
Abstract
Background
Anticonvulsant hypersensitivity syndrome is a rare adverse drug reaction associated with aromatic anticonvulsant drugs. This syndrome can range from mild cutaneous rash to drug reaction with eosinophilia and systemic symptoms that include fever, rash, lymphadenopathy, pancytopenia, and involvement of multiple internal organs. We aimed to report this case in the literature and make physicians aware of the uncommon symptoms of this syndrome when they prescribe antiepileptic medications in particular.
Case presentation
A 14-year-old Middle Eastern female patient from Iran with free past medical and allergic history was admitted to hospital because of fever, rash, lymphadenopathy, and pancytopenia after taking anticonvulsants due to new-onset seizure. High fever and cutaneous rash along with lymphadenopathy following administration of anticonvulsant medications that could not be explained by other causes alerted the physician to the possibility of this syndrome. Our investigation revealed no further diagnosis and 1 week after discontinuation of the drugs, her symptoms were resolved. Anticonvulsant hypersensitivity syndrome is a diagnosis of exclusion and immediate discontinuation of the suspicious drugs is necessary. Hence, early recognition can prevent permanent multiorgan damage.
Conclusions
Chlorpheniramine as a simple treatment was provided for this syndrome.
Similar content being viewed by others


Background
Anticonvulsant hypersensitivity syndrome (ACHS) is a rare but potentially fatal complication that is associated with aromatic antiepileptic drugs [1,2,3,4]. Women are more susceptible to this drug reaction, which indicates the role of sex hormones in the pathogenesis of ACHS [3, 5, 6].
Carbamazepine and phenytoin are the most common causes of ACHS, followed by lamotrigine [7].
Case presentation
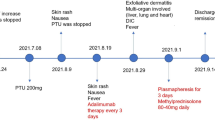
A 14-year-old Middle Eastern girl from Iran was admitted to Sina Medical Research and Training Hospital on 13 January 2019 because of fever and rash. She had been well until 4 weeks earlier, when she developed new-onset generalized tonic-clonic seizure with normal brain MRI and magnetic resonance venography (MRV).
An electroencephalogram (EEG) showed epileptic waves; thus, she was treated with phenobarbital and lamotrigine. Since then she had no more seizures. At time of admission (4 weeks after seizure treatment), she had fever and itchy skin rash, anorexia, nausea malaise, and fatigue of 1-week duration. She was otherwise fit and well with no significant past medical history of note and no history of familial disease.
On physical examination, she was alert, oriented, and conscious. She had multiple firm, non-tender, right and left cervical and inguinal lymph nodes as well as an erythematous maculopapular rash on her chest, abdomen, back, and upper limbs without palm and sole involvement that was highlighted with fever; the rash was inconspicuous when fever subsided.
Lungs, heart, abdomen, extremities, and neurologic examinations were normal. There was no hepatomegaly or splenomegaly.
Her vital signs on the first day of admission were: blood pressure, 105/65; body temperature, 38.7 axillary; pulse rate, 115; and respiratory rate, 20. Blood investigation immediately after admission revealed pancytopenia: white blood cells (WBC), 3140 cells/mm3; hemoglobin (Hb), 11.8 g/dl; and platelets (PLT), 118,000.
Cervical and abdominal ultrasounds were done and showed multiple lymph nodes in right posterior (5 × 23 mm), anterior (4 × 25 mm) cervical triangle, posterior occipital (6 × 15 mm), submandibular (5 × 14 mm) with echogenic hilum, left posterior (4 × 8, 4 × 11, 4 × 18 mm), anterior (2 × 11, 3 × 10 mm) cervical triangle with echogenic hilum, and submandibular (3 × 14 mm) without echogenic hilum with normal size of liver and spleen. A computed tomography (CT) scan of her chest (pulmonary and mediastinal) and abdomen was normal with bilateral inguinal lymphadenopathy. Cardiac echocardiography and electrocardiography (EKG) were both normal. Laboratory workup was summarized in Tables 1 and 2.
Our first diagnosis was ACHS based on fever, rash, lymphadenopathy, and pancytopenia after taking anticonvulsants, so a neurology consult was done to change phenobarbital and lamotrigine to levetiracetam. Our differential diagnoses were viral infections, collagen vascular disease, Kikuchi-Fujimoto disease, and hematologic malignancy; all of which were ruled out (Tables 1 and 2). During her first week of hospitalization, our patient had daily intermittent fever with spikes in the mornings and at nights up to 39.5–40 °C which responded to parenteral acetaminophen. Furthermore, her lactate dehydrogenase (LDH) level increased, whereas WBC and PLT decreased. Laboratory evaluation revealed no further diagnosis.
Moreover, a peripheral blood smear (PBS), which was reported by an oncologist, was normal without malignancy. On the eighth hospital day, she underwent cervical lymph node excisional biopsy with respect to oncologist’s recommendation and she was given chlorpheniramine 4 mg every 12 hours after returning from the operating room. The next day, her fever and rash completely resolved and she got well.
A brief report of the lymph node biopsy by the pathologist was as follows: Two lymph node tissues with architectural distortion and depletion in germinal centers and diffuse infiltration of the histiocytes in the parenchyma and some mature lymphocytes. Two vague granuloma formations composed of epithelioid cells aggregate, surrounded by a rim of lymphocytes were noted. There were a few (scattered) large cells with vesicular nuclei and high nuclear cytoplasmic (N/C) ratio, which were more probably immunoblasts. There were also foci of necrosis and necrotic debris in the background. Therefore, immunohistochemistry (IHC) was recommended.
The IHC results for PAX5, CD5, CD30, CD68, and Ki-67 were not in favor of lymphoma. According to the pathologist’s point of view, necrotizing lymphadenitis was a possible diagnosis.
On the 16th hospital day, our patient was discharged while receiving levetiracetam and clonazepam. She was visited10 days after discharge. She had been in a good clinical condition without any problem or fever. Her latest laboratory investigation revealed: WBC, 4260 cells/mm3 (with normal eosinophil count as outlined in Table 1); Hb, 12 g/dl; PLT, 267,000; LDH, 388 IU/L; erythrocyte sedimentation rate (ESR), 23 mm/hour; and C-reactive protein (CRP), negative.
Discussion and conclusions
ACHS, which is a rare but serious and potentially fatal complication, is associated with aromatic antiepileptic drugs, including phenytoin. ACHS occurs in 1 in 1000 to 1 in 10,000 patients treated with aromatic antiepileptic drugs such as carbamazepine, phenytoin, lamotrigine, oxcarbazepine, and phenobarbital, as well as allopurinol and the sulfonamides. This syndrome has a fatality rate of 10% [1,2,3,4].
Drug reaction to phenytoin was first recognized by Meritt and Putnam in the early 1930s. Then ACHS was described for the first time in 1950s [3, 4]. ACHS is a triad of fever, rash, and internal organ involvement [3]; cases are often underreported and unrecognized [1, 3].
Since women, especially fertile women, are more susceptible to this drug reaction, female sex hormones might have a role in the pathogenesis of ACHS [3, 5]. This tendency has previously been reported for lamotrigine [6].
ACHS has an autosomal dominant inheritance pattern. It is a type IV T cell-mediated delayed hypersensitivity reaction with high susceptibility between siblings and other first-degree relatives of affected patients [2, 3].
Studies showed that carbamazepine and phenytoin are the most common causes of ACHS, and lamotrigine is the next most common etiologic agent [7]. A high rate of cross-reactivity among aromatic antiepileptic drugs is of concern and it might be as high as 80% [4].
Factors which have been associated with ACHS are the human leukocyte antigen HLA-A*3101, HLA-B*1502, human herpesvirus 6 (HHV6), human herpesvirus 7 (HHV7), cytomegalovirus (CMV), and Epstein–Barr virus (EBV) [1, 3, 8,9,10,11].
Drug reaction with eosinophilia and systemic symptoms (DRESS) or drug-induced hypersensitivity syndrome (DIHS) are used as equivalent to ACHS, that is, a severe idiosyncratic reaction characterized by various features but symptoms classically include fever, rash, lymphadenopathy, pharyngitis which is common, and possibly other organ involvement [1,2,3,4, 7]. When severe, the syndrome can include hepatitis, megaloblastic anemia, rhabdomyolysis, and arteritis.
In most patients, the reaction begins 2 to 6 weeks after the initiation of the mentioned medications [1,2,3]. Early diagnosis is vital because of the high mortality rate [2, 12], and early recognition can prevent permanent multiorgan damage [2]. Diagnosis of ACHS is based on a history of antiepileptic drugs exposure and clinical symptoms. Signs and symptoms usually occur within 3 months of initiating treatment with an anticonvulsant, most often within 2 to 6 weeks [2]. When ACHS is suspicious due to signs and symptoms that occur typically 2–6 weeks after the initiation of antiepileptic drugs, discontinuation of them is essential [2, 4].
The optimal treatment approach is controversial [12]. Treatments include antihistamines (H1-receptor blockers), epinephrine, glucocorticoids, anabolic steroids, anti-gonadotropic agents, and airway management, depending on the severity of the condition [2, 4, 12]. N-acetylcysteine may be efficacious in hepatitis [13]. Some studies reported severe diseases and prolonged hospital stay for skin drug-related complications [12]. In patients who were treated with intravenous immunoglobulin and systemic corticosteroids, the length of hospitalization was short and all recovered without complication [12, 14].
Complications of DRESS based on age can be divided into two major types: in young patients, autoimmune diseases, specifically Graves’ disease, type 1 diabetes mellitus, and autoimmune hemolytic anemia may occur prominently; older patients are more susceptible to end-organ failure such as chronic renal failure [15].
The most probable causes of lymphadenopathy and pancytopenia according to our patient history and physical examination were drug reaction, lymphoproliferative disorders, brucellosis, human immunodeficiency virus (HIV), EBV, CMV, hepatitis B, hepatitis C, parvovirus B19, and rheumatologic diseases, especially systemic lupus erythematosus (SLE); all of which were ruled out based on the laboratory workup. Because of absence of severe anemia and arthralgia, parvovirus B19 was excluded. Toxoplasmosis causes fever and lymphadenopathy but its serology was negative in our patient.
ACHS was our first probable diagnosis because our patient initially had a seizure without other systemic symptoms, she was treated with antiepileptic drugs and 3 weeks later she presented with fever, rash, lymphadenopathy, and pancytopenia. At her first complete blood cell count, no eosinophilia and/or monocytosis and/or atypical lymphocytosis were detected, which is a major component of antiepileptic hypersensitivity syndrome (AHS)/DRESS syndrome. Another diagnosis was rapidly progressive lymphoproliferative disease but PBS and ESR were normal, although it could not be completely ruled out.
According to symptoms, age, sex and Asian ethnicity, Kikuchi-Fujimoto disease was considered. Kikuchi-Fujimoto disease is a rare histiocytic necrotizing lymphadenitis with a benign course and unknown etiology; it is characterized by cervical lymphadenopathy and fever. Histopathology of the involved lymph nodes differentiated it from other diseases that it mimics. In this disease, a complete blood cell count is often normal, although anemia, leukopenia, thrombocytopenia, and pancytopenia have been reported. High-level LDH and normal or elevated ESR are other nonspecific findings.
Our patient initially had a seizure and 3 weeks after initiation of phenobarbital and lamotrigine her systemic symptoms began; after discontinuation of all drugs and administration of chlorpheniramine 4 mg for two doses her symptoms were completely resolved.
Pathological study ruled out malignant etiology, so a diagnosis of ACHS was confirmed. Our patient was then discharged. She had no problems at the following visit.
According to the reports of autoimmune diseases subsequent to ACHS, our patient would be followed-up with thyroid function tests and the possibility of diabetes mellitus in the future.
Since genetic factors may predispose individuals to ACHS, it may occur in our patient’s siblings after taking anticonvulsants. Therefore, her parents became aware of this issue. Given that her history of seizure had remained a dilemma, it was better to follow-up for rheumatologic diseases, especially SLE.
In conclusion, ACHS is a rare genetic disease, which may occur after taking anticonvulsant drugs. Physicians should be aware of various symptoms of this syndrome to recognize it in patients with fever, leukopenia, rash, and lymphadenopathy after taking these drugs. Accordingly, they should pursue a quick therapeutic action, including discontinuing medications and prescribing antihistamines.
Availability of data and materials
All data and materials related to this report are accessible at any time upon request.
Abbreviations
- ACHS:
-
Anticonvulsant hypersensitivity syndrome
- CMV:
-
Cytomegalovirus
- DRESS:
-
Drug reaction with eosinophilia and systemic symptoms
- EBV:
-
Epstein–Barr virus
- ESR:
-
Erythrocyte sedimentation rate
- Hb:
-
Hemoglobin
- IHC:
-
Immunohistochemistry
- PBS:
-
Peripheral blood smear
- PLT:
-
Platelets
- SLE:
-
Systemic lupus erythematosus
- WBC:
-
White blood cells
References
Schweitzer I. Anticonvulsant hypersensitivity syndrome: a rare and serious complication. Med J Aus. 2011;194(11):609–10.
Mason ME. Anticonvulsant hypersensitivity syndrome: recognizing the signs and symptoms. Resid Staff Physician. 2007;53:29–36.
Ghannam M, Mansour S, Nabulsi A, Abdoh QJC. Anticonvulsant hypersensitivity syndrome after phenytoin administration in an adolescent patient: a case report and review of literature. Clin Mol Allergy. 2017;15(1):14.
Mehta M, Shah J, Khakhkhar T, Shah R, Hemavathi KJ. Anticonvulsant hypersensitivity syndrome associated with carbamazepine administration: case series. J Pharmacol Pharmacother. 2014;5(1):59–62.
JA DS. Sex hormones and glucocorticoids: interactions with the immune system. Ann N Y Acad Sci. 1999;876(1):102–18.
Wong IC, Mawer GE, JW S. Factors influencing the incidence of lamotrigine-related skin rash. Ann Pharmacother. 1999;33(10):1037–42.
Mansur AT, Pekcan Yaşar Ş, F G. Anticonvulsant hypersensitivity syndrome: clinical and laboratory features. Int J Dermatol. 2008;47(11):1184–9.
Pritchett JC, Nanau RM, MG N. The link between hypersensitivity syndrome reaction development and human herpes virus-6 reactivation. Int J Hepatol. 2012;2012 https://doi.org/10.1155/2012/723062.
Ö Ö. Development of antiepileptic hypersensitivity syndrome after phenytoin treatment. Turk J Pediatr. 2010;52(1):111–2.
Matsuda K, Ohnuma T, Fukuta M, Kawai M, Suzuki T, Ogawa H, et al. Case reports and literature review: the association between reactivation of human herpes virus-6 and peripheral white blood cell count in patients with carbamazepine-induced hypersensitivity syndrome. Prog Neuro-Psychopharmacol Biol Psychiatry. 2006;30(4):751–4.
Descamps V, S R-R. DRESS syndrome. Joint Bone Spine. 2014;81(1):15–21.
Prais D, Straussberg R, Amir J, Nussinovitch M, L H. Treatment of anticonvulsant hypersensitivity syndrome with intravenous immunoglobulins and corticosteroids. J Child Neurol. 2006;21(5):380–4.
Harrison PM, Wendon JA, Gimson AE, Alexander GJ, R W. Improvement by acetylcysteine of hemodynamics and oxygen transport in fulminant hepatic failure. N Engl J Med. 1991;324(26):1852–7.
Mostella J, Pieroni R, Jones R, CK F. Anticonvulsant hypersensitivity syndrome: treatment with corticosteroids and intravenous immunoglobulin. South Med J. 2004;97(3):319–21.
Chen Y-C, Chang C-Y, Cho Y-T, Chiu H-C, Chu C-Y. Long-term sequelae of drug reaction with eosinophilia and systemic symptoms: a retrospective cohort study from Taiwan. J Am Acad Dermatol. 2013;68(3):459–65.
Acknowledgements
The authors express their thanks to Dr Maryam Nouhi, a pathologist, who interpreted the result of pathology in our patient with ACHS.
Author information
Authors and Affiliations
Contributions
FRG, MV, BN, ZB, and PP were involved in patient care, review of literature, and writing of the manuscript. FRG, MV, and RRG prepared the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This case report does not involve any active intervention on patients; therefore, ethics approval is not applicable.
Consent for publication
Written informed consent has been obtained from the patient next of kin for publication of this case report and any accompanying images.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Varshochi, M., Ravanbakhsh Gavgani, R., Naghili, B. et al. A 14-year-old female with fever, rash, lymphadenopathy, and pancytopenia: a case report. J Med Case Reports 14, 20 (2020). https://doi.org/10.1186/s13256-019-2286-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-019-2286-2