
Abstract
Background
Germline pathogenic variants in the cadherin-1 (CDH1) gene cause a predisposition to hereditary diffuse gastric cancer (HDGC). We report an HDGC case in Vietnam and identify a novel mutation in the CDH1 gene.
Case presentation
A 28-year-old Vietnamese man was diagnosed with HDGC and a novel mutation at c.639G>A. All exons of CDH1 were sequenced in his pedigree, which revealed the c.639G>A mutation in the proband, his father, and uncle. The patient refused treatment and died 4 months after diagnosis. Endoscopic surveillance of the father and the uncle showed structural abnormalities in the father.
Conclusion
In cases of HDGC, identification of the CDH1 gene mutation is very important for better counseling and more effective strategies to prevent the development of diseases, such as prophylactic gastrectomy for family members with genetic mutations.
Similar content being viewed by others

Background
Hereditary diffuse gastric cancer (HDGC) is a diffuse-type autosomal dominant familial syndrome caused by a germline mutation in the cadherin-1 gene (CDH1) [1]. The CDH1 gene is located on chromosome 16q22.1 and is composed of 16 exons that transcribe the E-cadherin protein [2]. The syndrome is associated with a lifetime risk for mutation carriers of 70% in men and 56% for women, and a 42% risk for lobular breast cancer (LBC) in women [3]. Patients with gastric cancer and germline CDH1 mutations have a lower survival rate than patients who meet the HDGC criteria but do not have the CDH1 mutations [4]. Early detection of HDGC is also challenging because early-stage signet ring cell carcinoma is difficult to visualize using gastroduodenoscopy with random biopsies [5]. Therefore, the follow-up and management of carriers of a pathogenic germline CDH1 mutation, especially in the family proband, is necessary to prevent their death from invasive carcinoma [6]. To date, there has been no report of CDH1 or its mutation in Vietnamese patients. In this report, we present a novel c.639G>A (p.W213*) mutation in a Vietnamese man with HDGC.
Case presentation
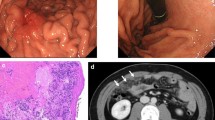
In September 2016, a 28-year-old Vietnamese male patient visited the National Geriatric Hospital in Hanoi due to epigastric pain, weight loss, and painful swollen lymph nodes in his neck for the past 5 months. He had no history of gastric disease. The subsequent workup included a gastroscopy that revealed an ulcer in the body of the stomach measuring more than 3 cm in diameter. The biopsy from gastroscopy was assessed for Helicobacter pylori by the Campylobacter-like organism (CLO) test and histopathological examination. Histopathology results for the ulcer were negative for H. pylori and revealed stage IV diffuse-type gastric cancer (DGC), characterized by signet ring cells. Unfortunately, for financial reasons, the patient refused additional diagnosis including neck lymph node biopsy, computed tomography (CT) scan, and positron emission tomography (PET)/CT to evaluate distant metastasis, and also refused to follow up on further treatments. The patient's health deteriorated, and he died 4 months post-diagnosis.
The patient’s family history did not show DGC. However, because of the clinicopathological presentation including early onset, severe clinical symptoms, and histopathological test results showing diffuse-type characteristics, we performed genetic screening for mutations in the CDH1 gene by E-cadherin protein expression.
With the patient’s consent, we used the Exgene™ Blood SV kit (GeneAll Biotechnology, Korea) to extract DNA from the whole blood sample according to the instruction manual. This was followed by polymerase chain reaction (PCR) amplification and DNA sequencing using both forward and reverse primers for all exons and introns of the CDH1 gene. The primers were designed using National Center for Biotechnology Information (NCBI) Primer-BLAST [Basic Local Alignment Search Tool] and the QIAGEN CLC Main Workbench (version 8.1.2.) and were reanalyzed using the OligoAnalyzer tool. After the polymerase chain reaction (PCR), we separated the DNA by size using gel electrophoresis (1.5% agarose gel). Finally, we performed gel analysis using the UVP GelDoc-It2 Imaging System (Fisher Scientific). The determination of DNA sequences for all samples was done on Applied Biosystems ABI 3500 (JP0 and ABI 3730xl; Thermo Fisher) deoxyribonuclease acid (DNA) analyzers, followed by gene analysis using BioEdit (version 2.0) and ApE (version 1.0) software.
We found a novel heterozygous nonsense mutation that resulted in a premature termination codon (PTC) UGA at p.213 in exon 5 (CDH1 c.639G>A, p.W213) (Fig. 1). The mutation shortened the CDH1 gene length from 882 aa to 213 aa. The immunohistochemical staining of the patient’s tissue sample showed decreased expression of E-cadherin (Fig. 2).

Analysis of exon 5 deoxyribonucleic acid sequencing; c.639G>A, p.W213* mutation was located. Patient B4 was the proband

Histopathology staining from the biopsy of the patient's stomach through gastroscopy. a Hematoxylin and eosin (H&E) staining with poorly cohesive carcinoma, characterized by signet ring cells. The distribution of cells is diffuse; they can form small groups of bands of gland-like structures with a conspicuous desmoplastic reaction of the stroma. b Immunohistochemical staining showed decreased E-cadherin expression. Positive staining is shown in brown. Representative images of E-cadherin staining demonstrated lower expression of E-cadherin
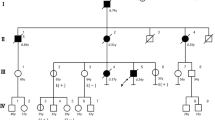
We collected blood samples from the patient’s family members (11 participants) for mutation screening in the CDH1 gene exon 5. The participants included three first cousins once removed (III-1, III-2, III-3), two uncles (III-4, III-5), the father (III-6), the mother (III-7), a male first cousin (IV-1), a female first cousin (IV-2), an older sister (IV-3), and a younger brother (IV-7) (Fig. 3). At the time of testing, no participants showed any symptoms of HDGC. Upon analysis of the pedigree, we found the patient’s parents to be healthy, and there was no family history of the disease. The DNA sequencing in CDH1 exon 5 showed a similar mutation (c.639G>A, p.W213*) in two members of the family: the father (III-6, 59 years old) and an uncle (III-4, 65 years old), with both heterozygous. Gastroscopy performed on the patient’s father revealed inflammation and antral gastritis. Six-core biopsy specimens showed chronic gastritis, intestinal metaplasia, and low-grade dysplasia. High iron diamine staining for metaplasia typing was negative.

Patient’s pedigree showing two members of the family carrying the cadherin-1 c.639G>A mutation
Discussion and conclusions
The reported patient was diagnosed with HDGC stage IV at a young age, characterized by severe clinical manifestations, which matched the latest guideline for diagnosis of HDGC [7]. The novel c.639G>A nonsense mutation reduced the expression of the E-cadherin protein due to the premature stop codon in the coding region of exon 5. Enrique and colleagues reported a heterozygous mutation c.1531C>T (p.Gln511*) which caused a PTC in a 22-year-old male patient with HDGC stage IV. Similar to the present case, the patient’s family history showed no sign of HDGC, and he died shortly after the diagnosis [8]. Another novel CDH1 mutation (c.1612delG), discovered by Caggiari and colleagues in 2017, also resulted in a PTC at codon 556 (p.Asp538Thrfs*19) in the extracellular fourth repeat region of a 41-year-old man with HDGC. In this case, the disease progressed rapidly to peritoneal, pancreatic, and hepatic metastasis despite 3 months of targeted therapy [9]. Both studies demonstrated that CDH1 nonsense mutations are associated with more severe clinical presentation and poor prognosis in patients with early-onset HDGC.
While none of the patient’s relatives had HDGC, we found the same CDH1 c.639G>A mutation in the tissue of the patient’s father and uncle, who were both elderly. The results from six-core biopsy and gastroscopy of the father showed the absence of cancer cells. The mechanism behind the formation of signet ring cell carcinoma in carriers of the c.639G>A mutation is unclear; however, we speculate that environmental factors may play a role in the process.
In recent years, HDGC incidence has been increasing in Asia, the United States, and Europe, despite being a rare inherited condition [10]. The rapid disease progression, difficulty in early diagnosis, and highly metastatic nature contribute to the poor prognosis, especially in patients with early-onset disease. A study by van der Post et al. in 2015 showed a lower survival rate for gastric cancer patients with a CDH1 mutation compared to non-carriers, with 5-year survival rates of 4% and 13%, respectively [4]. There are several problems regarding the diagnosis of gastric cancer such as difficulty in detecting cancer tissue in the gastric mucosa or even after biopsy. This indicates the need for total surveillance of individuals carrying a CDH1 mutation, especially those with a family history of gastric cancer. This includes periodic checkups or prophylactic total gastrectomy (PTG), which is recommended for CDH1 mutation carriers. In a systematic review of the histopathological diagnosis of gastric cancer, 87% of cases were detected from prophylactic gastrectomy [8, 11]. However, postoperative complications following PTG such as diarrhea, fatigue, acid reflux, and internal bleeding should be anticipated. Furthermore, considering the uniqueness of the healthcare system and the mindset of Vietnamese people, providing explanations and counseling to CDH1 carriers regarding PTG has proven to be challenging, mainly because of the absence of disease and/or symptoms. Thus, further research is needed to fully elucidate the effects of PTG in people with CDH1 mutations and those with family members having HDGC.
The limitation of the study was that the patient did not attend treatment after being diagnosed with HDGC. Therefore, we did not have the opportunity to evaluate and classify the disease stages according to the TNM Classification of Malignant Tumors and could not evaluate the response to therapy. As a result, we were unable to thoroughly analyze the relationship between the patient's clinical, subclinical, and genetic changes.
Case study research in Vietnam concerning CDH1 mutations in stomach cancer is rare, and there is a lack of emphasis on genetic screening in HDGC diagnosis. Early identification of CDH1 mutations in HDGC patients and their family members is a significant factor enabling better counseling and preventive strategies such as PTG against the disease in mutation carriers. More importantly, it provides recommendations for the development of disease prevention strategies and formulation of more specific governmental health policies to support the treatment of Vietnamese patients in extraordinary circumstances in the future.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- HDGC:
-
Hereditary diffuse gastric cancer
- DGC:
-
Diffuse-type gastric cancer
- CDH1:
-
Cadherin-1
- PTG:
-
Prophylactic total gastrectomy
- PTC:
-
Premature termination codon
References
Black MD, et al. Hereditary diffuse gastric cancer associated with E-cadherin germline mutation: a case report. Hawaii J Med Public Health. 2014;73(7):204–7.
Shenoy S. CDH1 (E-Cadherin) mutation and gastric cancer: genetics, molecular mechanisms and guidelines for management. Cancer Manag Res. 2019;11:10477–86.
Hansford S, et al. Hereditary diffuse gastric cancer syndrome: CDH1 mutations and beyond. JAMA Oncol. 2015;1(1):23–32.
van der Post RS, et al. Accuracy of hereditary diffuse gastric cancer testing criteria and outcomes in patients with a germline mutation in CDH1. Gastroenterology. 2015;149(4):897-906.e19.
Fujita H, et al. Endoscopic surveillance of patients with hereditary diffuse gastric cancer: biopsy recommendations after topographic distribution of cancer foci in a series of 10 CDH1-mutated gastrectomies. Am J SurgPathol. 2012;36(11):1709–17.
van der Post RS, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet. 2015;52(6):361–74.
Fitzgerald RC, et al. Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. J Med Genet. 2010;47(7):436–44.
Norero E, et al. Identification of c.1531C>T pathogenic variant in the CDH1 gene as a novel germline mutation of hereditary diffuse gastric cancer. Int J MolSci. 2019;20(20):4980.
Caggiari L, et al. A new mutation of the CDH1 gene in a patient with an aggressive signet-ring cell carcinoma of the stomach. Cancer BiolTher. 2018;19(4):254–9.
Taghavi S, et al. Prognostic significance of signet ring gastric cancer. J ClinOncol. 2012;30(28):3493–8.
Seevaratnam R, et al. A systematic review of the indications for genetic testing and prophylactic gastrectomy among patients with hereditary diffuse gastric cancer. Gastric Cancer. 2012;15(Suppl 1):S153–63.
Acknowledgements
We thank the patients and family for the kind cooperation and consent for assessment and publishing of the data. We also thank our colleagues at the National Geriatric Hospital in Hanoi who helped us with diagnosis and clinical data evaluations.
Funding
This research is funded by the Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 106-YS.02-2015.37.
Author information
Authors and Affiliations
Contributions
DD: Contributed ideas for research, coordinated all research activities. VT: Contributed to writing the manuscript. HN: Selected participants and collected samples, interviewed, gene analysis. AV: Selected participants and collected samples, interviewed, gene analysis. HN: Selected and collected the patient’s tissue, contributed in reading histopathology results, H&E and immunohistochemical staining. HD: Prepared, formatted, and contributed to writing the manuscript. BT: Performed biopsy, gastroscopy, and H. pylori test and evaluated test results. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethical Committee of Hanoi Medical University. The study only proceeded with the agreement of participation by oral consent.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dang, D.N.T., Nguyen, H.T.T., Ngo, H.D. et al. A novel stop codon mutation in exon 5 (c.639G>A) of the cadherin-1 gene in a Vietnamese man with hereditary diffuse gastric cancer: a case report. J Med Case Reports 15, 263 (2021). https://doi.org/10.1186/s13256-021-02837-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-021-02837-y