
Abstract
Background
Taking advantage of cellular paracrine mechanisms, the secretome of adipose-derived stem cells (ADSCs) and adipose tissue has been demonstrated to induce tissue repair and regeneration in various ischemic and impaired conditions. However, these cell-based therapies have been hindered by issues, such as inherent safety and cost-efficiency for clinical applications. In this study, we prepared a liquid cell-free extract from human adipose tissue [adipose liquid extract (ALE)] and evaluated its potential therapeutic efficacy.
Methods
ALE was prepared from human subcutaneous adipose tissue using a rapid and physical approach, and the protein components in ALE were identified using mass spectrometry analysis. In vivo, the therapeutic effect of this agent was investigated on wound healing in C57BL/6 mice, and wound healing rate, vessel density, and neo-adipocyte formation in wounded skins were measured at days 3, 7, 11, and 14. In vitro, the effect of ALE on the viability of human ADSCs, tube formation of human umbilical vein endothelial cells (HUVECs), and adipogenic differentiation of ADSCs were tested.
Results
The results demonstrated that ALE contained a variety of growth factors and did not affect cell viability. ALE-treated wounds exhibited accelerated wound healing with increased vessel density and formation of neo-adipocytes compared to that of control wounds. Moreover, when added as a cell culture supplement, ALE effectively induced tube formation of HUVECs and lipid accumulation in ADSCs. ALE-treated ADSCs also exhibited elevated levels of adipogenic gene expression.
Conclusions
ALE is a novel growth-rich therapeutic agent that is cell-free and easy to produce. Besides, it is also able to induce angiogenesis and adipogenesis both in vitro and in vivo, thus indicating that it could be used for wound repair and soft tissue regeneration.
Similar content being viewed by others

Background
Tissue repair and regeneration are complex biological processes that occur throughout human life. Therapeutic induction of damaged tissue/organs requires provision of instructive bioactive signals, and the most crucial among these signals are growth factors [1]. Growth factors are important biomolecules that instruct cell behavior (for example, cell migration, survival, adhesion, proliferation, and differentiation) and guide tissue regenerative process [2,3,4]. Till date, employment of exogenous growth factor products as therapeutic agents for clinical use is limited so far as the ease/cost of production, specificity of action, and suboptimal efficacy are concerned [5]. Hence, there is an urgent need for new technologies and strategies that may render growth factors more amenable to clinical treatment.
Taking advantage of paracrine secretory function of cells, developments in growth factors therapies have been reported to make use of cell-derived secretome, which is the complex set of biomolecules secreted during short-term culture in vitro [6]. The cell-derived secretome technology is a rapidly advancing field and has shown a successful outcome in stimulating tissue repair and regeneration both in vitro and in vivo [7,8,9,10]. Furthermore, a secretome extract harvested from cultured adipose tissue was shown to contain various angiogenic and adipogenic factors that were able to induce angiogenesis and adipogenesis in vitro [11]. Although the secretome of these cultured cells/tissue exhibits efficient therapeutic efficacy, there are several hurdles that are needed to be addressed before translating this therapy from bench-to-bedside. Examples of these drawbacks include isolation of cells using enzymatic digestion that increases the risk of biological contamination, the secretome harvested from cultured cells/tissues requires considerable cell expansion, specific laboratory equipment, and good manufacturing practice, which in turn increase the financial burden [12].
Adipose tissue has currently gained significant importance since it serves as an active endocrine organ and an abundant source of biologically active substances [13,14,15]. The study by Tonnard et al. demonstrated that the human lipoaspirates can be physically emulsified into a therapeutic liquid suspension (e.g., Nanofat) for skin rejuvenation [16]; recent studies have demonstrated that Nanofat contains a number of growth factors that are able to induce angiogenesis in vivo [17, 18]. Our research team utilized a similar physical method to process human lipoaspirates [19]. After centrifugation, we found that the processed lipoaspirates were separated into four parts (from top to bottom: lipid portion, lipid-devoid adipose tissue, liquid portion, and cell/tissue debris). The lipid-devoid adipose tissue, namely, extracellular matrix/stromal vascular fraction gel (SVF-gel), was found to have therapeutic ability in inducing angiogenesis and adipogenesis in vivo [20,21,22,23,24]. Unfortunately, the study of the liquid portion acquired from these physically processed lipoaspirates has been ignored. We speculated that this adipose tissue-derived liquid portion may contain a variety of bioactive factors that may exhibit therapeutic ability in regenerative medicine.
In this study, we described a novel method to obtain this adipose tissue-derived liquid portion, the “adipose liquid extract” (ALE), and evaluated its therapeutic effect on wound healing in mice. The protein components and osmotic pressure of ALE were measured, and the effect of ALE on cell viability was evaluated. More importantly, the effect of ALE on angiogenesis and adipogenesis was evaluated both in vitro and in vitro.
Methods
Ethics statement
All the procedures including animal studies were approved by Southern Medical University Institutional Review Board and the Nanfang Hospital Animal Ethics Committee and conducted in accordance with the ethical standards of the National Health and Medical Research Council (China).
ALE preparation and osmotic pressure measurement
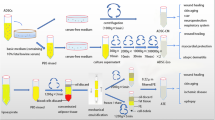
Human adipose tissues were harvested from female patients undergoing abdominal liposuction in the Plastic Surgery Department of Nanfang Hospital, after taking their informed consent. Detailed procedure of ALE preparation is shown in Fig. 1. First, the harvested adipose tissue was allowed to stand for 10 min in ice water. The liquid portion was discarded and adipose tissue layer collected. The obtained adipose tissue was then washed with phosphate-buffered saline (PBS) and centrifuged at 1200g for 3 min to remove the remaining blood constituents. The concentrated adipose tissue was mixed with an equal volume of PBS and then physically emulsified by repeated transfer between two 10-mL syringes connected by a female-to-female Luer-Lock connector with an internal diameter of 1.4 mm. Transfer of the adipose tissue was carried out for 1, 3, 5, 7, or 10 min at a constant rate (10 mL/s). After centrifugation at 2000g for 5 min, the liquid portion (ALE) from emulsified adipose tissue was collected and passed through a 0.20-μm syringe filter (Xiboshi, DIONEX, USA) to remove cell and tissue debris. The osmotic pressure of ALE was measured using Fiske® 210 Micro Osmometer and the sample stored at − 40 °C until use. ALE emulsified for 1 and 10 min were subjected to mass spectrometric analysis to investigate the effect of time variations on protein yield of ALE. All the five ALE samples were used for cell viability assay and osmotic pressure measurements, while only the ALE emulsified for 1 min was used for all the remaining experiments.

Schematic overview of ALE preparation
Mass spectrometry identification and database searches
The ALE samples were first digested by trypsin for subsequent liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis as described before [25]. The tryptic peptide products were then separated by reverse-phase liquid chromatography using nano LC system (DIONEX Thermo Scientific). Label-free LC-MS/MS identification was subsequently conducted using a Q Exactive Plus (Thermo Fisher Scientific, Marietta, OH, USA) equipped with a self-packed column (Thermo Fisher Scientific, Acclaim PepMap RSLC 50 μm × 15 cm, nano viper, P/N164943) at a flow rate of 300 nL/min according to previous study [26]. Label-free experiments’ data was analyzed using MaxQuant program with high-resolution instruments supported by Andromeda as a database search engine for peptide identification [27]. Label-free quantitation (LFQ) was performed as previously described [28]. LFQ intensity values were used for protein quantification, and protein information was searched against the human database (Uniprot_HomoSapiens_161584_20180123).
Gene Ontology (GO) analysis of ALE was performed to classify all identified proteins into two categories (biological process and molecular function) using Blast2GO v.2 software [29] and web database (www.geneontology.org). Functional annotation of proteins related to angiogenesis and adipogenesis was identified. Ingenuity Pathway Analysis (IPA) analysis of identified proteins in ALE was performed to obtain protein location information through a web database (https://www.qiagenbioinformatics.com).
Growth factor measurement
High sensitivity enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA) was used to quantify the levels of basic fibroblast growth factor (bFGF), epidermal growth factor (EGF), transforming growth factor-β1 (TGF-β1), and vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), and platelet-derived growth factor (PDGF) in ALE according to the manufacturer’s instructions.
Cell viability assay
Primary human adipose-derived stem cells (ADSCs) were isolated from adipose tissue as described previously [30, 31]. Briefly, fresh adipose tissue was washed and digested in 0.075% collagenase solution for 40 min on a shaker at 37 °C. The digested tissue was centrifuged at 180g for 5 min and then filtered to remove large debris. Next, the cellular pellet (SVF) was re-suspended in an erythrocyte lysis buffer and centrifuged at 180g for another 5 min. Finally, the SVF was cultured in human ADSC complete growth medium (HUXMD-90011, Cyagen, China). Passage 3 ADSCs were used in subsequent experiments.
ADSCs were treated with the five ALE samples to assess the effect of ALE on cell viability. Briefly, the ADSCs (5 × 103 cells/well) were incubated in a 96-well microplate at 37 °C for 24 h to permit cell adhesion and were later treated with each of the five ALE samples or PBS (control group). The relative cell viability present at the 12, 24, and 36 h time points was estimated using a Cell Counting Kit-8 assay kit (CCK-8, Dojindo, Japan) according to the manufacturer’s instructions. Absorbance was measured using a plate reader at 450 nm, and the OD values in different groups were used to analyze cell viability. These experiments were repeated three times.
Angiogenic induction ability of ALE in vitro
Human umbilical vein endothelial cells (HUVECs) were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured in endothelial cell medium (1001, ScienCell, USA). Tube formation assays were conducted by treating HUVECs with ALE to assess the angiogenesis-inducing ability of ALE. In brief, frozen Matrigel (356230; Becton, Dickinson & Co., Franklin Lakes, NJ, USA) was thawed overnight at 4 °C, and all experimental equipment were cooled to − 20 °C before use. The Matrigel (diluted 1:1 in PBS) was transferred to a 24-well plate and solidified by incubation at 37 °C and 5% CO2 for 30 min before seeding of the cells. HUVECs (5 × 105 per well) were then seeded on the Matrigel and treated with 2 mL medium (1:1, ALE to endothelial cell basal medium). HUVECs treated with angiogenic factors [20 ng/mL vascular endothelial growth factor (VEGF) and 5 ng/mL basic fibroblast growth factor (bFGF)] served as a positive control, while cells treated with 2 mL medium (1:1, PBS to endothelial cell medium) were used as a negative control. After 3, 9, and 20 h, tube formation of HUVECs was observed and photographed using a Nikon E200 microscope (Nikon Corp., Tokyo, Japan). Angiogenesis is evaluated by counting the number of tubular structures from six fields of each well. Tube formation assays were repeated three times.
Adipogenic induction ability of ALE in vitro
ADSCs were treated with ALE to assess the adipogenic potential of ALE. In brief, ADSCs (1 × 105 per well) were plated on 6-well plates and cultured overnight at 37 °C and 5% CO2. The next day, cells in each well were rinsed and treated with 2 mL medium (1:1, ALE to human ADSC complete growth medium). ADSCs treated with adipogenesis kit (HUXMD-90031, Cyagen Biosciences, Guangzhou, China) served as the positive control, and cells treated with 2 mL medium (1:1, PBS to human ADSC complete growth medium) were used as the negative control. The medium in all the groups was changed every 3 days. Adipogenic differentiation of ADSCs was examined for lipid accumulation by Oil Red O (ORO; Sigma-Aldrich, USA) staining. Cells were photographed using a Nikon E200 microscope and collected for gene expression analysis at days 4, 7, 14, 21, and 28. Adipogenic induction assays were repeated three times.
Therapeutic effect of ALE in vivo
The therapeutic effect of ALE in vivo was evaluated by the application of ALE in a wound healing model of mice. Female C57BL/6 mice, 4- to 6-week-old (n = 20), were purchased from the Southern Medical University Laboratory Animal Center and were maintained in microisolator cages at the Animal Experiment Center of Nanfang Hospital. For wound healing experiments, mice were anesthetized and two circular full-thickness excisional skin wounds of 8-mm radius were made on the dorsum of mice. A silicone ring was then placed around the perimeter of the wound and secured with 6-0 sutures to prevent wound contraction. Wounds were randomly treated either with 200 μL ALE or 200 μL PBS (control group) in each mouse. After the treatments, the wounds were covered with Tegaderm sterile dressing (3 M Healthcare, St Paul, Minn.). Treatments were administered to the wounds every 2 days, and sterile dressings were changed every day. Pictures of the wounds were taken, and the skins around the wounds as well as health skins were harvested for further analysis on days 3, 7, 11, and 14 after surgery. The wound area was quantified using ImageJ software, and the wound healing was expressed as follows: residual wound area/original wound area × 100.
Histological assessment and immunohistochemical staining
Fresh skin samples were fixed in formalin for histological assessment and immunohistochemical staining. Briefly, formalin-fixed samples were embedded and sliced into 5-μm sections. For histological assessment, tissue sections were deparaffinized in xylene, rehydrated through graded alcohol in phosphate-buffered saline (PBS), and then stained with a hematoxylin-eosin (HE) working solution. For immunohistochemical analysis, sections were dewaxed and rehydrated, then incubated in 3% H2O2 to block endogenous peroxidase. Immunohistochemical staining was performed using antibodies against CD31 (1:25, ab28364, Abcam, Cambridge, UK) and perilipin (1:500, GP29; Progen, Heidelberg, Germany), followed by secondary antibody. Angiogenesis and adipogenesis were evaluated by counting the number of brown-labeled vessel-like structures and brown-labeled adipocytes, respectively, from six fields of each slide.
Quantitative reverse-transcription polymerase chain reaction (qRT-PCR)
Total RNA in ADSCs was extracted and measured using qRT-PCR. Primers used for adipogenesis-specific genes were from peroxisome proliferator-activated receptor gamma (PPAR-γ) and CCAAT/enhancer-binding protein alpha (CEBP-α), master regulators for the induction of adipogenic differentiation. RNA was isolated from the samples using Trizol and SYBR Green quantitative PCR SuperMix (Thermo Fisher Scientific, Marietta, USA), followed by reverse transcription. The complementary DNA samples were measured using customized TaqMan Array Plates (Thermo Fisher Scientific, Marietta, USA). Relative expressions were calculated by the cycle threshold method using GAPDH as an endogenous reference gene.
Statistical analysis
Data are expressed as mean ± SD. Results were analyzed using SPSS 22.0 software. An independent t test and least significant difference post hoc analysis were used to compare two and three groups at a single time point, respectively, and one-way analysis of variance (ANOVA) was used to compare groups at all time points. OD values from different ALE samples were subjected to one-way ANOVA followed by Tukey’s multiple comparison test to evaluate the cell viability with ALE treatment. Values of p < 0.05 were considered statistically significant.
Results
Proteomic profiles and functional categories of identified proteins in ALE
ALE samples (1 min processing time and 10 min processing time) were analyzed by label-free LC-MS/MS and Maxquant software to acquire their quantitative proteomic profiles. The mass spectrometry analysis showed that totally 1975 human proteins were reliably quantified (1742 in 1 min processed ALE and 1769 in 10 min processed ALE). Interestingly, over 87% of the identified proteins in the two groups overlapped with one another (1552 out of 1742 in the 1 min processed group and 1552 out of 1769 in the 10 min processed group) (Fig. 2a). IPA analysis showed that the majority of ALE proteins were from the cytoplasm, followed by the nucleus, and finally from the extracellular space and plasma membrane (Fig. 2b, c).

Mass spectrometry-based quantitative proteomic analysis of ALE and classification of identified proteins. a Expression profiling of proteins in 1 min ALE and 10 ALE. b Subcellular distribution of proteins in 1 min ALE. c Subcellular distribution of proteins in 10 min ALE. d Biological processes and molecular function categories of identified proteins in ALE
The GO analysis of biological processes indicated that the proteins in ALE were mainly involved in the cellular process, biological regulation, and metabolic process. In addition, the three most abundant classes of the molecular functions in ALE were binding, catalytic activity, and molecular function regulators (Fig. 2d). Functional annotation revealed that 67 proteins were involved in angiogenesis (Table 1) and 25 proteins were involved in adipogenesis (Table 2). Elisa assay was carried out to further measure the level of growth factors in ALE. High levels of growth factors (bFGF, EGF, TGF-β1, VEGF, HGF, and PDGF) were detected in the ALE, and the expression level of each factor are presented in Table 3.
ALE did not affect cell viability
Osmotic pressure measurement showed that there were no significant differences among all the five ALE samples and the values remained in the normal physiological range (280–310 mOsm/kg) (Fig. 3a).

Osmotic pressure measurement and cell viability assay. a Osmotic pressure measurement of five ALE samples. b Cell viability assessment of ADSCs with five ALE samples’ treatment
The viability of ALE-treated ADSCs was evaluated at 12, 24, and 36 h. Although the ALE samples seemed to lower the cell viability than that in the control group at 12 h, the difference was not significant. Similarly, we did not observe any significant variation in the viability of ADSCs by treating with all the five ALE samples at 24 and 36 h (Fig. 3b).
ALE enhanced wound healing in mice
Figure 4a shows representative images of the full-thickness wound beds treated with either ALE or PBS in mice. Histological observations showed that inflammatory cell infiltration could be observed in both the groups on day 3. On day 7, the ALE-treated wounds showed re-epithelialization, while almost no re-epithelialization could be observed in the control group. On day 14, wounds in the ALE-treated group exhibited increased skin appendage numbers compared to those in the control group (Fig. 4b).

Therapeutic effect of ALE on wounds. a Representative images of wound beds treated with either ALE or PBS in mice. b HE staining of wounded skins. c Quantification of the rate of wound healing. *P < 0.05. Scale bars = 200 μm
Statistical analysis of the rate of wound healing revealed that the healing of wounds was significantly faster in the ALE-treated group than in the control group from day 7 to day 14 (Fig. 4c). Moreover, the ALE-treated group nearly achieved complete wound healing by day 14, whereas the control group still had unhealed wounds at that point of time.
ALE induced angiogenesis in vitro and in vivo
The angiogenic induction ability of ALE was tested by culturing HUVECs in ALE-supplemented medium. In the presence of ALE, tubular structures started to develop at 3 h, and ALE stimulated a time-dependent induction of tubule formation in the assay (Fig. 5a). The production of typical tubular structures was very similar to that in the positive control group at 20 h. However, tubular structures were hardly detected in the negative control group throughout the assay (Fig. 5b). Quantification analysis revealed that the number of tubular structures was significantly higher in both the ALE-treated group and the positive group than in the negative group (Fig. 5c).

Tubule formation of HUVECs in the presence of ALE. a Tubule formation of HUVECs with ALE treatment at different time points. b Tubule formation of HUVECs in positive control and negative control groups at 20 h. c Quantification of tubular structures in all groups at 20 h. ***P < 0.001. Scale bars = 200 μm
The angiogenic potential of ALE was further evaluated in a mouse wound healing model. Immunohistochemical staining for angiogenesis marker, CD31, in mouse skin samples revealed that the CD31+ neo-vessels were most frequently observed in both groups from day 7 to day 14 (Fig. 6a), while only a small number of neo-vessels were observed in healthy skins at all time points. Quantification of angiogenesis revealed that the number of CD31+ vessels was significantly higher in the ALE-treated group than in the control group at all time points (Fig. 6b).

Angiogenesis in vivo. a Immunostaining of CD31+ vessels (black arrows) in wounded skins. b Quantification of CD31+ vessels. *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars = 400 μm
ALE induced adipogenesis in vitro and in vivo
The adipogenic induction potential of ALE was tested by culturing human ADSCs in ALE-supplemented medium up to 28 days. ALE was shown to induce lipid accumulation in ADSCs in the culture, the effect being more extensive when longer culture times were used (Fig. 7a). The relative gene expression of adipogenesis markers, PPAR-γ and CEBP-α, in ALE-treated ADSCs exhibited a time-dependent increase from days 4 to 21 and a decrease from days 21 to 28. Statistical analysis revealed that the relative expression of the two adipogenesis markers was significantly higher in the ALE-treated ADSCs than in the negative control from days 14 to 28 (Fig. 7b).

Adipogenic differentiation of ADSCs in the presence of ALE. a ORO staining of ADSCs. b The relative mRNA expression levels of PPAR-γ and CEBP-α in ADSCs. *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars = 200 μm
In vivo results revealed that perilipin+ adipocytes could be observed in the skin of ALE-treated wounds at day 7, and frequently observed from day 11 to day 14, while adipocytes appeared scattered in control skins from day 11 to day 14 (Fig. 8a). Moreover, quantification of adipogenesis revealed that the number of perilipin+ adipocytes was significantly higher in the ALE-treated group than in the control group at all time points (Fig. 8b).

Adipogenesis in vivo. a Immunostaining of perilipin+ adipocytes (black arrows) in wounded skins. b Quantification of perilipin+ adipocytes. ***P < 0.001. Scale bars = 400 μm
Discussion
Growth factor therapy holds a great promise in regenerative medicine applications. Ideally, therapeutic growth factors should also be cost beneficial, easy to prepare, safe, and reproducible (Table 3). In this study, we prepared a cell-free, growth factor-rich, adipose liquid extract using an easy and rapid method. ALE was shown to induce tube formation of HUVECs and adipogenic differentiation of ADSCs in vitro. More importantly, ALE-treated wounds were more angiogenic, adipogenic, and normalized than the control wounds in mice. Further analysis revealed that ALE, despite the differing processing time periods of the adipose tissue, had similar protein yield, and osmotic pressure values of their liquid extracts were within the normal physiological range. Cell viability assay demonstrated that ALE did not affect cell viability at different processing time points. Thus, 1 min processing of adipose tissue is time-saving and convenient, and therefore, recommended as a standard ALE isolation procedure.
In this study, we demonstrated that the liquid extract from human adipose tissue has a unique capacity of inducing angiogenesis and adipogenesis. In vitro, ALE was shown to efficiently induce tubule formation of HUVECs. Interestingly, ALE treatment exerted a similar proangiogenic ability compared to that of the positive group (VEGF and bFGF treatment) in vitro. The strong proangiogenic ability of ALE could be explained by the presence of multiple angiogenic factors, and a combination of these factors likely acts synergistically to induce angiogenesis [32, 33]. Angiogenesis is a critical process of wound healing [34], and the development of wounds is mainly attributed to the deficiencies of endogenous growth factors [35, 36]. The progression of angiogenesis is controlled by a wide variety of angiogenic factors, which may play important roles in endothelial cell recruitment, proliferation, and differentiation [37]. These processes rely on the cross-talk between various angiogenic growth factors to initiate neo-angiogenesis, which may account for the early angiogenic response of ALE treatment in the skin wounds. Consequently, the enhanced angiogenesis in ALE-treated wounds may have contributed to the healing of such wounds. ALE was shown to induce adipogenic differentiation of ADSCs less than 14 days after treatment. The formation of adipocytes from precursor stem cells involves a complex and highly orchestrated program of gene expression [38]. Important adipogenic genes, PPARγ and CEBPα, were significantly increased in the ALE-treated ADSCs compared with those in the control. Adipogenesis was further observed in the skin of wounds after ALE treatment, indicating that ALE is efficacious in inducing adipogenesis. Interestingly, ALE induced a higher PPARγ and CEBPα expression in ADSCs compared to the standard adipogenic medium. Adipogenic medium is consist of five adipogenesis-related contents (glutamine, insulin, 3-isobutyl-1-methylxanthine, rosiglitazone, dexamethasone), while ALE contains 25 adipogenesis-related factors. Among all these factors, adiponectin, sulfotransferase, and aldehyde dehydrogenase have been proven to contribute to adipogenesis [39,40,41]. Therefore, we speculated that the presence of multiple adipogenesis-related factors and a combination of these factors may likely act synergistically to induce adipogenesis, leading to higher adipogenic marker expression in ALE-treated cells compared to the standard adipogenic medium. Moreover, a previous study by Uriel et al. showed that secretory factors of cultured adipose tissue induced higher PPARγ and even fourfold higher triglyceride accumulation of ADSCs compared with adipogenic medium treatment [11]. Although they did not clarify the exact mechanisms underlying this phenomenon in their study, it could be demonstrated that adipose-derived factors is efficient for reproduction and modeling of natural adipogenesis. However, more work should be done to clarify the exact mechanisms why higher adipogenic markers were expressed in ALE-treated cells compared to standard adipogenic medium in our future study. Collectively, the proangiogenic and proadipogenic features of ALE suggest that this agent could be used for overcoming the challenge of vascularization and further adipogenesis in soft tissue engineering. Proteomic analysis was conducted to further evaluate the responsible factors for the angiogenic and adipogenic effect of ALE, and the results revealed that among the 1742 proteins detected, 67 were angiogenesis-related and 25 adipogenesis-related. Thus, we can speculate that the proangiogenic and the proadipogenic effect of ALE is attributed to the presence of a combination of various angiogenesis- and adipogenesis-related factors.
Adipose tissue is a rich and very convenient source of cells for regenerative medicine therapeutic approaches. In the past decade, the therapeutic effects of ADSCs and adipose tissue have been widely studied [9, 42,43,44]. The underlying mechanisms of these therapies were attributed to the paracrine growth factors released by various cellular components. Recently, adipose derivatives, such as Nanofat and SVF-gel, containing active SVF components, were shown to exert a therapeutic effect on wounds as reported in multiple basic studies and clinical trials [21, 22, 24, 45]. Nevertheless, ALE therapy is different from these cell-based therapies. The mechanical processing of the adipose tissue breaks most of the mature adipocytes as well as many SVF cell components and cell culture process is not included. Therefore, the growth factors in ALE are not cellular secretome components. Recently, adipose tissue was recognized as an endocrine organ and an important source of biologically active substances possessing local and/or systemic action, such as adipokines [46]. Components of the adipose extracellular matrix, such as glycosaminoglycans and proteoglycans possess the ability to bind soluble growth factors and cytokines, which allow the ECM to serve as a reservoir of these biologically active factors [47, 48]. A recent mass spectrometric analysis of adipose ECM revealed that it contains a variety of matricellular proteins and adhesion glycoproteins known to be key modulators of cell functions, as well as a range of growth factors [49]. Therefore, from our study, it is reasonable to speculate that the mechanical emulsification of adipose tissue led to a rapid rise in bioactive factors that were originally stored in cells and ECM of adipose tissue, and these factors were then collected as ALE after centrifugation of the emulsified adipose tissue.
In 2017, our research team reported a novel adipose tissue derivative, SVF-gel [50]. SVF-gel is particularly rich in viable SVF cells and adipose-derived ECM and has been used in the treatment of hypertrophic scars [51], soft tissue defect [52], and chronic wound healing [24] in recent years. Despite the satisfactory therapeutic effect, SVF-gel is essentially a mature adipocyte-free, SVF cell-enriched adipose tissue, and can be only used for autotransplantation. Compared with SVF-gel and other cell-based therapies, the advantages of using ALE can be summarized as follows: (1) ALE does not contain cells and therefore can be assumed to be nonimmunogenic, which may facilitate allogenic or xenogeneic use of ALE in clinics. The therapeutic effect of human ALE on mice wound healing demonstrated in this study may support the xenogeneic use of ALE in the future. (2) The challenges of stem cell-based therapy, such as poor survival of administered cells or potential tumor development, could be overcome by using ALE. (3) Mass production of ALE has become possible, as the popularity of liposuction has increased globally and lipoaspirates can be easily collected from clinics. (4) ALE is prepared from adipose tissue via a purely physical approach without chemical and biological contamination, largely reducing safety and financial burdens. (5) The preparation of ALE is relatively rapid and simple which allows this procedure to be carried out in an operation room during surgery for autologous use. (6) The cell-free and growth factor-rich properties of ALE suggests that ALE could be easily sterilized, stored, transported, and serve as a potential “off-the-shelf” product for clinical use. Despite the advantages mentioned above, there are still several limitations remaining to be addressed before clinical application of ALE. First, the rates of in vitro degradation and inactivation of growth factors in ALE need to be estimated to help optimize in vivo therapeutic strategies concerning treatment doses and frequencies. Second, because ALE was directly injected in the wound bed, the release of growth factors could not be controlled precisely in this study. Therefore, the use of a controlled release system that combines a delivery vehicle and ALE to make the release orderly and continuous would be preferable. Third, in this study, ALE was obtained only from human subcutaneous adipose tissue; further studies on ALE obtained from other types of adipose tissues (e.g., visceral adipose tissue) and adipose tissue from other species might deepen our understanding of these therapeutic agents and would probably extend their applications. Finally, ALE mainly consists of water-soluble components and the study of lipid-soluble components from adipose tissue has been ignored; therefore, studies related to adipose-derived lipid-soluble component would be valuable in the future.
Conclusion
This study demonstrates a rapid and simple physical method to prepare a cell-free and growth factor-rich liquid extract from human subcutaneous adipose tissue. ALE contains a wide variety of proangiogenic and proadipogenic factors and efficiently induces angiogenesis and adipogenesis both in vitro and in vivo. Thus, ALE could serve as a novel therapeutic agent in regenerative medicine applications.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ADSCs:
-
Adipose-derived stem cells)
- ALE:
-
Adipose liquid extract
- ANOVA:
-
Analysis of variance
- ATCC:
-
American Type Culture Collection
- bFGF:
-
Basic fibroblast growth factor
- CCK-8:
-
Cell Counting Kit-8
- CEBP-α:
-
CCAAT/enhancer-binding protein alpha
- EGF:
-
Epidermal growth factor
- GO:
-
Gene Ontology
- HE:
-
Hematoxylin-eosin
- HGF:
-
Hepatocyte growth factor
- HUVECs:
-
Human umbilical vein endothelial cells
- IPA:
-
Ingenuity Pathway Analysis
- LC-MS/MS:
-
Liquid chromatography tandem mass spectrometry
- LFQ:
-
Label-free quantitation
- ORO:
-
Oil Red O
- PBS:
-
Phosphate-buffered saline
- PDGF:
-
Platelet-derived growth factor
- PPAR-γ:
-
Peroxisome proliferator-activated receptor gamma
- qRT-PCR:
-
Quantitative reverse-transcription polymerase chain reaction
- SVF-gel:
-
Extracellular matrix/stromal vascular fraction gel
- TGF-β1:
-
Transforming growth factor-β1
- VEGF:
-
Vascular endothelial growth factor
References
Moss AJ, Sharma S, Brindle NPJ. Rational design and protein engineering of growth factors for regenerative medicine and tissue engineering: figure 1. Biochem Soc Trans. 2009;37(4):717–21.
Liu L, et al. Controlled release of growth factors for regenerative medicine. Curr Pharm Des. 2015;21(12):1627.
Mitchell AC, et al. Engineering growth factors for regenerative medicine applications. Acta Biomater. 2016;30:1–12.
Atanasova M, Whitty A. Understanding cytokine and growth factor receptor activation mechanisms. Crit Rev Biochem Mol Biol. 2012;47(6):502–30.
Lee K, Silva EA, Mooney DJ. Growth factor delivery-based tissue engineering: general approaches and a review of recent developments. J R Soc Interface. 2011;8(55):153–70.
Jayaraman P, et al. Stem cells conditioned medium: a new approach to skin wound healing management. Cell Biol Int. 2013;37(10):1122–8.
Kwon SH, et al. Conditioned medium of adipose-derived stromal cell culture in three-dimensional bioreactors for enhanced wound healing. J Surg Res. 2015;194(1):8–17.
Kim MH, et al. Galectin-1 from conditioned medium of three-dimensional culture of adipose-derived stem cells accelerates migration and proliferation of human keratinocytes and fibroblasts. Wound Repair Regen. 2017;Suppl 1:S9–S18.
Jun E, et al. Hypoxic conditioned medium from human amniotic fluid-derived mesenchymal stem cells accelerates skin wound healing through TGF-β/SMAD2 and PI3K/Akt pathways. Int J Mol Sci. 2014;15(1):605–28.
Walter MNM, et al. Mesenchymal stem cell-conditioned medium accelerates skin wound healing: an in vitro study of fibroblast and keratinocyte scratch assays. Exp Cell Res. 2010;316(7):1271–81.
Sarkanen J, et al. Human adipose tissue extract induces angiogenesis and adipogenesis in vitro. Tissue Eng A. 2012;18(1–2):17–25.
Halme DG, Kessler DA. FDA regulation of stem-cell–based therapies. N Engl J Med. 2006;355(16):1730–5.
Poulos SP, Hausman DB, Hausman GJ. The development and endocrine functions of adipose tissue. Mol Cell Endocrinol. 2010;323(1):20–34.
Trayhurn P, Beattie JH. Physiological role of adipose tissue: white adipose tissue as an endocrine and secretory organ. Proc Nutr Soc. 2001;60(3):329–39.
Trayhurn P. Endocrine and signalling role of adipose tissue: new perspectives on fat. Acta Physiol Scand. 2005;184(4):285–93.
Tonnard P, et al. Nanofat grafting: basic research and clinical applications. Plast Reconstr Surg. 2013;132(4):1017–26.
Xu P, et al. Nanofat increases dermis thickness and neovascularization in photoaged nude mouse skin. Aesthet Plast Surg. 2018;42(2):343–51.
Yu Q, et al. Co-transplantation of Nanofat enhances neovascularization and fat graft survival in nude mice. Aesthet Surg J. 2018;38(6):667–75.
Yao Y, et al. Adipose extracellular matrix/stromal vascular fraction gel. Plast Reconstr Surg. 2017;139(4):867–79.
Zhang Y, et al. Improved long-term volume retention of stromal vascular fraction gel grafting with enhanced angiogenesis and adipogenesis. Plast Reconstr Surg. 2018;141(5):676e–86e.
Sun M, et al. Adipose extracellular matrix/stromal vascular fraction gel secretes angiogenic factors and enhances skin wound healing in a murine model. Biomed Res Int. 2017;2017:3105780.
Deng C, et al. Extracellular matrix/stromal vascular fraction gel conditioned medium accelerates wound healing in a murine model. Wound Repair Regen. 2017;25(6):923–32.
Zhang P, et al. Ischemic flap survival improvement by composition-selective fat grafting with novel adipose tissue derived product - stromal vascular fraction gel. Biochem Biophys Res Commun. 2018;495(3):2249–56.
Deng C, et al. Treatment of human chronic wounds with autologous extracellular matrix/stromal vascular fraction gel: a STROBE-compliant study. Medicine (Baltimore). 2018;97(32):e11667.
Wiśniewski JR, et al. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J Proteome Res. 2010;9(6):3280–9.
Hauck SM, et al. Deciphering membrane-associated molecular processes in target tissue of autoimmune uveitis by label-free quantitative mass spectrometry. Mol Cell Proteomics. 2010;9(10):2292–305.
Cox J, et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10(4):1794–805.
Luber CA, et al. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity. 2010;32(2):279–89.
Gotz S, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36(10):3420–35.
He Y, et al. The combination of tissue dissection and external volume expansion generates large volumes of adipose tissue. Plast Reconstr Surg. 2017;139(4):888e–99e.
Philips BJ, et al. Prevalence of endogenous CD34+ adipose stem cells predicts human fat graft retention in a xenograft model. Plast Reconstr Surg. 2013;132(4):845–58.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307.
Herbert SP, Stainier DY. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol. 2011;12(9):551–64.
Tonnesen MG, Feng X, Clark RA. Angiogenesis in wound healing. J Investig Dermatol Symp Proc. 2000;5(1):40–6.
Beer HD, Longaker MT, Werner S. Reduced expression of PDGF and PDGF receptors during impaired wound healing. J Invest Dermatol. 1997;109(2):132–8.
Frank S, et al. Regulation of vascular endothelial growth factor expression in cultured keratinocytes. Implications for normal and impaired wound healing. J Biol Chem. 1995;270(21):12607–13.
Reinke JM, Sorg H. Wound repair and regeneration. Eur Surg Res. 2012;49(1):35–43.
Lowe CE, O'Rahilly S, Rochford JJ. Adipogenesis at a glance. J Cell Sci. 2011;124(Pt 16:2681–6.
Ihunnah CA, et al. Estrogen sulfotransferase/SULT1E1 promotes human adipogenesis. Mol Cell Biol. 2014;34(9):1682–94.
Fu Y, et al. Adiponectin promotes adipocyte differentiation, insulin sensitivity, and lipid accumulation. J Lipid Res. 2005;46(7):1369–79.
Yu YH, et al. PKC-ALDH2 pathway plays a novel role in adipocyte differentiation. PLoS One. 2016;11(8):e0161993.
Zhao L, Johnson T, Liu D. Therapeutic angiogenesis of adipose-derived stem cells for ischemic diseases. Stem Cell Res Ther. 2017;8(1):125.
Han SK, Kim HR, Kim WK. The treatment of diabetic foot ulcers with uncultured, processed lipoaspirate cells: a pilot study. Wound Repair Regen. 2010;18(4):342–8.
Lopez J, et al. Cytokine-rich adipose tissue extract production from water-assisted lipoaspirate: methodology for clinical use. BioRes Open Access. 2016;5(1):269–78.
Tonnard P, et al. Nanofat grafting. Plast Reconstr Surg. 2013;132(4):1017–26.
Wozniak SE, et al. Adipose tissue: the new endocrine organ? A review article. Dig Dis Sci. 2009;54(9):1847–56.
Smaniotto S, et al. Combined role of extracellular matrix and chemokines on peripheral lymphocyte migration in growth hormone transgenic mice. Brain Behav Immun. 2010;24(3):451–61.
Kim EJ, et al. Injectable and thermosensitive soluble extracellular matrix and methylcellulose hydrogels for stem cell delivery in skin wounds. Biomacromolecules. 2016;17(1):4–11.
Kuljanin M, et al. Collagenase treatment enhances proteomic coverage of low-abundance proteins in decellularized matrix bioscaffolds. Biomaterials. 2017;144:130–43.
Yao Y, et al. Adipose extracellular matrix/stromal vascular fraction gel: a novel adipose tissue-derived injectable for stem cell therapy. Plast Reconstr Surg. 2017;139(4):867–79.
Wang J, et al. Mechanical micronization of lipoaspirates for the treatment of hypertrophic scars. Stem Cell Res Ther. 2019;10(1):42.
Yao Y, et al. Adipose stromal vascular fraction gel grafting: a new method for tissue volumization and rejuvenation. Dermatol Surg. 2018;44(10):1278–86.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Nature Science Foundation of China (81471881, 81660323, 81801921).
Author information
Authors and Affiliations
Contributions
FL and CD contributed to the conception and study design. JX helped with the in vitro experiments and data collection. YH and YW helped with the in vivo experiments. YH contributed to the data analysis and manuscript writing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was conducted along the guidelines set by the Southern Medical University Institutional Review Board and the Nanfang Hospital Animal Ethics Committee.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
He, Y., Xia, J., Chen, H. et al. Human adipose liquid extract induces angiogenesis and adipogenesis: a novel cell-free therapeutic agent. Stem Cell Res Ther 10, 252 (2019). https://doi.org/10.1186/s13287-019-1356-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-019-1356-0