
Abstract
Background
It is not uncommon that an infant with a disease of unknown etiology is presented to a physician. Facial dysmorphic features lead to a different diagnosis. It is a challenge to link the presentation to the newfound diagnosis.
Case presentation
A 37-day-old Yemenite Jewish girl was presented to our institution with a clinical picture of pseudohypoaldosteronism due to abnormal facial features and a psychomotor developmental delay. Further investigation led to the diagnosis of CDK13-related disorder. According to the literature, CDK13 has a key role in the cell cycle, but no interference with the aldosterone signaling pathway or electrolyte balance was described. No mutations in the previously described gene NR3C2 (cytogenetic location 4q31.23), encoding the mineralocorticoid receptor, were found. Although the clinical presentation corresponded to pseudohypoaldosteronism type 1, we could not genetically confirm this.
Conclusions
Probably pseudohypoaldosteronism was a coincidental finding in this girl with a CDK13 mutation, but because only limited information is known about CDK13-related disorders, further investigation could be more informative to clarify this presentation.
Similar content being viewed by others

Introduction
Water balance and potassium homeostasis are critically regulated by the aldosterone hormone. Aldosterone binds to the mineralocorticoid receptor, which is expressed in the distal tubules of the kidney, the salivary and sweat glands, the intestinal epithelium, and in the airway epithelium in the lung and heart. Aldosterone binding to the mineralocorticoid receptor results in increased expression of the epithelial sodium channel (ENaC) and the Na+-K+-ATPase in principal cells of the distal tubule in nephrons, and potassium accumulated in principal cells is secreted into the urine by the renal outer medullary potassium channel. Aldosterone also mediates proton secretion into the urine by induction of H+-K+-ATPase in intercalated cells, leading to a net sodium-water reabsorption and potassium and proton secretion [1,2,3]. Pseudohypoaldosteronism (PHA) is a heterogeneous disorder characterized by salt wasting resulting from target organ unresponsiveness to mineralocorticoids. Because the principal effect of aldosterone is to promote potassium and hydrogen secretion, the condition typically presents in the neonatal period with hyperkalemia, metabolic acidosis, and an elevated plasma aldosterone concentration because of loss of negative feedback. We present a case of an infant with the classic presentation of PHA but clinical features that raised suspicion of other problems.
Case presentation
A 37-day-old Yemenite Jewish girl was seen in the emergency department of our institution for ongoing restlessness and feeding refusal. The girl was born at term by uncomplicated vaginal delivery after a normal pregnancy. Besides folic acid, no other medications had been used. Her birth weight was 2.435 kg (standard deviation [SD], − 1.8); her length was 50 cm (SD, 0.3); and her head circumference was 34 cm (SD, − 1.4). The result of her physical examination was reported as normal. She was first child of healthy, nonconsanguineous Yemenite Jewish parents. Her family history was unremarkable. During the first weeks of life, she failed to gain weight, and gastroesophageal reflux disease was suspected. Empirically, Enfamil AR (Mead Johnson & Co., Chicago, IL, USA) and a proton pump inhibitor (omeprazole 10 mg twice daily) were started with moderate response.
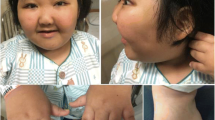
At the emergency department, the girl was presented in stable condition with vital signs appropriate for her age (pulse 128 beats/minute, blood pressure 90/50 mmHg, respiratory rate 30 breaths/minute, normal peripheral perfusion) and normal hydration status. Her weight was 3.340 kg (SD, − 2.0); her length was 52 cm (SD, − 1.4); and her head circumference was 36 cm (SD, − 1.5). Her physical examination revealed craniofacial dysmorphism (low-set ears, micrognathia, flat midface, broad nasal bridge, telecanthus, epicanthus, thin upper vermillion) and hypotonia, but the remainder of her examination was unremarkable. No skin or musculoskeletal (including skull and vertebral column) abnormalities were noted. The result of her eye examination was normal.
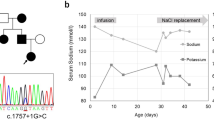
Laboratory tests (see Table 1) revealed hyperkalemic metabolic acidosis. Because the patient’s serum anion gap was normal and she had no gastrointestinal loss of bicarbonate, type 4 renal tubular acidosis was the primary working diagnosis. Her elevated ammonia can be explained by hyperkalemia causing reduction of ammonia generation in the proximal tubule leading to impaired ammonia excretion responsible for the metabolic acidosis [4]. Hyperkalemic metabolic acidosis gave rise to the suspicion of congenital adrenal hyperplasia (CAH). Further testing revealed elevated levels of renin and aldosterone but normal levels of 17-hydroxyprogesterone and 21-α-hydroxylase. The result of a synacthen test to exclude late-onset CAH was normal. Radiologic evaluation showed no abnormalities of the kidneys and the adrenal glands. When CAH was ruled out, hyperkalemic metabolic acidosis together with high levels of renin and aldosterone made the diagnosis of PHA type 1 very likely. Treatment with kayexalate was initiated with gradual normalization of the potassium levels, and it was continued five times per week until it was stopped when the girl reached the age of 2 years.
Considering the dysmorphic physical signs along with metabolic disturbances, we suspected a congenital anomaly. Chromosome analysis revealed a normal 46XX karyotype. Echocardiography and magnetic resonance imaging (MRI) of the brain (including cervical spine) revealed no abnormalities. The patient was followed closely in the pediatric nephrology and neurology clinics.
Whole-genome sequencing was requested, which revealed a missense mutation (c.2525A>G [p.Asn842Ser]) in the cyclin-dependent kinase 13 gene (CDK13) located on 7p14.1. Genetic counseling was provided, and the parents underwent genetic testing, which revealed normal CDK13 genes. Advice for prenatal diagnosis was not given, owing to the very low risk of recurrence.
Over time, the girl’s psychomotor development was clearly delayed. At the age of 8 years, she is still hypotonic. The result of further neurologic examination was normal. Her head circumference is 50 cm (SD, − 1.3); her length is 123 cm (SD, − 0.8); and her weight is 21 kg (SD, − 1.4). She has had no seizures. She has moderate mental retardation and behavior problems. Her current medications are risperidone, methylphenidate, and clonidine. She has a normal diet and eats independently, but she needs assistance with many general daily activities. She receives appropriate education. She is regularly seen by a neurologist and a psychiatrist in the outpatient clinic.
Discussion
PHA is a rare, inherited salt-wasting disorder that was first described by Cheek and Perry in 1958 as a defective renal tubular response to mineralocorticoids in infancy [5]. This clinical syndrome is caused by end-organ resistance of aldosterone, resulting in hyperkalemic metabolic acidosis with high serum aldosterone levels. PHA can be primary (hereditary) or secondary (acquired) [6].
The primary form is subclassified into type 1 and type 2. PHA type 1 can be divided into a renal form (autosomal dominant inheritance) and a systemic form (autosomal recessive inheritance). The renal form is more frequent and caused by inactivating mutations in the NR3C2 gene encoding the aldosterone receptor [7], resulting in an absence of binding of aldosterone to the receptor [8]. To date, more than 100 mutations in this gene have been reported [2, 9,10,11,12,13]. The salt loss is restricted to the kidneys and is biochemically accompanied by hyperkalemic metabolic acidosis with elevated plasma renin and aldosterone levels. Generally, the clinical expression of this condition can vary widely. It typically has a mild course followed by spontaneous remission over time [14, 15]. The systemic form is caused by mutations in the SSCNN1A, SCNN1B, and SCNN1G genes encoding for the ENaC α, β, and γ subunits, respectively, resulting in impairment in channel activity. Mutations in the α subunit are the most common and could involve the ENaC of kidneys (cortical collecting tubules), respiratory airways, colon, and salivary and sweat glands. PHA type 2 (Gordon syndrome or familial hyperkalemic hypertension) shows consistent features of hypertension, hyperkalemia, hyperchloremia, and mild metabolic acidosis. The reabsorption of sodium and the renal function are normal. The renin is low, whereas the aldosterone level can be normal or mildly elevated. In this autosomal dominant type, mutations affecting WNK4 kinase have been reported and are important in the regulation of ion transport in the kidney, behaving like a gain of function in the thiazide-sensitive NaCl cotransporter.
The secondary form (also termed “type 3”) may be caused by medications or may be seen with acute pyelonephritis, urologic malformations, and obstructive uropathy [16]. This secondary PHA is always transient.
In the presence of hyponatremia, hyperkalemia, and nongap metabolic acidosis, possible signs of adrenal insufficiency, it is important to exclude CAH. In cases of atypical external genitalia, an urgent karyotype must be done to determine the gender. To differentiate from PHA type 1, investigations should include plasma renin activity, aldosterone, 17-hydroxyprogesterone, dehydroepiandrosterone, cortisol levels, renal ultrasound scan, and a urine steroid profile [6].
To date, CDK13-related disorder has been reported in 44 patients [17], and although there is a wide phenotypic heterogeneity [18], all patients are characterized with developmental delay (motor and speech domains) and variable craniofacial features (hypertelorism, flat midface, broad nasal bridge, small mouth, low-set ears); most show mild to moderate intellectual disability. Other common findings are feeding difficulties (gastroesophageal reflux, velopharyngeal dysfunction), structural brain abnormalities (hypoplastic or absent corpus callosum, periventricular leukomalacia or gliosis, Chiari malformations), and structural heart defects (most commonly atrial or ventricular septal defects) [17]. Bostwick et al. reported three patients with renal abnormalities (duplicated or dilated collecting systems, fused renal ectopia) [18]. To the best of our knowledge, no metabolic abnormalities or electrolyte disturbances have been reported to be related to CDK13 gene mutations.
Cyclin-dependent kinases are a family of serine threonine kinases and are involved in several key cellular processes that include regulation of the cell cycle, transcription, RNA splicing, neurogenesis, and apoptosis [19]. CDK13 is involved in pre-mRNA splicing and is a component of the perinucleolar compartment, which is primarily detected in solid tissue-derived cancer cells and rarely present in normal cells [20].
CDK13-related disorder is inherited in an autosomal dominant manner, and all cases are the result of a de novo CDK13 pathogenic variant. No individuals with CDK13-related disorder are known to reproduce [21]. Although no clinical diagnostic criteria have been published, the clinical findings of developmental delay and facial dysmorphic features are suggestive, and the diagnosis of CDK13-related disorder primarily depends on molecular genetic testing. Of the 44 patients reported, 36 were found to have missense mutation affecting the protein kinase domain of CDK13. Of these 36, 20 affected amino acid residue 842, and 18 had the same mutation (p.Asn842Ser) as our patient [17]. Structural anomalies of the heart and brain are common in the patients with mutations affecting amino acid residue 842, although this was not the case in our patient [17]. Workup for suspected CDK13 gene mutations should include diligent physical examination; MRI of the brain; echocardiography; and consultations with a neurologist, cardiologist, dietitian, and geneticist. More extensive research may be required (such as radiology of the spine). The follow-up is very individual-dependent but includes neurological follow-up, as well as support in daily life, such as suitable schooling and proper nutrition [22].
According to the literature, CDK13 has a key role in the cell cycle, but no interference with the aldosterone signaling pathway or electrolyte balance has been described.
No mutations in the previously described NR3C2 gene (cytogenetic location 4q31.23), encoding the mineralocorticoid receptor, were found in our patient. Although the clinical presentation corresponded to PHA type 1, we could not genetically confirm this.
Conclusion
We report a case of a child presenting with PHA with a CDK13 gene mutation. It is possible that the PHA at presentation was a coincidental finding in this girl with a CDK13 mutation, but because only limited information is known about CDK13-related disorders, further investigation could be more informative to clarify this.
Availability of data and materials
The data are noted in the report. Additional information can be requested from the corresponding author.
References
Pascual-Le Tallec L, Lombes M. The mineralocorticoid receptor: a journey exploring its diversity and specificity of action. Mol Endocrinol. 2005;19:2211–21.
Riepe FG. Clinical and molecular features of type 1 pseudohypoaldosteronism. Horm Res. 2009;72:1–9.
Zennaro MC, Hubert EL, Fernandes-Rosa FL. Aldosterone resistance: structural and functional considerations and new perspectives. Mol Cell Endocrinol. 2012;350:206–15.
Harris AN, Grimm PR, Lee HW, Delpire E, Fang L, Verlander JW, Welling PA, Weiner ID. Mechanism of hyperkalemia-induced metabolic acidosis. J Am Soc Nephrol. 2018;29:1411–25.
Cheek DB, Perry JW. A salt wasting syndrome in infancy. Arch Dis Child. 1958;33:252–6.
Amin N, Alvi NS, Barth JH, Field HP, Finlay E, Tyerman K, Frazer S, Savill G, Wright NP, Makaya T, Mushtaq T. Pseudohypoaldosteronism type 1: clinical features and management in infancy. Endocrinol Diabetes Metab Case Rep. 2013;2013:130010.
Geller DS, Zhang J, Zennaro MC, Vallo-Boado A, Rodriguez-Soriano J, Furu L, Haws R, Metzger D, Botelho B, Karaviti L, et al. Autosomal dominant pseudohypoaldosteronism type 1: mechanisms, evidence for neonatal lethality, and phenotypic expression in adults. J Am Soc Nephrol. 2006;17:1429–36.
Zennaro MC, Borensztein P, Jeunemaitre X, Armanini D, Corvol P, Soubrier F. Molecular characterization of the mineralocorticoid receptor in pseudohypoaldosteronism. Steroids. 1995;60:164–7.
Hatta Y, Nakamura A, Hara S, Kamijo T, Iwata J, Hamajima T, Abe M, Okada M, Ushio M, Tsuyuki K, Tajima T. Clinical and molecular analysis of six Japanese patients with a renal form of pseudohypoaldosteronism type 1. Endocr J. 2013;60:299–304.
Kawashima Sonoyama Y, Tajima T, Fujimoto M, Hasegawa A, Miyahara N, Nishimura R, Hashida Y, Hayashi A, Hanaki K, Kanzaki S. A novel frameshift mutation in NR3C2 leads to decreased expression of mineralocorticoid receptor: a family with renal pseudohypoaldosteronism type 1. Endocr J. 2017;64:83–90.
Morikawa S, Komatsu N, Sakata S, Nakamura-Utsunomiya A, Okada S, Tajima T. Two Japanese patients with the renal form of pseudohypoaldosteronism type 1 caused by mutations of NR3C2. Clin Pediatr Endocrinol. 2015;24:135–8.
Tajima T, Kitagawa H, Yokoya S, Tachibana K, Adachi M, Nakae J, Suwa S, Katoh S, Fujieda K. A novel missense mutation of mineralocorticoid receptor gene in one Japanese family with a renal form of pseudohypoaldosteronism type 1. J Clin Endocrinol Metab. 2000;85:4690–4.
Tsunogai T, Miyata I, Kotake S, Matsuura R, Takagi K, Nanba H, Takahata N, Tajima T, Wada Y. A novel NR3C2 mutation in a Japanese patient with the renal form of pseudohypoaldosteronism type 1. Clin Pediatr Endocrinol. 2016;25:111–4.
Belot A, Ranchin B, Fichtner C, Pujo L, Rossier BC, Liutkus A, Morlat C, Nicolino M, Zennaro MC, Cochat P. Pseudohypoaldosteronisms, report on a 10-patient series. Nephrol Dial Transplant. 2008;23:1636–41.
Dillon MJ, Leonard JV, Buckler JM, Ogilvie D, Lillystone D, Honour JW, Shackleton CH. Pseudohypoaldosteronism. Arch Dis Child. 1980;55:427–34.
Manipriya R, Umamaheswari B, Prakash A, Binu N. Rare cause of hyperkalemia in the newborn period: report of two cases of pseudohypoaldosteronism type 1. Indian J Nephrol. 2018;28:69–72.
Hamilton MJ, Suri M. CDK13-related disorder. Adv Genet. 2019;103:163–82.
Bostwick BL, McLean S, Posey JE, Streff HE, Gripp KW, Blesson A, Powell-Hamilton N, Tusi J, Stevenson DA, Farrelly E, et al. Phenotypic and molecular characterisation of CDK13-related congenital heart defects, dysmorphic facial features and intellectual developmental disorders. Genome Med. 2017;9:73.
Cao L, Chen F, Yang X, Xu W, Xie J, Yu L. Phylogenetic analysis of CDK and cyclin proteins in premetazoan lineages. BMC Evol Biol. 2014;14:10.
Even Y, Escande ML, Fayet C, Geneviere AM. cdk13, a kinase involved in pre-mrna splicing, is a component of the perinucleolar compartment. PLoS One. 2016;11:e0149184.
Bostwick B. CDK13-related disorder. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews. Seattle, WA: University of Washington; 1993.
van den Akker WMR, Brummelman I, Martis LM, Timmermans RN, Pfundt R, Kleefstra T, Willemsen MH, Gerkes EH, Herkert JC, van Essen AJ, et al. De novo variants in CDK13 associated with syndromic ID/DD: molecular and clinical delineation of 15 individuals and a further review. Clin Genet. 2018;93:1000–7.
Acknowledgements
Not applicable.
Funding
No funding was secured for this study.
Author information
Authors and Affiliations
Contributions
RY was responsible for the data collection and obtaining consent and was the main author of the manuscript. AA was responsible for part of the Discussion section. AKK was responsible for part of the Discussion section and for reviewing the paper. MvdA was responsible for writing and finalizing the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient’s legal guardian(s) for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yakubov, R., Ayman, A., Kremer, A.K. et al. One-month-old girl presenting with pseudohypoaldosteronism leading to the diagnosis of CDK13-related disorder: a case report and review of the literature. J Med Case Reports 13, 386 (2019). https://doi.org/10.1186/s13256-019-2319-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-019-2319-x