
Abstract
Background
Say–Barber–Biesecker–Young–Simpson (SBBYS) (OMIM #603736, Ohdo syndrome variant) is a rare type of severe blepharophimosis intellectual disability syndrome, which is generally characterized by a global developmental delay, distinctive facial features, and intellectual disability with multiple congenital anomalies, including skeletal involvement, missing, or underdeveloped kneecaps, and genital anomalies, in affected males. It has been shown that mutations in the KAT6B gene, which is a lysine acetyltransferase-encoding gene, have been associated with SBBYS syndrome. All the known variants are dominant de novo mutations that result in protein truncation.
Case presentation
A 14-year-old Iranian Azeri boy with an intellectual disability, distinct dysmorphic facial features such as open-mouth expression, sparse medial eyebrows, widely spaced upward-slanted eyes, epicanthal folds, broad nasal bridge, low-set ears, anteverted ears, short philtrum, hypertelorism, microphthalmia is presented in this case study. Cryptorchidism was reported. Neurologically, the patient presented with poor eye contact, hypotonia, and speech difficulties. In the skeletal X-ray, underdeveloped kneecaps with some new features were observed.
Conclusion
We present the first case of SBBYS syndrome in association with some new anomaly features in the Iranian population. Based on this diagnosis, we could provide the patient with a suitable plan of management as well as appropriate genetic counseling for his family.
Similar content being viewed by others


Introduction
Heterozygous de novo mutations in the KAT6B gene, which encodes the histone acetyltransferase, can cause two syndromes with mental retardation and congenital abnormalities, known as Genitopatellar syndrome (GPS; OMIM #606170) and Say–Barber–Biesecker–Young–Simpson syndrome (SBBYSS, OMIM #603736) [1]. The GPS is a severe disease characterized by aplasia, renal anomalies, external genital abnormalities, flexion deformities of the extremities, facial dysmorphism, patellar hypoplasia, microcephaly, agenesis of the corpus callosum (ACC), and severe psychomotor retardation [2,3,4]. On the other hand, the SBBYSS (a variant of Ohdo syndrome) is a crippling disorder, which is diagnosed based on the presence of distinct facial dysmorphic characteristics, including mask-like face, ptosis, and blepharophimosis. In addition, hypotonia, big toes and long thumbs, dental malformations, feeding problems, hypoplastic or dislocated patellae, hearing impairment, cryptorchidism, congenital heart diseases, severe intellectual impairment, and developmental delay may also be observed in patients with SBBYSS [5, 6]. The majority of the patients reported with SBBYSS were sporadic, but at least three generations affected have been reported in a family [7].
In 1986, SBBYSS was reported by Ohdo et al. [8] and then later described in detail as a variant of Ohdo's syndrome by Say and Barber [9], Young and Simpson [10], and Biesecker [11]. SBBYSS is a rare genetic disorder, which has been reported in less than 20 cases in the literature [12]. It is caused by different types of mutations in the KAT6B gene [MIM 605880] on chromosomal region 10q22, which encodes a highly conserved lysine acetyltransferase 6B, a member of the MYST (SAS/MOZ) family [13], which plays a remarkable role in chromatin modification, leading to the SBBYSS features and phenotypes.
Because of clinical overlap between GPS and SBBYSS syndromes, the KAT6B-related disease spectrum is a subject of interest to researchers [14]. Herein, we are going to report a 14-year-old developmental delay boy with cognitive impairment, significant facial dysmorphisms, undescended testes and bilateral hypoplasis of patellae as a new case of SBBYSS in Iranian population, which is caused by de novo mutation in KAT6B gene.
Case presentation
Evaluation of clinical features in the proband
The proband is an Azeri boy of 14 years old from a healthy and non-consanguineous couple. Intrauterine growth retardation was identified in the pregnancy of the proband. He was born at 36 weeks by cesarean section due to no fetal movement, with Apgar scores 9/10. At birth, he was 2.97 kg (50th percentile, − 0.45 SD) and his cranial circumference was 33 cm (10th percentile, − 0.61 SD). The subject began to sit at four years of age and made his first unaided steps when he was six. He could take some steps alone and walk with an increased lift base. At nine years of age, he was referred to a medical genetic counseling center and diagnosed with blepharophimosis syndrome because of blepharophimosis and intellectual impairment. He showed a few episodes of loss of consciousness at six years old, but his electroencephalogram (EEG) was normal. He presented a non-symptomatic large atrial septal defect, ASD (ostium secundum atrial septal defects).
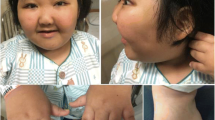
On physical examination at 9 years old, the patient occipitofrontal circumference (OFC) was 52cm (~ 50th centile) but had dysmorphic facial features including flat occiput, triangular long face, frontal bossing, protruding tongue that caused an open mouth expression, a thin upper lip, sparse medial eyebrows, thin eyebrow, epicanthal folds, widely spaced upward-slanted eyes, broad nasal bridge, low-set ears, anteverted ears, short philtrum, hypertelorism, ptosis, microphthalmia, hypoplastic teeth. Congenital urogenital anomaly as cryptorchidism without any renal anomalies was diagnosed. The patient was tall stature and had first (big) toe/long thumbs with patella missing but the most significant abnormal findings in skeletal and joint X-ray, which have not been reported so far, included symmetric bilateral coxa valga, hypoplastic iliac wings and scoliosis, increased diameter of the lumbar vertebral canal, genu valgum, pes planus and sloping forehead with a large brain capacity (Fig. 1A).

A Phenotypic characteristics of the patient; B Brain MRI of the patient. Imaging of the brain shows inappropriate myelination and disturbed white matter integrity; C Genetic test results. Sanger sequencing reveals a c.3147G>A; p.P1049P de novo heterogeneous mutation in this patient
From the neurological point of view, he presented with global developmental delay, intellectual disability, poor eye contact, hypotonia and speech difficulties. Brain MRI (magnetic resonance imaging) showed no evidence of corpus callosum agenesis, however inappropriate myelination and disturbed white matter integrity was the significant findings (Fig. 1B). There was no sphincter control. Now, the patient is taken care of mental disability care institute on physiotherapy and speech therapy, due to inability of parents to take care and behavioral disturbances and ADHD (attention deficit-hyperactivity disorder) traits, respectively. In Table 1, we compared the clinical presentations of the present patient with that of some patients in previous reports.
DNA extraction and quality control
In the present study, written informed consent was obtained from the parents of a 14-year-old male with SBBYS syndrome; then, he was included in the study. The blood sample was collected, and DNA was extracted by standard salting-out method. Finally, the integrity and purity of DNA were analyzed by agarose gel electrophoresis and Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), respectively [15].
Whole exome sequencing and data analysis
After extraction of Genomic DNA from the proband, it was processed for whole-exome sequencing (WES) using the SureSelect Human All ExonV6 kit. The DNA was fragmented, and blunt ends were created. Adapter oligonucleotides were ligated to DNA fragments, followed by enrichment via PCR. Purification was performed using the AMPure XP system, and quantification was done using the Agilent Bioanalyzer 2100. The captured library was sequenced on a NovaSeq 6000 Illumina sequencer with an average coverage of 100X. Data quality was assessed using FastQC and NGS QC toolkits. High-quality reads were aligned to the human reference genome (GRCh37.p13/hg19) using the Burrows-Wheeler Alignment tool. Picard tools were employed to mark and remove duplicated reads, and the SAM file was converted to BAM format. Base quality score recalibration was performed using GATK. Variants were filtered based on criteria such as minor allele coverage ≥ 10, variants with coverage ≥ 15, and call quality ≥ 20. ANNOVAR was used for genetic variant annotation. Annotated variants were further filtered and selected for analysis.
This process enabled the identification and analysis of genetic variants in the proband's genomic DNA sample [16].
Functional analysis of the detected mutation
The pathogenicity of the identified variant was assessed through in silico analysis using web server Mutation Taster. Prediction of the structure of wild-type and mutant protein was done by SWISS-MODEL, the SWISS-MODEL web server (https://swissmodel.expasy.org/). To predict whether the identified variant would escape NMD, the nonsense-mediated mRNA decay (NMD) ESC predictor was used (https://nmdprediction.shinyapps.io/nmdescpredictor/). Also, the STRING database was used to predict interactions among proteins.
Sanger sequencing for validation of variants
After WES, Sanger sequencing was used to validate the variant through specific primers. According to the reference genomic sequences from the Human Genome from GenBank in NCBI, primer pairs were designed for the candidate loci (Fig. 1C). We identified a de novo heterozygous mutation of guanine to adenine NM_012330.4(KAT6B):c.3147G>A (p.Pro1049 =) using WES. Sanger sequencing subsequently confirmed this heterozygous variant identified by WES in this individual. The mutation could not be detected in the parental DNA by Sanger sequencing (Fig. 1C), suggesting its de novo occurrence. The results of sequence analysis revealed that the c.3147G>A; p.P1049P transition at codon 1,049 was a synonymous mutation (p.Pro1049 =) in the exon 16 of the KAT6B gene.
In addition, KAT6B protein is a 231,378 Da protein with 2,073 aa. Functional annotation of KAT6B by DAVID displayed that this protein expresses in different human tissues. Considerably, it has high quantities in the immune cells, uterus, thymus, fetal brain, and other tissues according to the (http://biogps.org). Also, KAT6B protein has been identified in different diseases such as genitopatellar syndrome, ohdo syndrome, Sbbyss variant, monocytic leukemia, and blepharophimosis. Furthermore, the NMD web server represented this premature codon likely led to nonsense-mediated degradation (Fig. 2A). According to the InterProScan as a tool for the analysis of protein domain, KAT6B encodes a histone acetyltransferase protein with the N-terminus which is involved in transcriptional activation and the C-terminus, which play a role in transcriptional repression.

A The result of nonsense-mediated mRNA decay prediction in the patient by the NMD Esc Predictor Web Site. B Protein–protein interaction network of KAT6B gene. C Prediction of the effect of p.P1049P mutation on the protein structure and chains using Swiss model
The analysis of interaction among proteins by STRING indicated that KAT6B protein creates a network with about some different gene products (Fig. 2B). Also, this protein plays an important role in some biological pathways through three important activities, including histone acetylation, nucleosome assembly, and regulation of transcription (https://www.ebi.ac.uk/QuickGO/GTerm). The SWISS-MODEL server displayed this mutation resulted in a truncated protein (Fig. 2C).
Discussion
Say–Barber–Biesecker–Young–Simpson (SBBYS) is a rare genetic abnormality due to the mutations in the KAT6B gene. The KAT6B encodes a histone-acetyl transferase, which is a highly conserved part of the MOZ/MORF complex. In addition to the activity of acetyltransferase, C-terminal and N-terminal ends of the encoded protein have transcriptional repression and transcriptional activation activity, respectively. This protein is essential for RUNX2-dependent transcriptional regulation and can be involved in the growth of the brain. Because of the clinical overlapping and similarities, some researchers proposed that KAT6B causes two allelic GPS and SBBYS syndromes, whereas others have suggested them as two different disorders [14, 17]. Although SBBYSS and GPS have been described as two distinct genetic disorders, they have some overlapping phenotypes, including genital abnormalities, hearing loss, global developmental delay, hypotonia, congenital heart defects, intellectual disability, and thyroid anomalies [17]. Lemire et al. [18] suggested some features might raise for KAT6B-related anomalies. Patients with two major phenotypes or one major phenotype and two minor phenotypes presumably have a KAT6B-related disorder. The main phenotypes of SBBYSS are described by mask-like facial appearance, long thumbs/great toes, blepharophimosis, dacryostenosis, and ptosis. However, the distinct features of GPS are characterized by agenesis of the corpus callosum, club feet, patellar hypoplasia/agenesis, and flexion contractures of the hips and knees [19]. Nevertheless, it is noticeable that Mendez et al. [20] reported a patient with long thumbs/halluces and dysmorphic face as the major features, suggesting the identification of SBBYSS, while he had a corpus callosum agenesis, which is a major phenotype of GPS. Also, Niida Yo et al. [21] reported a case with KAT6B mutation that was formerly reported in SBBYSS cases, but he also had some features, including genital anomalies, laryngomalacia, and developmental delay that confirmed GPS. Our patient has some distinct major features, including dysmorphic facial and blepharophimosis with big toes and long thumbs, suggesting the SBBYSS. In addition, we identified some new skeletal abnormality features and phenotypes, including symmetric bilateral coxa valga, hypoplastic iliac wings and scoliosis, and increased diameter of the lumbar vertebral canal, genu valgum, and pes planus that had not been reported in previous cases to date altogether. It is noticeable that KAT6B-related disease spectrum, especially GPS and SBBYSS have clinical similarities and overlapping in their phenotypes, and making a clear genotype–phenotype correlation is very challenging, if not possible. Although an appropriate genotype–phenotype correlation is required for accurate diagnosis, a clear correlation has not yet been defined. We hope that these new phenotypes and features help make appropriate and credible genotype–phenotype correlations to identify KAT6B-related disorders.
Furthermore, our results are comparable with two different studies. The mutation has been previously reported as a pathogenic variant by Gannon et al. and Yilmaz et al. in six patients with SBBYSS [1, 22]. By using Alamut 2.2 prediction program, Gannon et al. proposed that c.3147G>A; p.P1049P mutation could lead to a cryptic splice acceptor site, which could be followed by protein truncation. They suggested that this mutation could be disease-causing; however, no experimental evidence was available in support of aberrant splicing. Clinically, they indicated that all three patients with that splice site variant were fitted better with SBBYSS [22]. In another study, to experimentally evaluate this aberrant splice site change, Yilmaz et al. extracted total RNA from blood sample of patient and their healthy parents. Then, reverse transcriptase PCR (RT-PCR) was applied with specific primers for amplifying the part of exons 15 and 16 of the KAT6B gene, followed by sequencing analyses of PCR products. They showed that aberrant splicing of KAT6B could result in an out-of-frame deletion of 127 bp in exon 16, which was followed by a premature stop codon (p.Ala1008Argfs 62) [1].
Our genetic findings confirm the diagnosis of SBBYSS in the 14-year-old boy due to a known de novo heterozygous mutation in KAT6B gene. Since already seven unrelated individuals reported c.3147G>A; p. p.P1049P is actually the most common KAT6B mutation, which is detected in individuals with SBBYSS. In addition, our results are in line with previous studies that suggested that the position of the mutations in KAT6B is associated with the phenotypes of the patients. Previous studies indicated that most mutations in KAT6B were in exon 18, which is the last exon of KAT6B and encodes the highly conserved C-terminal acidic and transcriptional activation domains [7, 17]. Gannon et al. [22] suggested that while GPS mutations occurred more proximally in the exon 18, losing both acidic and transcriptional activation domains led to gain-of-function mutation and affected the critical binding sites of the protein. SBBYSS mutations happened more distally in the same exon, lacking the transcriptional activation domain and preserving the acidic domain that led to loss-of-function mutation. Also, it is noticeable that recent studies reported mutations more proximal in exons 15, 16, and 17 with the typical phenotypes of SBBYSS [1]. As a result, screening of exon 16 along with exons 15, 17, and 18 have to be tested in targeted mutation detection methods for diagnosis of the KAT6B-related spectrum abnormalities due to its recurrence [23].
Conclusion
In the current case study, a de novo heterozygous mutation was detected in the KAT6B gene in an Iranian male manifesting typical SBBYS symptoms. The c.3147G > A; p.P1049P transition at codon 1,049 was a synonymous mutation, which resulted in a cryptic splice acceptor site. Our patient is the first case report due to KAT6B alteration in Iranian patients. Records of further evidence and patients with GPS and SBBYSS phenotypes can support accurate identification and describe genotype–phenotype correlation.
Data availability
Input data for the analyses are available from the corresponding authors on request.
References
Yilmaz R, Beleza-Meireles A, Price S, Oliveira R, Kubisch C, Clayton-Smith J, et al. A recurrent synonymous KAT6B mutation causes Say-Barber-Biesecker/Young-Simpson syndrome by inducing aberrant splicing. Am J Med Genet A. 2015;167(12):3006–10.
Cormier-Daire V, Chauvet M-L, Lyonnet S, Briard M-L, Munnich A, Le Merrer M. Genitopatellar syndrome: a new condition comprising absent patellae, scrotal hypoplasia, renal anomalies, facial dysmorphism, and mental retardation. J Med Genet. 2000;37(7):520–4.
Reardon W. Genitopatellar syndrome: a recognizable phenotype. Am J Med Genet. 2002;111(3):313–5.
Armstrong L, Clarke J. Report of a new case of “genitopatellar” syndrome which challenges the importance of absent patellae as a defining feature. J Med Genet. 2002;39(12):933–4.
Verloes A, Bremond-Gignac D, Isidor B, David A, Baumann C, Leroy MA, et al. Blepharophimosis-mental retardation (BMR) syndromes: a proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am J Med Genet A. 2006;140(12):1285–96.
Campeau PM, Lu JT, Dawson BC, Fokkema IF, Robertson SP, Gibbs RA, et al. The KAT6B-related disorders genitopatellar syndrome and Ohdo/SBBYS syndrome have distinct clinical features reflecting distinct molecular mechanisms. Hum Mutat. 2012;33(11):1520–5.
Kim YR, Park JB, Lee YJ, Hong MJ, Kim HT, Kim HJ. Identifying the KAT6B mutation via diagnostic exome sequencing to diagnose Say–Barber–Biesecker–Young–Simpson syndrome in three generations of a family. Ann Rehabil Med. 2017;41(3):505.
Ohdo S, Madokoro H, Sonoda T, Hayakawa K. Mental retardation associated with congenital heart disease, blepharophimosis, blepharoptosis, and hypoplastic teeth. J Med Genet. 1986;23(3):242–4.
Say B, Barber N. Mental retardation with blepharophimosis. J Med Genet. 1987;24(8):511.
Young I, Simpson K. Unknown syndrome: abnormal facies, congenital heart defects, hypothyroidism, and severe retardation. J Med Genet. 1987;24(11):715–6.
Biesecker LG. The Ohdo blepharophimosis syndrome: a third case. J Med Genet. 1991;28(2):131–4.
Mundama M, Ayong S, Rossillon R. Management of patella dislocation in Say–Barber–Biesecker–Young–Simpson’s Syndrome: a report of two cases. Case Rep Orthop. 2018;2018:6415713.
Campeau PM, Kim JC, Lu JT, Schwartzentruber JA, Abdul-Rahman OA, Schlaubitz S, et al. Mutations in KAT6B, encoding a histone acetyltransferase, cause Genitopatellar syndrome. Am J Human Genet. 2012;90(2):282–9.
Radvanszky J, Hyblova M, Durovcikova D, Hikkelova M, Fiedler E, Kadasi L, et al. Complex phenotypes blur conventional borders between Say–Barber–Biesecker–Young–Simpson syndrome and genitopatellar syndrome. Clin Genet. 2017;91(2):339–43.
AbbaspourRodbaneh E, Panahi M, Rahimi B, Mokabber H, Farajollahi R, Davarnia B. GJB2 mutations in Iranian Azeri population with autosomal recessive nonsyndromic hearing loss (ARNSHL): first report of c. 238 C>A mutation in Iran. J Clin Lab Anal. 2021;35(11):e24024.
Nasirshalal M, Panahi M, Javanshir N, Salmani H. Identification of a novel heterozygous mutation in the MITF gene in an Iranian family with Waardenburg syndrome type II using next-generation sequencing. J Clin Lab Anal. 2021;35(6): e23792.
Vlckova M, Simandlova M, Zimmermann P, Stranecky V, Hartmannova H, Hodanova K, et al. A patient showing features of both SBBYSS and GPS supports the concept of a KAT6B-related disease spectrum, with mutations in mid-exon 18 possibly leading to combined phenotypes. Eur J Med Genet. 2015;58(10):550–5.
Lemire G, Campeau PM, Lee BH. KAT6B disorders. 2020.
Shin JH, Lim HH, Gang MH, Kim SY, Yang S-s, Chang M-y. A neonate with Say–Barber–Biesecker–Young–Simpson syndrome with a novel pathogenic mutation in KAT6B gene: a case report. J Genet Med. 2021;18(2):147–51.
Mendez R, Delea M, Dain L, Rittler M. A novel pathogenic frameshift variant of KAT6B identified by clinical exome sequencing in a newborn with the Say–Barber–Biesecker–Young–Simpson syndrome. Clin Dysmorphol. 2020;29(1):42–5.
Niida Y, Mitani Y, Kuroda M, Yokoi A, Nakagawa H, Kato A. A Say–Barber–Biesecker–Young–Simpson variant of Ohdo syndrome with a KAT6B 10-base pair palindromic duplication: a recurrent mutation causing a severe phenotype mixed with genitopatellar syndrome. Congenit Anom. 2017;57(3):86–8.
Gannon T, Perveen R, Schlecht H, Ramsden S, Anderson B, Kerr B, et al. Further delineation of the KAT6B molecular and phenotypic spectrum. Eur J Hum Genet. 2015;23(9):1165–70.
Clayton-Smith J, O’Sullivan J, Daly S, Bhaskar S, Day R, Anderson B, et al. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am J Hum Genet. 2011;89(5):675–81.
Marangi G, Di Giacomo MC, Lattante S, Orteschi D, Patrizi S, Doronzio PN, et al. A novel truncating variant within exon 7 of KAT6B associated with features of both Say-Barber-Bieseker-Young-Simpson syndrome and genitopatellar syndrome: further evidence of a continuum in the clinical spectrum of KAT6B-related disorders. Am J Med Genet Part A. 2018;176(2):455–9.
Sun X, Luo X, Lin L, Wang S, Wang C, Yuan F, et al. Clinical features and underlying mechanisms of KAT6B disease in a Chinese boy. Mol Genet Genom Med. 2023;2023:e2202.
Acknowledgements
We thank the patient’s family for their participation in this study.
Funding
The authors are thankful to Behzad Davarnia and the MyGene genetic diagnostic laboratory for supporting this project.
Author information
Authors and Affiliations
Contributions
BD, MP and BR contributed to WES filtering, bioinformatics analysis, data interpretation regarding whole-exome sequencing. EAB also have great contribution in WES analysis. HA and RF contributed effectively to the plan regarding patient diagnosis and diagnostic imaging procedures. FJ is an important role in the preparation of the first format of MS.These authors also contributed to this work equally and wrote the manuscript. BD supervised, reviewed, and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Ethics Committee of Ardabil University of Medical Sciences approved the study.
Consent for publication
Written informed consent was obtained from the patient’s legal guardian(s) for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interest
The authors declare no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Davarnia, B., Panahi, M., Rahimi, B. et al. De novo KAT6B mutation causes Say–Barber–Biesecker–Young–Simpson variant of Ohdo syndrome in an Iranian boy: a case report. J Med Case Reports 18, 4 (2024). https://doi.org/10.1186/s13256-023-04237-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-023-04237-w