression profiles reveal that chondrogenic progenitor cells and synovial cells are closely related. J Orthop Res. 2014;32(8):981–8.
Lee SY, Miwa M, Sakai Y, Kuroda R, Oe K, Niikura T, et al. Isolation and characterization of connective tissue progenitor cells derived from human fracture-induced hemarthrosis in vitro. J Orthop Res. 2008;26(2):190–9.
Klimczak A, Kozlowska U. Mesenchymal stromal cells and tissue-specific progenitor cells: their role in tissue homeostasis. Stem Cells Int. 2016;2016:4285215.
Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult β cells does not involve specialized progenitors. Dev Cell. 2007;12(5):817–26.
Glickstein SB, Monaghan JA, Koeller HB, Jones TK, Ross ME. Cyclin D2 is critical for intermediate progenitor cell proliferation in the embryonic cortex. J Neurosci. 2009;29(30):9614–24.
Pauklin S, Madrigal P, Bertero A, Vallier L. Initiation of stem cell differentiation involves cell cycle-dependent regulation of developmental genes by Cyclin D. Genes Dev. 2016;30(4):421–33.
Fang J, Hall BK. Differential expression of neural cell adhesion molecule (NCAM) during osteogenesis and secondary chondrogenesis in the embryonic chick. Int J Dev Biol. 1995;39(3):519–28.
Widelitz RB, Jiang TX, Murray BA, Chuong CM. Adhesion molecules in skeletogenesis: II. Neural cell adhesion molecules mediate precartilaginous mesenchymal condensations and enhance chondrogenesis. J Cell Physiol. 1993;156(2):399–411.
Gigout A, Jolicoeur M, Nelea M, Raynal N, Farndale R, Buschmann MD. Chondrocyte aggregation in suspension culture is GFOGER-GPP- and beta1 integrin-dependent. J Biol Chem. 2008;283(46):31522–30.
Metsuyanim S, Harari-Steinberg O, Buzhor E, Omer D, Pode-Shakked N, Ben-Hur H, et al. Expression of stem cell markers in the human fetal kidney. PLoS One. 2009;4(8):e6709.
Pode-Shakked N, Metsuyanim S, Rom-Gross E, Mor Y, Fridman E, Goldstein I, et al. Developmental tumourigenesis: NCAM as a putative marker for the malignant renal stem/progenitor cell population. J Cell Mol Med. 2009;13(8b):1792–808.
Lanctot PM, Gage FH, Varki AP. The glycans of stem cells. Curr Opin Chem Biol. 2007;11(4):373–80.
Blaney DE, Vitters E, van Lent P, van de Loo F, van den Berg W, van der Kraan P. Elevated extracellular matrix production and degradation upon bone morphogenetic protein-2 (BMP-2) stimulation point toward a role for BMP-2 in cartilage repair and remodeling. Arthritis Res Ther. 2007;9(5):R102.
Zhu J, Thakolwiboon S, Liu X, Zhang M, Lubman DM. Overexpression of CD90 (Thy-1) in pancreatic adenocarcinoma present in the tumor microenvironment. PLoS One. 2014;9(12):e115507.
Fausther M, Sheung N, Saiman Y, Bansal MB, Dranoff JA. Activated hepatic stellate cells upregulate transcription of ecto-5′-nucleotidase/CD73 via specific SP1 and SMAD promoter elements. Am J Physiol Gastrointest Liver Physiol. 2012;303(8):G904–14.
Shamis Y, Silva EA, Hewitt KJ, Brudno Y, Levenberg S, Mooney DJ, et al. Fibroblasts derived from human pluripotent stem cells activate angiogenic responses in vitro and in vivo. PLoS One. 2013;8(12):e83755.
Quan C, Cho MK, Shao Y, Mianecki LE, Liao E, Perry D, et al. Dermal fibroblast expression of stromal cell-derived factor-1 (SDF-1) promotes epidermal keratinocyte proliferation in normal and diseased skin. Protein Cell. 2015;6(12):890–903.
Sharma M, Afrin F, Satija N, Tripathi RP, Gangenahalli GU. Stromal-derived factor-1/CXCR4 signaling: indispensable role in homing and engraftment of hematopoietic stem cells in bone marrow. Stem Cells Dev. 2011;20(6):933–46.
Dar A, Schajnovitz A, Lapid K, Kalinkovich A, Itkin T, Ludin A, et al. Rapid mobilization of hematopoietic progenitors by AMD3100 and catecholamines is mediated by CXCR4-dependent SDF-1 release from bone marrow stromal cells. Leukemia. 2011;25(8):1286–96.
Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–88.
Nie Y, Han Y-C, Zou Y-R. CXCR4 is required for the quiescence of primitive hematopoietic cells. J Exp Med. 2008;205(4):777–83.
Acknowledgements
We thank Ms. Kavitha Saw for providing patient nasoseptal cartilage samples for this study.
Funding
Research reported in this study was supported by the Medical Research Council (MR/N002431/1), The Oakgrove Medical Charitable Trust, ABMU Health Board, the Royal College of Surgeons of England, The British Association of Plastic, Reconstructive and Aesthetic Surgeons (BAPRAS), the Fulbright Commission and the St David’s Foundation.
Author information
Authors and Affiliations
Contributions
ZMJ and ISW conceived the study. ZMJ and AA were responsible for designing and performing experiments, analysing data and interpreting results. ZMJ, AA, INS and SB wrote the manuscript. SB performed the proliferation experiments and interpreted results. ILP, CAT and ISW critically reviewed and edited the manuscript. ISW provided funding and coordinated the project. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Human chondrocytes were isolated from nasoseptal cartilage samples collected during routine septorhinoplasties that would have otherwise been discarded following informed consent and approval by the South Wales Research and Ethics Committee (IRAS ID 99202).
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1 : Supplementary Table S1.
Detailed information on fluorescent dye and excitation and emission wavelengths of the antibodies used for flow cytometry. AF, Alexa Fluor; APC, allophycocyanin; BV, brilliant violet; FITC, fluorescein isothiocyanate; PE, phycoerythrin; PerCP, peridinin-chlorophyll protein complex.
Additional file 2 : Supplementary Figure S1.
Detailed information on the contents of the RT2 Profiler™ Human Stem Cell PCR Array as supplied by Qiagen, including gene description and position in array.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jessop, Z.M., Al-Sabah, A., Simoes, I.N. et al. Isolation and characterisation of nasoseptal cartilage stem/progenitor cells and their role in the chondrogenic niche. Stem Cell Res Ther 11, 177 (2020). https://doi.org/10.1186/s13287-020-01663-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-020-01663-1