
Abstract
During tumorigenesis and development, participation of the tumor microenvironment is not negligible. As an important component in the tumor microenvironment, mesenchymal stem cells (MSCs) have been corroborated to mediate proliferation, metastasis, and drug resistance in many cancers, including osteosarcoma. What’s more, because of tumor site tropism, MSCs can be engineered to be loaded with therapeutic agents so that drugs can be precisely delivered to tumor lesions. In this review, we mainly discuss recent advances concerning the functions of MSCs in osteosarcoma and their possible clinical applications in the future.
Similar content being viewed by others


Background
Osteosarcoma (OS) is a malignant bone tumor and usually afflicts people in adolescence. Nowadays, neoadjuvant chemotherapy combined with conservative surgical resection and post-operative adjuvant chemotherapy is the preferential treatment for OS patients. However, because of distant metastasis and chemotherapy resistance, efficacy is still unsatisfactory. Local tumor microenvironments (TMEs) comprise extracellular matrix and cellular components [1] and, over the years, the TME has been documented to be firmly associated with the initiation and progression of OS and to contribute its poor prognosis. In the TME, non-tumor cells such as fibroblasts, endothelial cells, immune cells, and MSCs play important roles in tumor development [2]. Notably, a growing body of evidence has implied the involvement of MSCs in OS progression.
MSCs are non-hematopoietic precursor cells and have been found in many human tissues, such as bone marrow, adipose tissue, peripheral blood, placenta, and umbilical cord [3, 4]. They also exhibit an ability to self-renew and differentiate into a variety of mesenchymal cell lineages, like osteocytes, chondrocytes, and adipocytes [5].
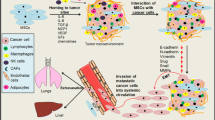
Recently, MSCs have been considered to participate in wound healing [6, 7]. As “wounds that never heal”, tumors are also conjectured to be influenced by MSCs. Prior reports have provided results that although they share similar characteristics with regard to morphology and multilineage differentiation capability, MSCs from tumor tissue (T-MSCs) and MSCs from normal tissue (N-MSCs) have different impacts on tumor development [8]. For instance, in Ewing sarcoma, T-MSCs exhibit a significantly greater proliferative capacity than N-MSCs [9]. Under the influence of tumor cells and chronic TME, circling N-MSCs migrate to tumor lesions and are “educated” to be T-MSCs that support the growth of tumor. Moreover, after MSCs are recruited to the tumor lesions, they can obtain a specific tumor-associated fibroblast (TAF) phenotype and encourage tumor cell growth [10]. A large amount of cytokines secreted by the TME and tumor cells are involved in this process, including interleukin (IL)-6, transforming growth factor (TGF)-β, stromal-derived factor (SDF)-1, tumor necrosis factor (TNF)-α, interferon (IFN)-γ, macrophage inhibition factor (MIF), and IL-1α [8, 11] (Fig. 1). In this review, we discuss the role of MSCs in OS and their potential treatment applications based on the findings from recent studies.

Different effects exerted by the TME on naïve MSCs (N-MSC). Under the effects of some cytokines (SDF-1, MIF, TGF-β, and so on), N-MSCs are recruited to the TME. Through the paracrine network in the TME, N-MSCs undergo a series of functional transformations. On one hand, INF-γ, TNF-α, and IL-1α strengthen the tumor growth-promoting effects of MSCs; On the other hand, INF-γ, TNF-α, and TGF-β enhance the ability of MSCs to promote tumor metastasis. Furthermore, MSCs can differentiate into cancer-associated fibroblasts (CAF) under the stimulation of TGF-β
MSCs and the biogenesis of OS
OS is characterized by a variety of histopathologic subtypes, suggesting the hypothesis that MSCs may be the origin of OS cells. According to previous investigations, mutation of related genes [12], the differentiation stage of MSCs [13], the tissue source of MSCs [13], and the bone environment [14] can influence the malignant transformation of MSCs.
In vivo research found that the overexpression of c-MYC with loss of Ink4a/Arf could induce the malignant transformation from bone marrow-derived MSCs (BM-MSCs) to OS [15]. Wang et al. conducted research to evaluate the impact of mutation of c-MYC, Ras, Rb, and P53 on the transformation of MSCs [12]. The results demonstrated that Rb silencing and c-MYC overexpression in MSCs were associated with OS tumorigenesis [12]. Under other oncogenic stresses, however, BM-MSCs expressing the FUS-CHOP fusion gene were reported to develop as liposarcoma [16]. As such, the mesenchymal tumor histological type may be firmly linked with the type of oncogenic stress.
Except for undifferentiated MSCs, osteogenic lineage-committed progenitor cells can also become the origin for OS. It was documented that undifferentiated p53−/− and Rb−/− p53−/− BM-MSCs led to the generation of leiomyosarcoma in vivo [13]. Interestingly, after BM-MSCs differentiated to the osteogenic lineage and were treated with p53 and Rb genes excised, they developed into tumors displaying clear OS histological features [13]. Moreover, when researchers prolonged the time of differentiation toward the osteogenic lineage, the degree of OS differentiation increased as well [13]. All these findings imply that it may be much more appropriate to classify the cellular origin of OS as “a MSC population” that is generated from MSCs induced by a specific oncogenic stress in a specific differentiation stage. The researchers also reported that none of the Rb−/− p53−/− ASC-derived osteogenic progenitors and undifferentiated ASCs (adipose derived MSCs) developed to be OS-like tumors in vivo [13]. Accordingly, the generation of OS may also rely on the tissue source of MSCs. Of course, we cannot exclude the possibility that ASCs with other transforming mutations will develop to be OS.
The bone environment is yet another factor affecting differentiation. In one study, the MSCs isolated from OS were described to become the progenitor cells of OS after they had been injected into nude mice [17]. In vitro, however, the MSCs could still differentiate into osteoblasts, chondrocytes, and adipocytes. Rubio et al. [14] inoculated GFP-tagged wild type, p53−/−, RB−/−, and p53−/− RB−/− MSCs into NSG immunodeficient mice to investigate whether bone environment would influence the transformation of MSCs. They provided evidence that only p53−/− and p53−/− RB−/− MSCs developed into OS-like tumors in the recipients’ bone. At the same time, tumoral positions far away from the host bone exhibited a different pathology, with the tumor resembling leiomyosarcoma much more than OS [14]. These results show that the host bone environment might be another factor in programming the sarcoma phenotype of transformed MSCs. Bone morphogenetic protein (BMP)-2 and calcium substrates are abundant in the bone milieu [14]. BMP-2 was shown to affect the osteoblastic differentiation of p53−/− RB−/− MSCs through autocrine activation of WNT ligands [18], thereby leading to OS phenotype development. Calcium substrates can also contribute to the development of p53−/− RB−/− MSC osteogenesis [19]. In light of this, researchers established a ceramic bone-like environment model to analyze the impact of the calcified extracellular matrix on the osteogenesis and sarcomagenesis potential of p53−/− RB−/− MSCs [14]. In the absence of BMP-2, the tumor showed a small area of osteogenic differentiation that just surrounded the ceramic material; however, when BMP-2 was added to the ceramic-fibrin structure, the OS-like tumor occupied a much larger area [14]. Consequently, bone environment signals can influence the differentiation of distinct sarcoma phenotypes.
Promotion of OS growth
Proliferation
Cancers require a sufficient energy supply for development. What’s more, they have to escape autologous immune surveillance that is usually initiated by NK cells and CD8+ T cells. Tumor cells are able to secret copious cytokines, like SDF-1, IL-6, and platelet-derived growth factor (PDGF), that can induce MSCs to migrate to tumor tissue [8]. On one hand, cross-talk between MSCs and tumor cells favors angiogenesis and finally creates a network of new vessels in tumor tissue. On the other hand, some cytokines secreted by MSCs, such as TGF-β, can blunt the antitumor immune responses and facilitate tumor immune escape [20].
Mohseny et al. [21] sorted three different types of low-passage MSCs (normal MSCs), high-passage MSCs (transformed MSCs), and tumorigenic MSCs (representative of OS-producing cells) through diverse times of passage. By analyzing whole-genome expression, they found that, in transformed and tumorigenic MSCs, expression of a T-cell response-associated gene (Fut-7) and major histocompatibility complex-related genes (H2-K1 and H2-D1) was reduced, while the pro-angiogenic gene Est1 was upregulated [21]. These results indicate that early OS is endowed with angiogenesis and immunosuppressive properties. Vascular endothelial growth factor (VEGF) is a classic cytokine that can activate the PI3K-Akt pathway and contribute to angiogenesis [22]. Some other cytokines such as IL-6 and Monocyte chemoattractant protein (MCP)-1 are also linked with the PI3K-Akt pathway [23]. The growth-promoting effect of IL-6 and VEGF secreted by MSCs has been verified in many studies when MSCs are co-cultured with OS cells [24,25,26]. As a form of intercellular communication, extracellular vesicles’ (EVs) effects on recipient cells are usually dependent on their cargos. In recent studies on OS-derived EVs, exosome-like EVs released by OS could be internalized by MSCs and the educated MSCs promoted the growth and metastasis of OS [27]. Follow-up studies implied the involvement of an inflammatory loop in which TGF-β in OS EVs induced the production of IL-6 in MSCs, activated the IL-6/STAT signaling axis in OS cells, and promoted the growth of OS in vivo [27]. Furthermore, OS can also inhibit the osteogenic differentiation of MSCs, strengthening the pro-tumor effects of MSCs [28].
Metastasis and migration
Lung is the preferred metastatic target organ in OS. Via interclonal co-operation, highly metastatic clonal variants of OS can induce poorly metastatic clonal variants of the same cell line into a migratory and invasive phenotype [29]. During the process of horizontal phenotypic transfer, EVs wrapped with metastasis-associated proteins were released by highly metastatic OS clonal variants and internalized by poorly metastatic clonal variants. The results show that OS with high heterogeneity can strengthen its metastasis via unidirectional EV-mediated interclonal cooperation. The lung tropism of EVs also implies that lung is the preferred target organ for the establishment of a pre-metastatic niche [29].
Emerging evidence has demonstrated the metastasis-promoting effects of MSCs on OS. Under the stimulation of human MSCs, pulmonary metastasis was promoted and some cytokines such as CCL5 (CC chemokine ligand5) and VEGF may be responsible for the migration [25, 30]. Another study documented that VEGF secreted by MSCs promoted the metastasis through elevating the expression of CXCR4 (CXC chemokine receptor4) in OS cells [26]. The hypoxic environment in cancer results from the rapid proliferation of tumor cells [31]. A recent investigation demonstrated that hypoxia together with some chemoattractants such as IL-6 and MCP-1 could recruit MSCs to tumor lesions. Via the cross-talk between MSCs and tumor cells, the secretions, e.g., CCL5, placenta growth factor (PIGF) and CXCL10 promoted the metastasis of malignant cells [32]. When the hypoxia persisted, the metabolic process of the cells switched to continuous anaerobic glycolysis, and subsequently induced tumor-associated extracellular acidification [32]. In another study, short-term acidification was described to convert MSCs to T-MSCs [33]. Moreover, via the activation of the NF-κB pathway, T-MSCs secreted some clonogenicity-related and migration-related factors such as CCL-5 [33]. This study demonstrated that tumoral acidosis was crucial in the migration, which was usually ignored in the past.
In the model of cancer stem cell (CSC) theory, CSCs originate from stem cells or precursor cells with epigenetic mutations. They can form and sustain tumors and are resistant to traditional therapies [34]. Many studies have provided evidence for the existence of stem-like properities in OS [35, 36]. Under the influence of MSCs, OS cells can also acquire the phenotype of stem cells [24, 33]. What’s more, CSCs can stimulate MSCs to secret TGF-β1, which induces NF-kB gene activation in MSCs and subsequent IL-6 secretion by MSCs [24]. After that, the expression of some prometastatic genes, including intercellular adhesion molecule-1 and MET, is elevated [24]. The cellular interaction based on cytokines is critically important, and some investigations also showed the importance of horizontal gene transfer among cells for tumor aggressiveness. An investigation demonstrated Ki-ras gene transfer from 143B-GFP cells (OS cells with high metastatic potential) to MNNG/HOS-RFP cells (OS cells with low metastatic potential) in vivo. The metastasis of MNNG/HOS-RFP was enhanced in response [37]. Whether gene transfer between OS cells and MSCs cells promotes OS needs to be further explored.
Drug resistance
MSCs can foster the stemness of tumor cells. Through the following mechanisms, CSCs can strengthen their tolerance to chemotherapeutics: first, upregulated expression of drug efflux pumps (Pgp and ABCG2); second, more active ALDH (aldehyde dehydrogenase), which has been demonstrated to confer resistance to cyclophosphamide; third, increased expression of pro-survival BCL-2 (B-cell lymphoma-2) protein family members; and last but not least, strengthened DNA damage repair [38]. Furthermore, cross-talk between tumor cells and MSCs can also protect tumor cells from lethal [39]. Many products of MSCs, such as SDF-1, contribute to the drug resistance of tumors [39].
With regard to short-term acidification, MSCs were shown to secret some pro-inflammatory factors such as IL-6 and IL-8 and fostered the stemness of HOS cells [33]. As a result, OS cell sensitivity to DXR was reduced [33]. Another study showed that MSCs promoted the survival of OS cells treated with cisplatin and DOX [40]. Subsequent investigations documented that, via the activation of STAT3, IL-6 secreted by MSCs promoted the expression of MDR-1 (Multi-drug resistance gene 1) and MRP (Multidrug resistance-associated protein), which work as a drug efflux pump [40]. Collectively, MSCs play an important role in drug resistance of OS, through increasing the expression of multi-drug resistance genes or inducing the OS cells’ stemness in a paracrine pathway.
Inhibition of OS growth
Even though many researchers have reported that MSCs support OS growth, a few studies have also shown that MSCs suppress OS growth. Wharton’s jelly-derived MSCs (WJ-MSCs) are homogeneous populations of MSCs isolated from the gelatinous Wharton’s jelly in the umbilical cord. When OS cells (MG-63) were cultured in human WJ-MSC-conditioned medium, their growth, including proliferation and migration in vitro, was inhibited [41].
Taken together, the previous studies on the effects of MSCs on tumor development have demonstrated contradicting results. Even though the reasons for these contradictions are yet to be explored, it can be inferred that the results are affected by many aspects, including the TME, the source of MSCs, the type of tumor, the methodologies of the experiments, and the animal model. As a special type of MSC, WJ-MSCs have been reported to attenuate the growth of many tumors, including OS [41]. According to this, WJ-MSCs may be safe for future clinical applications. The details of some roles of MSCs in OS are shown in Table 1.
The prospects of MSCs in clinical applications
Delivering agents to OS tumors
MSCs have tropism toward tumor stroma. Taking advantage of this characteristic, we can load MSCs with therapeutic agents and utilize their anti-cancer effects more effectively. In a study, murine ASCs were transfected with the full-length human TRAIL (TNF-related apoptosis-inducing ligand) gene, which has been reported to induce death in OS [42]. The experiment in vitro showed that TRAIL delivered by MSCs significantly induced the apoptosis of SAOS2 via the activation of caspase-8 [3]. After MSCs were transfected with adenoviruses that carried the osteoprotegerin gene, they were also reported to suppress the growth of OS and bone destruction [43]. Except for being transfected with some pro-apoptosis genes, MSCs can also be modified to deliver medicine. Generally speaking, the cargos MSCs carry should possess at least the following four characteristics: 1, strong toxicity towards tumor tissue; 2, non-deleterious to the MSCs during migration; 3, not affected by the MSCs; 4, not affect the biological properties of the MSCs.
Traditional chemical drugs as cargos in MSCs would possibly ruin the MSCs, which will make the drug delivery fail. Because of this, photodynamic therapy may be a good choice. Duchi et al. [44] loaded a photosensitizer (TPPS) into core-shell PMMA nanoparticles (FNPs) and incorporated the compound into MSCs. When the human OS cell line U2OS-RFP-TUBA1B was co-cultured with MSCs that were loaded with TPPS-FNPs in vitro, the MSCs released reactive oxygen species and killed the surrounding tumor cells under the stimulation of photoactivation. In the process, the MSCs were also induced to die and the safe clearance of MSCs was ensured [44]. Apart from cytotoxic agents and viral vectors, anti-angiogenic agents and immunostimulatory agents have also been reported to be transported to tumor lesions by MSCs [45]. Promising procedures for using MSCs as delivery vehicles are shown in Fig. 2 [46, 47]. Compared with other drug carriers, such as liposomes and magnetic beads, MSCs possess particular advantages, including specific targeting, easy availability, and good immunological compatibility [45].

Promising methods for using MSCs as delivery vehicles. (i) Transduct MSCs with retroviral vectors that carry therapeutic RNA: capture the MSCs from the patient, modify MSCs by transduction with retroviral vectors at the first passage, culture the transducted MSCs for several days, select the cells with puromycin, expand the transducted and selected cells to the clinical necessary dose, and finally administer them to the patient. (ii) Load MSCs with nanoparticles incorporated with drugs: load chemotherapeutic agents in nanoparticles, uptake the nanoparticles with MSCs, and administer the MSCs into patients
As a highly heterogeneous disease, OS usually exhibits different phenotypes in different patients. Identifying the specific phenotypes as therapeutic targets may lead to a better prognosis. Recently, researchers performed molecular profiling in two cases of OS. Even though they used sirolimus + crizotinib and abraxane + celecoxib to match the P1K3CA, c-MET, and SPARC and COX2 mutations in patient 1 and used temsirolimus, sorafenib, and bevacizumab to match NF2, PDGFRα, and TP53 mutations in patient 2, the prognosis of the two patients was still bad [48]. However, the result doesn’t mean that personalized targeted therapy is not feasible for the treatment of OS, and MSCs may exhibit their advantages in carrying targeted drugs or anti-cancer genes for personalized medicine.
Targeting upstream and downstream modulators of MSCs
Through the exchange of cytokines between MSCs and OS cells, cross-talk can be established to mediate the development of OS. Via targeting upstream and downstream modulators of MSCs, we can block the cross-talk and inhibit tumor progression. The SDF-1/CXCR4 axis is well-known for its pleiotropic role in tumor development [49]. Some antagonists of the CXCR4 receptor, such as AMD3100, have been developed to disturb the cross-talk [49]. Scientists employed a novel antagonist of CXCR4 named ‘Peptide R’ to inhibit BM-MSC-mediated OS [49]. The results showed that Peptide R suppressed the migration and invasiveness of U2OS through ceasing the Ras/ERK and PI3K/Akt activation that were mediated by BM-MSCs [49]. Some new medicines, such as the IDO inhibitor 1-methyl-DL-tryptophan (1-MT), can enhance the activity of antitumor T cells [50]. Even though no investigations have been carried out on the effects of IDO inhibitors on OS, strategies that reverse the immunosuppression mediated by MSCs may be promising for combination with other immunotherapies.
Promoting bone reconstruction in OS patients
After large segment resection of tumor, OS patients are usually confronted with many challenges in the process of bone reconstruction, such as infection and local recurrence. MSCs have the ability to migrate to the wound and promote wound healing through transforming the inflammatory microenvironment [51]. With the maturity of 3D printing technique, personalized bone reconstruction supported by MSCs is developing [52]. Therefore, MSCs have potential for therapeutic use in limb salvage after tumor resection. To study their feasibility in the clinical setting, researchers inoculated mice with DLM8-luc OS cells and performed resection of the primary tumor when the OS model had developed [53]. After that, they separated the mice into three ASC treatment groups (the mice received intravenous MSCs, MSCs at the surgical area, or no MSCs). By comparing the number of pulmonary nodules, the time to first evidence of metastasis, and the size of the recurrent OS, the researchers reported that the mice that received intravenous MSCs healed much faster than the other two groups, while the development of local recurrence and the size of recurrent tumors showed no apparent difference in the three groups [53]. So it might be promising to use MSCs to augment bone healing in patients with lesions after surgery.
Nevertheless, we have to be aware of possible relapse after the administration of MSCs. Recently, Perrot and colleagues [54] reported an unexpected late local recurrence of human OS that occurred at the site of autologous fat grafts 13 years after the initial pathology and 18 months after a lipofilling procedure. The results of their further experimental model implied the possible OS-promotion role of MSC-like cells that were contained in the fatty tissues [54]. In another study, researchers addressed that SAOS2 OS cells could maintain human ASCs in a “stemness” state which would exacerbate OS growth and aggressiveness [55]. In this manner, the remaining OS cells after resection may develop quickly under the stimulation of the MSC-like cells in the autologous fat grafts. Combined with the OS-promoting effects of MSCs we introduced above, we have to realize that the clinical applications of MSCs for OS treatment must be carefully performed after deciding on the originating tissue of MSCs, the number of MSCs, the time of MSC injection, and so on. Only when no risk of OS promotion is detected in animal experiments will MSCs meet the regulatory requirements [56].
Conclusions and prospects
Nowadays, more and more researchers are focusing on the effects of MSCs on wounds and tumors. In this review, we discuss the role of MSCs in OS and the potential applications of MSCs in OS treatment. One of the promising clinical applications of MSCs is as a carrier to deliver anti-cancer agents. However, we have to consider the consequence that the recruited MSCs may exhibit their OS-promoting effects. Under this circumstance, it is indispensable for us to evaluate the anti-cancer agents’ OS-inhibiting effects and MSCs’ OS-promoting effects. Another promising application is to target upstream or downstream modulators to block the cross-talk between OS cells and MSCs. Yet, the complexity and multiplicity of the communication networks make it difficult to find an effective antagonist. AMD3100 (inhibiting CXCR4) [49] and 1-MT (inhibiting indoleamine 2,3-dioxygenase) [50] are current frequently used antagonists. Although these antagonists haven’t been used for the treatment of OS, investigations on them are ongoing, and the potential of MSCs may be massive in the future.
Abbreviations
- ASC:
-
Adipose tissue-derived MSC
- BM-MSC:
-
Bone marrow MSC
- BMP:
-
Bone morphogenetic protein
- CSC:
-
Cancer stem cell
- IFN:
-
Interferon
- IL:
-
Interleukin
- MCP:
-
Monocyte chemoattractant protein
- MIF:
-
Macrophage inhibition factor
- MSC:
-
Mesenchymal stem cell
- N-MSC:
-
Normal tissue MSC
- OS:
-
Osteosarcoma
- PDGF:
-
Platelet derived growth factor
- PIGF:
-
Placenta growth factor
- SDF:
-
Stromal-derived factor
- TAF:
-
Tumor-associated fibroblast
- TGF:
-
Transforming growth factor
- TME:
-
Tumor microenvironment
- T-MSC:
-
Tumor tissue MSC
- TNF:
-
Tumor necrosis factor
- VEGF:
-
Vascular endothelial growth factor
- WJ-MSCs:
-
Wharton’s jelly derived MSC.
References
Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, Gong Z, Zhang S, Zhou J, Cao K, Li X, Xiong W, Li G, Zeng Z, Guo C. Role of tumor microenvironment in tumorigenesis. J Cancer. 2017;8:761–73.
Ye J, Wu D, Wu P, Chen Z, Huang J. The cancer stem cell niche: cross talk between cancer stem cells and their microenvironment. Tumor Biol. 2014;35:3945–51.
Hass R, Kasper C, Bohm S, Jacobs R. Different populations and sources of human mesenchymal stem cells (MSC): a comparison of adult and neonatal tissue-derived MSC. Cell Commun Signal. 2011;9:12.
Viswanathan S, Keating A, Deans R, Hematti P, Prockop D, Stroncek DF, Stacey G, Weiss DJ, Mason C, Rao MS. Soliciting strategies for developing cell-based reference materials to advance mesenchymal stromal cell research and clinical translation. Stem Cells Dev. 2014;23:1157–67.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–7.
De Francesco F, Tirino V, Desiderio V, Ferraro G, D'Andrea F, Giuliano M, Libondi G, Pirozzi G, De Rosa A, Papaccio G. Human CD34/CD90 ASCs are capable of growing as sphere clusters, producing high levels of VEGF and forming capillaries. PLoS One. 2009;4, e6537.
Ferraro GA, De Francesco F, Nicoletti G, Paino F, Desiderio V, Tirino V, D'Andrea F. Human adipose CD34+ CD90+ stem cells and collagen scaffold constructs grafted in vivo fabricate loose connective and adipose tissues. J Cell Biochem. 2013;114:1039–49.
Sun Z, Wang S, Zhao RC. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J Hematol Oncol. 2014;7:14.
Johann PD, Vaegler M, Gieseke F, Mang P, Armeanu-Ebinger S, Kluba T, Handgretinger R, Muller I. Tumour stromal cells derived from paediatric malignancies display MSC-like properties and impair NK cell cytotoxicity. BMC Cancer. 2010;10:501.
De Wever O, Van Bockstal M, Mareel M, Hendrix A, Bracke M. Carcinoma-associated fibroblasts provide operational flexibility in metastasis. Semin Cancer Biol. 2014;25:33–46.
Lourenco S, Teixeira VH, Kalber T, Jose RJ, Floto RA, Janes SM. Macrophage migration inhibitory factor-CXCR4 is the dominant chemotactic axis in human mesenchymal stem cell recruitment to tumors. J Immunol. 1950;194(2015):3463–74.
Wang JY, Wu PQ, Chen PC, Lee CW, Chen WM, Hung SC. Generation of osteosarcomas from a combination of Rb silencing and c-Myc overexpression in human mesenchymal stem cells. Stem Cells Transl Med. 2016;6:512–16.
Rubio R, Gutierrez-Aranda I, Saez-Castillo AI, Labarga A, Rosu-Myles M, Gonzalez-Garcia S, Toribio ML, Menendez P, Rodriguez R. The differentiation stage of p53-Rb-deficient bone marrow mesenchymal stem cells imposes the phenotype of in vivo sarcoma development. Oncogene. 2013;32:4970–80.
Rubio R, Abarrategi A, Garcia-Castro J, Martinez-Cruzado L, Suarez C, Tornin J, Santos L, Astudillo A, Colmenero I, Mulero F, Rosu-Myles M, Menendez P, Rodriguez R. Bone environment is essential for osteosarcoma development from transformed mesenchymal stem cells. Stem Cells. 2014;32:1136–48.
Shimizu T, Ishikawa T, Sugihara E, Kuninaka S, Miyamoto T, Mabuchi Y, Matsuzaki Y, Tsunoda T, Miya F, Morioka H, Nakayama R, Kobayashi E, Toyama Y, Kawai A, Ichikawa H, Hasegawa T, Okada S, Ito T, Ikeda Y, Suda T, Saya H. c-MYC overexpression with loss of Ink4a/Arf transforms bone marrow stromal cells into osteosarcoma accompanied by loss of adipogenesis. Oncogene. 2010;29:5687–99.
Riggi N, Cironi L, Provero P, Suva ML, Stehle JC, Baumer K, Guillou L, Stamenkovic I. Expression of the FUS-CHOP fusion protein in primary mesenchymal progenitor cells gives rise to a model of myxoid liposarcoma. Cancer Res. 2006;66:7016–23.
Mohseny AB, Szuhai K, Romeo S, Buddingh EP, Briaire-de Bruijn I, de Jong D, van Pel M, Cleton-Jansen AM, Hogendoorn PC. Osteosarcoma originates from mesenchymal stem cells in consequence of aneuploidization and genomic loss of Cdkn2. J Pathol. 2009;219:294–305.
Rodriguez R, Rubio R, Menendez P. Modeling sarcomagenesis using multipotent mesenchymal stem cells. Cell Res. 2012;22:62–77.
Arinzeh TL, Tran T, McAlary J, Daculsi G. A comparative study of biphasic calcium phosphate ceramics for human mesenchymal stem-cell-induced bone formation. Biomaterials. 2005;26:3631–8.
Chang AI, Schwertschkow AH, Nolta JA, Wu J. Involvement of mesenchymal stem cells in cancer progression and metastases. Curr Cancer Drug Targets. 2015;15:88–98.
Mohseny AB, Xiao W, Carvalho R, Spaink HP, Hogendoorn PC, Cleton-Jansen AM. An osteosarcoma zebrafish model implicates Mmp-19 and Ets-1 as well as reduced host immune response in angiogenesis and migration. J Pathol. 2012;227:245–53.
Soler A, Angulo-Urarte A, Graupera M. PI3K at the crossroads of tumor angiogenesis signaling pathways. Mol Cell Oncol. 2015;2, e975624.
Hung SC, Pochampally RR, Chen SC, Hsu SC, Prockop DJ. Angiogenic effects of human multipotent stromal cell conditioned medium activate the PI3K-Akt pathway in hypoxic endothelial cells to inhibit apoptosis, increase survival, and stimulate angiogenesis. Stem Cells. 2007;25:2363–70.
Cortini M, Massa A, Avnet S, Bonuccelli G, Baldini N. Tumor-activated mesenchymal stromal cells promote osteosarcoma stemness and migratory potential via IL-6 secretion. PLoS One. 2016;11, e0166500.
Zhang P, Dong L, Long H, Yang TT, Zhou Y, Fan QY, Ma BA. Homologous mesenchymal stem cells promote the emergence and growth of pulmonary metastases of the rat osteosarcoma cell line UMR-106. Oncol Lett. 2014;8:127–32.
Zhang P, Dong L, Yan K, Long H, Yang TT, Dong MQ, Zhou Y, Fan QY, Ma BA. CXCR4-mediated osteosarcoma growth and pulmonary metastasis is promoted by mesenchymal stem cells through VEGF. Oncol Rep. 2013;30:1753–61.
Baglio SR, Lagerweij T, Perez Lanzon M, Xuan Ho D, Leveille N, Melo SA, Cleton-Jansen AM, Jordanova ES, Roncuzzi L, Greco M, van Eijndhoven MA, Grisendi G, Dominici M, Bonafede R, Lougheed S, de Gruijl TD, Zini N, Cervo S, Steffan A, Canzonieri V, Martson A, Maasalu K, Koks S, Wurdinger T, Baldini N, Pegtel DM, Blocking tumor-educated MSC paracrine activity halts osteosarcoma progression. Clin Cancer Res. 2017;23:3721–33.
Tu B, Peng ZX, Fan QM, Du L, Yan W, Tang TT. Osteosarcoma cells promote the production of pro-tumor cytokines in mesenchymal stem cells by inhibiting their osteogenic differentiation through the TGF-beta/Smad2/3 pathway. Exp Cell Res. 2014;320:164–73.
Macklin R, Wang H, Loo D, Martin S, Cumming A, Cai N, Lane R, Ponce NS, Topkas E, Inder K, Saunders NA, Endo-Munoz L. Extracellular vesicles secreted by highly metastatic clonal variants of osteosarcoma preferentially localize to the lungs and induce metastatic behaviour in poorly metastatic clones. Oncotarget. 2016;7:43570–87.
Xu WT, Bian ZY, Fan QM, Li G, Tang TT. Human mesenchymal stem cells (hMSCs) target osteosarcoma and promote its growth and pulmonary metastasis. Cancer Lett. 2009;281:32–41.
Gao T, Li JZ, Lu Y, Zhang CY, Li Q, Mao J, Li LH. The mechanism between epithelial mesenchymal transition in breast cancer and hypoxia microenvironment. Biomed Pharmacother. 2016;80:393–405.
Chaturvedi P, Gilkes DM, Wong CC, Kshitiz, Luo W, Zhang H, Wei H, Takano N, Schito L, Levchenko A, Semenza GL. Hypoxia-inducible factor-dependent breast cancer-mesenchymal stem cell bidirectional signaling promotes metastasis. J Clin Invest. 2013;123:189–205.
Avnet S, Di Pompo G, Chano T, Errani C, Ibrahim-Hashim A, Gillies RJ, Donati DM, Baldini N. Cancer-associated mesenchymal stroma fosters the stemness of osteosarcoma cells in response to intratumoral acidosis via NF-kappaB activation. Int J Cancer. 2017;140:1331–45.
Papaccio F, Paino F, Regad T, Papaccio G, Desiderio V, Tirino V. Cancer cells, cancer stem cells, and mesenchymal stem cells: influence in cancer development. Stem Cells Transl Med. 2017;6:2115–25.
Gibbs CP, Kukekov VG, Reith JD, Tchigrinova O, Suslov ON, Scott EW, Ghivizzani SC, Ignatova TN, Steindler DA. Stem-like cells in bone sarcomas: implications for tumorigenesis. Neoplasia. 2005;7:967–76.
Tirino V, Desiderio V, Paino F, De Rosa A, Papaccio F, Fazioli F, Pirozzi G, Papaccio G. Human primary bone sarcomas contain CD133+ cancer stem cells displaying high tumorigenicity in vivo. FASEB J. 2011;25:2022–30.
Tome Y, Tsuchiya H, Hayashi K, Yamauchi K, Sugimoto N, Kanaya F, Tomita K, Hoffman RM. In vivo gene transfer between interacting human osteosarcoma cell lines is associated with acquisition of enhanced metastatic potential. J Cell Biochem. 2009;108:362–7.
Abdullah LN, Chow EK. Mechanisms of chemoresistance in cancer stem cells. Clin Transl Med. 2013;2:3.
Lis R, Touboul C, Mirshahi P, Ali F, Mathew S, Nolan DJ, Maleki M, Abdalla SA, Raynaud CM, Querleu D, Al-Azwani E, Malek J, Mirshahi M, Rafii A. Tumor associated mesenchymal stem cells protects ovarian cancer cells from hyperthermia through CXCL12. Int J Cancer. 2011;128:715–25.
Tu B, Zhu J, Liu S, Wang L, Fan Q, Hao Y, Fan C, Tang TT. Mesenchymal stem cells promote osteosarcoma cell survival and drug resistance through activation of STAT3. Oncotarget. 2016;7:48296–308.
Gauthaman K, Yee FC, Cheyyatraivendran S, Biswas A, Choolani M, Bongso A. Human umbilical cord Wharton's jelly stem cell (hWJSC) extracts inhibit cancer cell growth in vitro. J Cell Biochem. 2012;113:2027–39.
Grisendi G, Spano C, D'Souza N, Rasini V, Veronesi E, Prapa M, Petrachi T, Piccinno S, Rossignoli F, Burns JS, Fiorcari S, Granchi D, Baldini N, Horwitz EM, Guarneri V, Conte P, Paolucci P, Dominici M. Mesenchymal progenitors expressing TRAIL induce apoptosis in sarcomas. Stem Cells. 2015;33:859–69.
Qiao B, Shui W, Cai L, Guo S, Jiang D. Human mesenchymal stem cells as delivery of osteoprotegerin gene: homing and therapeutic effect for osteosarcoma. Drug Des Dev Ther. 2015;9:969–76.
Duchi S, Sotgiu G, Lucarelli E, Ballestri M, Dozza B, Santi S, Guerrini A, Dambruoso P, Giannini S, Donati D, Ferroni C, Varchi G. Mesenchymal stem cells as delivery vehicle of porphyrin loaded nanoparticles: effective photoinduced in vitro killing of osteosarcoma. J Control Release. 2013;168:225–37.
Shah K. Mesenchymal stem cells engineered for cancer therapy. Adv Drug Deliv Rev. 2012;64:739–48.
Zhao Y, Tang S, Guo J, Alahdal M, Cao S, Yang Z, Zhang F, Shen Y, Sun M, Mo R, Zong L, Jin L. Targeted delivery of doxorubicin by nano-loaded mesenchymal stem cells for lung melanoma metastases therapy. Sci Rep. 2017;7:44758.
Niess H, von Einem JC, Thomas MN, Michl M, Angele MK, Huss R, Gunther C, Nelson PJ, Bruns CJ, Heinemann V. Treatment of advanced gastrointestinal tumors with genetically modified autologous mesenchymal stromal cells (TREAT-ME1): study protocol of a phase I/II clinical trial. BMC Cancer. 2015;15:237.
Subbiah V, Wagner MJ, McGuire MF, Sarwari NM, Devarajan E, Lewis VO, Westin S, Kato S, Brown RE, Anderson P. Personalized comprehensive molecular profiling of high risk osteosarcoma: Implications and limitations for precision medicine. Oncotarget. 2015;6:40642–54.
Fontanella R, Pelagalli A, Nardelli A, D'Alterio C, Ierano C, Cerchia L, Lucarelli E, Scala S, Zannetti A. A novel antagonist of CXCR4 prevents bone marrow-derived mesenchymal stem cell-mediated osteosarcoma and hepatocellular carcinoma cell migration and invasion. Cancer Lett. 2016;370:100–7.
Ling W, Zhang J, Yuan Z, Ren G, Zhang L, Chen X, Rabson AB, Roberts AI, Wang Y, Shi Y. Mesenchymal stem cells use IDO to regulate immunity in tumor microenvironment. Cancer Res. 2014;74:1576–87.
Kinnaird T, Stabile E, Burnett MS, Lee CW, Barr S, Fuchs S, Epstein SE. Marrow-derived stromal cells express genes encoding a broad spectrum of arteriogenic cytokines and promote in vitro and in vivo arteriogenesis through paracrine mechanisms. Circ Res. 2004;94:678–85.
Liu A, Xue GH, Sun M, Shao HF, Ma CY, Gao Q, Gou ZR, Yan SG, Liu YM, He Y. 3D printing surgical implants at the clinic: a experimental study on anterior cruciate ligament reconstruction. Sci Rep. 2016;6:21704.
Aanstoos ME, Regan DP, Rose RJ, Chubb LS, Ehrhart NP. Do mesenchymal stromal cells influence microscopic residual or metastatic osteosarcoma in a murine model? Clin Orthop Relat Res. 2016;474:707–15.
Perrot P, Rousseau J, Bouffaut AL, Redini F, Cassagnau E, Deschaseaux F, Heymann MF, Heymann D, Duteille F, Trichet V, Gouin F. Safety concern between autologous fat graft, mesenchymal stem cell and osteosarcoma recurrence. PLoS One. 2010;5, e10999.
Paino F, La Noce M, Di Nucci D, Nicoletti GF, Salzillo R, De Rosa A, Ferraro GA, Papaccio G, Desiderio V, Tirino V. Human adipose stem cell differentiation is highly affected by cancer cells both in vitro and in vivo: implication for autologous fat grafting. Cell Death Dis. 2017;8, e2568.
Fink Jr DW. FDA regulation of stem cell-based products. Science. 2009;324:1662–3.
Acknowledgements
This work was supported by the National Natural Science Foundation (81202115), Doctoral Innovation Fund of Shanghai Jiaotong University School of Medicine (number BXJ201732), Shanghai Municipal Human Resources and Social Security Bureau (0403 N14001), Shanghai Charity cancer research center (0703 N14012), and ShanghaiScience and Technology Commission (14140904000).
Funding
National Natural Science Foundation of China (81202115). Doctoral Innovation Fund of Shanghai Jiaotong University School of Medicine (number BXJ201732). Shanghai Municipal Human Resources and Social Security Bureau (0403 N14001), Shanghai Charity cancer research center (0703 N14012). ShanghaiScience and Technology Commission (14140904000).
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
YZ conceptualized the review, performed the literature search, and drafted most of the manuscript. GW, RC, YH, and ZC revised the article. ZC directed the review to be more focused and gave final approval for the article to be published. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Authors’ information
Ying Zheng and Ruiling Chen are undergraduates of Shanghai Jiao Tong University School of Medicine. Gangyang Wang is PhD of Shanghai Bone Tumor Institution. Yingqi Hua and Zhengdong Cai are professors of the research group.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zheng, Y., Wang, G., Chen, R. et al. Mesenchymal stem cells in the osteosarcoma microenvironment: their biological properties, influence on tumor growth, and therapeutic implications. Stem Cell Res Ther 9, 22 (2018). https://doi.org/10.1186/s13287-018-0780-x
Published:
DOI: https://doi.org/10.1186/s13287-018-0780-x