
Abstract
Background
PTEN is a well-established risk gene for autism spectrum disorder (ASD). Yet, little is known about how PTEN mutations and associated molecular processes influence neurobehavioral function in mutation carriers with (PTEN-ASD) and without ASD (PTEN no-ASD). The primary aim of the present study was to examine group differences in peripheral blood-derived PTEN pathway protein levels between PTEN-ASD, PTEN no-ASD, and idiopathic macrocephalic ASD patients (macro-ASD). Secondarily, associations between protein levels and neurobehavioral functions were examined in the full cohort.
Methods
Patients were recruited at four tertiary medical centers. Peripheral blood-derived protein levels from canonical PTEN pathways (PI3K/AKT and MAPK/ERK) were analyzed using Western blot analyses blinded to genotype and ASD status. Neurobehavioral measures included standardized assessments of global cognitive ability and multiple neurobehavioral domains. Analysis of variance models examined group differences in demographic, neurobehavioral, and protein measures. Bivariate correlations, structural models, and statistical learning procedures estimated associations between molecular and neurobehavioral variables. To complement patient data, Western blots for downstream proteins were generated to evaluate canonical PTEN pathways in the PTEN-m3m4 mouse model.
Results
Participants included 61 patients (25 PTEN-ASD, 16 PTEN no-ASD, and 20 macro-ASD). Decreased PTEN and S6 were observed in both PTEN mutation groups. Reductions in MnSOD and increases in P-S6 were observed in ASD groups. Elevated neural P-AKT/AKT and P-S6/S6 from PTEN murine models parallel our patient observations. Patient PTEN and AKT levels were independently associated with global cognitive ability, and p27 expression was associated with frontal sub-cortical functions. As a group, molecular measures added significant predictive value to several neurobehavioral domains over and above PTEN mutation status.
Limitations
Sample sizes were small, precluding within-group analyses. Protein and neurobehavioral data were limited to a single evaluation. A small number of patients were excluded with invalid protein data, and cognitively impaired patients had missing data on some assessments.
Conclusions
Several canonical PTEN pathway molecules appear to influence the presence of ASD and modify neurobehavioral function in PTEN mutation patients. Protein assays of the PTEN pathway may be useful for predicting neurobehavioral outcomes in PTEN patients. Future longitudinal analyses are needed to replicate these findings and evaluate within-group relationships between protein and neurobehavioral measures.
Trial registration
ClinicalTrials.gov Identifier NCT02461446
Similar content being viewed by others


Background
PTEN encodes a dual-specificity phosphatase that inhibits PI3K/AKT/mTOR and MAPK/ERK pathway activation [1] and is an important regulator of neural connectivity and plasticity [2]. A suggestive association between germline heterozygous PTEN mutations and autism spectrum disorder (ASD) was first identified through cases of ASD with extreme macrocephaly [3, 4]. Subsequent small cohort studies identified an enriched number of germline PTEN mutations in ASD cases with macrocephaly [5,6,7]. Larger clinical cohort studies of ASD and/or intellectual disability with macrocephaly also found an enrichment of PTEN mutations [8,9,10,11,12], resulting in a weighted average of 7% of macrocephalic ASD and translating to approximately 1% of all ASD cases [13]. Exome and genome sequencing studies of large ASD cohorts have confirmed PTEN as well-established risk gene for ASD and neurodevelopmental disorders [14,15,16,17,18,19,20], and recent case series have suggested that 25–50% of children with PTEN mutations are identified with ASD [21, 22].
The association between PTEN and ASD has been further supported by nearly two decades of mouse model studies [23,24,25,26,27,28,29]. These investigations have shown that PTEN loss impairs brain development and leads to aberrant brain connectivity [13], a well-replicated phenotype observed in idiopathic ASD cases [30,31,32]. A recent knock-in model that results in cytoplasmic predominance of PTEN was found to have expression changes in genes important for white matter development, along with social and motor deficits reminiscent of ASD cases with intact cognitive ability [28, 29]. Findings from this model also suggest that the impact on neurobehavior is highly variable, consistent with observations in human patients [33,34,35].
Discoveries from cohorts of human patients with PTEN mutations have generally mirrored the PTEN mouse literature. For example, an initial study of patients with germline heterozygous PTEN mutations and ASD identified major increases in structural white matter volumes as well as hypo-intensities on structural MRI images, consistent with neuropathology observed in mouse models [33]. In this cohort, PTEN loss was associated with white matter abnormalities, which, in turn, predicted greater cognitive deficits. More recent investigations of PTEN mutation patients with (PTEN-ASD) and without ASD (PTEN no-ASD) have observed reductions in frontal sub-cortical neurobehavioral functions in most cases. However, impairments in general cognitive ability are only common in PTEN-ASD. For most neurobehavioral measures, variability within and across patient groups was very large, with some patients functioning in the average or above average range in most neurocognitive domains and other patients showing moderate to severe global cognitive dysfunction [33,34,35]. Thus, it is important to understand what molecular factors may be driving variability in neurobehavioral function to identify potential treatment targets and inform individual patient prediction.
A key initial question to address is whether PTEN-related pathway proteins show differences in expression due to the presence of a PTEN mutation or the presence of ASD regardless of mutation status. In our small initial cohort, PTEN-ASD cases (n = 17) had significantly reduced PTEN levels [33], but the magnitude of reduction was modest, suggesting that the healthy allele may be partially compensating for the deleterious allele. Furthermore, it is unclear whether reductions in PTEN are specific to PTEN-ASD or would also be observed in PTEN no-ASD. Intriguingly, there were no significant group differences in AKT or ERK levels. Re-evaluating PTEN-related pathway protein levels in a new cohort is crucial to determining whether prior exploratory findings replicate and whether other canonical PTEN pathway proteins show abnormal expression patterns.
Variable phenotypes in PTEN mutation cases also imply that mutational status alone is not sufficient to predict outcome. Rather, the complexity of PTEN-influenced mechanisms, such as gene–gene and gene–environment interplay, may require multivariable modeling to predict neurobehavioral outcomes. This possibility is consistent with the growing recognition of the importance of primary and secondary factors in determining phenotypic outcomes in ASD cases, even when rare pathogenic variants are identified [36, 37]. An important advance of the present study is inclusion of PTEN no-ASD cases to evaluate whether combinations of protein levels may predict phenotypic outcomes beyond PTEN mutation status. The present study also compares key protein differences observed in human ASD and/or PTEN mutation cases with neural protein expression data from a knock-in murine PTEN model (m3m4).
The present study
The present study evaluated the influence of PTEN mutation and ASD diagnostic status on relevant pathway protein levels in peripheral blood. In addition to understanding pathophysiology, our study also begins the process of identifying peripheral markers that may reflect the brain. Peripheral PTEN levels were hypothesized to be lower in all PTEN mutation cases, and downstream signaling molecules such as P-S6 and P-AKT were predicted to be upregulated in individuals with germline PTEN mutations without ASD. No specific predictions were made for P-S6/S6/P-AKT levels in the ASD with or without PTEN mutation groups. The secondary purpose was to investigate cross-level relationships between pathway measures and neurobehavioral domains independent of mutational or diagnostic status. We hypothesized that PTEN and S6 levels would be significantly associated with global cognitive ability, but no specific predictions were made for other proteins or neurobehavioral measures. Finally, the study also explored whether protein levels, as a group, might predict neurobehavioral functioning beyond PTEN mutation status.
Methods
Participants
Participants in this study were recruited from four large tertiary medical centers (Cleveland Clinic, Boston Children’s Hospital, Stanford University, and University of California, Los Angeles) as part of an IRB-approved, ongoing, multicenter prospective study designed to examine the natural history of autism and germline heterozygous PTEN mutations. All potential study participants were screened by a clinical psychologist with expertise in ASD to determine whether they met Diagnostic and Statistical Manual of Mental Disorders—Fifth Edition (DSM-5) diagnostic criteria for ASD. All potential participants also underwent genetic testing to determine the presence/absence of a mutation in PTEN. Individuals were included in the study if they met the following criteria: (1) age 3–21 years; (2) confirmed diagnosis of ASD (based on consensus of expert clinician evaluation and Autism Diagnostic Observation Schedule-2) and/or a confirmed heterozygous mutation in PTEN; (3) English as primary communicative language; and (4) completion of baseline neuropsychological evaluation. In addition, individuals with ASD and macrocephaly, but without a PTEN mutation (Macro-ASD), had to have an orbitofrontal head circumference ≥ 98th percentile for their age to be included in the study. Informed consent for study participation was obtained from adult participants and/or a parent or legal guardian. Assent was obtained from all participants age 7 years and older who were cognitively able to provide the same.
A total of 84 participants met initial study inclusion criteria, and of these, 68 had PTEN pathway protein measurements with at least one neurobehavioral test result. However, 7 cases (2 PTEN-ASD, 3 PTEN no-ASD, and 2 Macro-ASD) had invalid protein measurement results that were more than 6 standard deviations beyond the remaining values and were therefore excluded. The final sample included 61 participants: 25 PTEN-ASD (ASD with macrocephaly, PTEN mutation), 16 PTEN-no ASD (no ASD, PTEN mutation), and 20 Macro-ASD (ASD with macrocephaly, no PTEN mutation).
PTEN genotyping
Germline genomic DNA was extracted in the Genomic Medicine Institute’s Genomic Medicine Biorepository. PCR-based LightCycler mutation scanning and semi-automated PCR-based Sanger sequencing (ABI3730xl in Genomics Core Facility) of exons 1 through 9 and flanking intronic regions of genomic DNA were performed as per routine in the Eng laboratory since 1997 to reveal germline intragenic mutations in exons 1–9 and in splice sites. All novel variants were checked for presence and frequency in 350 ancestry-matched, sex-matched population controls available in the Eng laboratory, ClinVar-PTEN, 1000G, and gnomAD.
PTEN pathway protein assays
Western blot analyses of lymphoblastoid cell total protein lysate were performed with the antibodies and techniques routinely used in the Eng laboratory, by investigators blinded to subject genotype and group [38]. Protein lysates were prepared according to Genomic Medicine Biorepository standards (https://www.lerner.ccf.org/gmi/gmb/). All lysates were quantified for protein content using BCA assays, and equalized for protein content, and 40 μg of protein per sample was loaded on a 10% TGX polyacrylamide gel. The separated proteins were transferred to a polyvinylidene difluoride (PVDF) membrane, and the membrane was blocked overnight in 3% normal goat serum at 4 °C. Membranes were then washed with Tris-buffered saline, containing 0.2% Tween-20 TBST, and incubated with experiment-specific primary antibodies diluted in TBST overnight at 4 °C. The following antibodies were used: PTEN (1:1000, clone 6H2.1, #ABM-2025, Cascade Bioscience, Winchester, MA), P-AKT-S473 (1:1000, #4060L, Cell Signaling) and total AKT (1:750, # 4691, Cell Signaling), P-ERK1/2 (1:1000, #4376, Cell Signaling) and total ERK1/2 (1:750, #9102, Cell Signaling), P-S6-S235/236 (1:2000, #2211, Cell Signaling) and total S6 (1:1000, #2217, Cell Signaling), p27 KIP1 (1:1000, #3686, Cell Signaling), EIF2 (1:1000, #9722, Cell Signaling), MnSOD (1:1000, #06-984, Millipore, Burlington, MA), GAPDH (1:10,000, #2118L, Cell Signaling), and Actin (1:10,000, sc-8432, Santa Cruz, Dallas, TX). We removed the primary antibody solution and performed three washes, for 10 min per wash, with TBST. Blots were probed with Goat anti-Rabbit or antimouse secondary antibodies conjugated with horse radish peroxidase (1:10,000 or 1:20,000, Southern Biotech, Birmingham, AL) diluted in TBST, for 2 h at room temperature. The PVDF membranes were washed three times, 10 min each in TBST, and imaged using the iBright FL1000 (Invitrogen, Carlsbad, CA). Using ImageJ (National Institute of Health, Bethesda, Maryland, 1995), we performed densitometry analysis on these images to quantify protein expression. Active PTEN results in decreased phosphorylation of AKT and ERK, upregulation of p27, and downregulation of cyclin D1 protein levels, resulting in decreased proliferation and increased apoptosis (canonical PTEN signaling—Fig. 1). All of these proteins have been shown to be consistent in peripheral protein lysates from 100 population controls [38]. Thus, protein studies should reveal the canonical functional effects of PTEN alterations.

Canonical PTEN pathway depicting PI3K/AKT/mTOR and MAPK/ERK signaling
Western blots using protein lysates from mouse brains and livers for murine downstream protein expression were performed as previously reported [39, 40]. The PTEN-m3m4 murine model has been reported [29, 39, 40].
Neurobehavioral measurements
Neurobehavioral assessment included a cognitive battery consisting of developmentally appropriate measures of global, verbal, and non-verbal cognitive ability, language, attention, working memory, and processing speed. Due to anticipated severe symptom and intellectual impairment in at least a subset of cases, cognitive ability, language, working memory, and processing speed measures were prioritized for completion. Parents/guardians completed questionnaires designed to measure adaptive function, motor difficulties, autism symptoms, and behavioral challenges [35]. The specific cognitive and behavioral measures used in this study are outlined in Additional file 1: Table S1. All measures were scored according to published test manuals using age- and/or sex-corrected norms as available. To reduce the number of analyses and focus on domains rather than specific measures, language, attention, and motor measures were combined into separate composite scores for each domain. A frontal sub-cortical composite was also created based on our prior data. This composite averaged the attention, working memory, processing speed, and motor measures into a single broad composite score.
Incomplete neurobehavioral data were largely due to a subset of patients who were unable to tolerate testing or due to time constraints during research visits. For those patients who could not complete a particular measure due to low functional capacity, the lowest possible score was assigned [33, 35]. Assessments that were incomplete due to time constraints were considered missing at random. Missing data (10%) were minimal for intelligence quotient (IQ), frontal sub-cortical composite, and language composite measures (< 4%) but higher for attention (36%) and processing speed (28%). Missing data were more likely to be present in younger children (r = 0.28–0.30), and attention data were more likely to be missing in PTEN patients (r = 0.31), potentially reflecting greater attention impairment leading to the test not being attempted. There were no relationships between missing data and sex, race, ASD diagnosis, or general cognitive ability (IQ). To address missing data, five imputed datasets were computed using the SPSS missing values analysis option with a fully conditional specification using Markov Chain Monte Carlo estimation. All variables were included in the multiple imputation process to satisfy the missing at random assumption, to reduce bias, and to increase the precision of imputed scores [41,42,43]. Results were highly consistent between the original and imputed datasets. For this reason, neurobehavioral results are presented using the first imputation dataset.
Statistical analyses
Frequency distributions and bivariate plots were evaluated, but no univariate or bivariate outliers or high leverage cases were detected. To characterize the sample, univariate analyses of variance and Chi-square analyses compared patient groups across demographic (age, sex, race/ethnicity), clinical (head circumference z-score, height, weight, heart rate, respiration, body temperature, and systolic and diastolic blood pressure), and neurobehavioral measurements. For clinical and neurobehavioral measurements, age and sex were included as covariates, as age and sex were not equivalent across groups and were correlated with clinical and neurobehavioral variables. Protein assay scores were compared across groups using analysis of variance, and the independent effects of PTEN mutation status and ASD diagnosis were evaluated in linear regression models. PTEN mutation (yes/no) and ASD (yes/no) were predictors with protein scores as dependent variables in separate analyses. For these analyses, raw protein scores were the primary measures, but we also explored ratio scores for P-AKT/AKT, P-ERK/ERK, and P-S6/S6.
To examine cross-level relationships between protein assays and neurobehavioral measures, we first examined the bivariate correlation between PTEN levels and global cognitive ability as we predicted a positive association based on our prior findings. Second, all correlations between protein assays and neurobehavioral measures were computed. To examine whether this matrix reflected random associations or whether the number of statistically significant correlations exceeded the expected number based on chance a sign test was computed. A significant value of this test would indicate that at least some of the significant observed correlations were not due to change and likely represent meaningful associations. Significant correlations were interpreted with reference to expected canonical influences of PTEN mutations. Observed relationships were then tested in structural (path) models to better understand these relationships. Specifically, mediational models were computed if any patterns of correlation suggest potential mediation by downstream (S6, EIF2A, p27) molecules of relationships between canonical upstream pathway proteins (PTEN, AKT, ERK) and neurobehavioral measures.
To evaluate whether PTEN-related pathway proteins improve prediction of neurobehavioral measures (as a group) over and above PTEN mutation status, a series of hierarchical linear regressions were computed with PTEN mutation (yes/no) entered in Step 1 and forward entry (p < 0.05, removal p > 0.10) of protein assay values in Step 2. A significant increment in R2 would indicate that protein measure improves prediction over and above mutation status. In this case, stepwise regression provides useful information about which protein measures enter the equation and provide a significant prediction of neurobehavioral outcomes. However, stepwise regression tends to overfit a dataset and will often produce unrealistically large predictive values. For this reason, we also computed a support vector machine regression model with PTEN mutation (yes/no) and all protein assay values entered as predictors of neurobehavioral measures. A fivefold cross-validation was used to evaluate predictive validity of each model. While less optimal than having a holdout test sample, which is not possible in this modest rare disease cohort, this approach should produce more realistic estimates of validity generalization.
Sensitivity power analysis indicated that a large effect size (f ≥ 0.41, d = 0.82) was required to have at least good power (1-β ≥ 0.80) to detect statistically significant group differences (p < 0.05, two-tailed). For correlational analyses, given a total sample size of 61, a correlation (r ≥|.31|) was required to have adequate power (1-β ≥ 0.70) to detect a significant association. Power to detect structural model parameters, including indirect (mediation) effects, is approximately equivalent to the power for bivariate correlations [44]. Following the a priori prediction that PTEN levels would be lower in mutation cases, a one-tailed p < 0.05 was used to evaluate this effect. For all other statistical tests, two-tailed p < 0.05 was used. Significant R2 (p < 0.05) from fivefold cross-validation was used to identify models with significant predictive validity for neurobehavioral measures. No correction was made for multiple comparisons given the small sample size and preliminary nature of these analyses. Statistical analyses were computed using SPSS version 27 [45]. Mediational models were computed using MPlus version 7.2 [46], following the analytic criteria described by Muthén, Muthén, and Asparouhov [44].
Results
Sample characteristics
Macro-ASD patients were significantly older than PTEN-ASD and PTEN no-ASD patients (Table 1). There were no significant differences in sex distributions, although PTEN no-ASD tended to have lower proportions of males than the other ASD groups, consistent with the known sex ratio in ASD [47]. There were no significant differences in race/ethnicity or any other clinical variable (all p > 0.08). All PTEN patients had significant macrocephaly. A summary of the germline PTEN variants observed in study groups, including fitness [48] and abundance [49] scores for missense mutations, is provided in Additional file 1: Table S2.
Protein levels
Significant group differences were observed for MnSOD, P-S6, and the ratio of P-S6/S6 (Table 2). The presence of genotype-independent ASD was associated with reductions in MnSOD and increases in P-S6 and P-S6/S6. PTEN mutation status was associated with reduced PTEN levels (as hypothesized), reduced S6 (Fig. 2), and increased P-S6/S6 ratio. Trends toward reduced p27 with the presence of genotype-independent ASD with PTEN mutation status were also observed. The magnitude of PTEN and ASD effects tended to be medium-sized (β =|.24–0.38|), even when simultaneously controlling for the other predictor.

Boxplots for PTEN, MnSOD, P-S6, and S6. *p < .05 for group comparisons. kD = kilo Dalton
Murine model PTEN pathway findings
PTEN-m3m4 murine models showed PI3K/AKT pathway activation (homozygous > heterozygous > wildtype), including increased phosphorylation of Akt and S6, in brain (Fig. 3) and liver (data not shown). However, variability was substantial, particularly in heterozygous mice. Together, these findings suggest canonical PTEN pathway activation, but also substantial variability within and across models, particularly in the heterozygous mice which most closely mirror heterozygosity in the human condition.

(Left) Western blots on murine brain tissue at 6 weeks of age showing levels of PI3K pathway mediators in a model of PTEN mutation. (Right) Quantification of signal intensity for each mediator normalized to Gapdh (n = 3 each)
Neurobehavioral differences
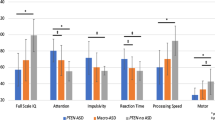
Consistent with our prior analyses [35], large group differences were observed across IQ (full-scale, verbal, and non-verbal), frontal sub-cortical, motor, and adaptive behavior measures (Additional file 1: Table S3). PTEN-ASD patients were impaired across all domains with severe impairments in IQ, frontal sub-cortical (attention, working memory, processing speed, motor), and adaptive functions. PTEN no-ASD had generally intact cognitive ability, but showed small average reductions in frontal sub-cortical measures (with the exception of working memory). PTEN no-ASD also had slightly elevated internalizing problems and autism traits relative to population expectation. These data support wide neurobehavioral variability in PTEN mutation cases, indicating that mutation status alone does not account for phenotypic presentation.
Cross-level relationships
Higher PTEN levels were significantly associated with lower general cognitive ability (r = -0.26, p = 0.024). Overall, the number of statistically significant correlations (p < 0.05 or p < 0.10) between the molecular and neurobehavioral measures was greater than would be expected by chance (p = 0.010 for p < 0.05 threshold and p = 0.003 for p < 0.10 threshold) (Additional file 1: Table S4). The major patterns observed were negative correlations between PTEN protein levels and IQ, language, frontal sub-cortical, working memory, and adaptive functions; positive correlations between total AKT and EIF2A levels and a highly overlapping set of neurobehavioral functions to that observed for PTEN; and positive correlations between p27 levels and frontal sub-cortical, attention, processing speed, and motor functions. For protein ratios, only 3 significant correlations were observed—a negative relationship between P-AKT/AKT ratio and autism traits and positive relationships between P-S6/S6 ratio and externalizing problems and repetitive behavior (Additional file 1: Table S5).
To further understand how PTEN, AKT, and EIF2A levels may interact with global IQ differences, a structural mediational model was computed with direct effects of PTEN and AKT on full-scale IQ and indirect effects through EIF2A being estimated. This model was chosen because of the well-known multiple functions of PTEN beyond AKT pathway regulation and the known downstream impact on EIF2A influencing its role in transcription. PTEN and AKT showed significant direct effects on full-scale IQ, but the indirect effects through EIF2A were small and not significant (Fig. 4). p27 significantly influenced frontal sub-cortical and motor function and marginally significantly influenced attention, when these relationships were simultaneously estimated (Additional file 1: Fig. S1).

Effects of PTEN and total AKT protein levels on full-scale IQ directly and indirectly via EIF2A
PTEN mutation status did not account for significant variance in any of the neurobehavioral measures. However, both in the regression and support vector models, protein measures significantly predicted IQ scores, language, frontal sub-cortical composite, and externalizing scores. Using fivefold cross-validation, the magnitude of predictive validity was medium-to-large (r = 0.26–0.47) across these domains (Table 3).
Discussion
PTEN levels were modestly but reliably reduced in PTEN-ASD and PTEN no-ASD groups, replicating and extending our prior findings from a PTEN-ASD only cohort [33]. This modest, but variable, reduction likely reflects the inclusion of many missense mutation cases where PTEN levels may not be substantially reduced. This may suggest that early developmental timing of alterations in PTEN protein levels is a key factor in determining neurobehavioral outcomes. It is also possible that the negative correlations observed between PTEN protein levels and cognitive ability and language reflect a dominant negative process operating in some cases where one mutant PTEN allele’s product negatively impacts the remaining wildtype PTEN protein to disrupt neurodevelopment. Specifically, in missense mutations where PTEN protein levels are not reduced, mutant PTEN may still bind substrate but not be able to act on it. This could trap substrate and prevent it from being catalyzed by wildtype PTEN. Additionally, PTEN functionally exists as dimers [50]. In heterozygotes, mutant PTEN dimerizes with wildtype PTEN protein and prevents the latter from working on substrate. If a dominant negative process is operating, it may suggest that early interventions that focus on degrading mutant PTEN could be a useful strategy in some cases. Recent work has demonstrated substantial variability in impact on PTEN protein stability and function across different missense mutations [48, 49, 51]. This suggests that the above-described pattern of modest reduction in PTEN levels may mask substantial variability, potentially explaining the observed heterogeneity of outcomes across PTEN mutation cases. Future work in large, longitudinal cohorts is needed to better understand the precise developmental timing and mechanisms—including variability in stability and function across missense mutations—leading to ASD and related neurobehavioral phenotypes in PTEN patients.
The direct relationships between PTEN and total AKT levels with cognitive ability indicate that the canonical PI3K/AKT/mTOR pathway may play an important role in neurobehavioral variation within PTEN mutation-positive patients [52]. This possibility is consistent with mouse model findings [28, 29], although both murine and human results suggest high variability in the effects on PI3K/AKT/mTOR activation [29, 33, 35]. In spite of this variability, the statistically reliable identification of canonical PI3K/AKT/mTOR activation and its relationship with cognitive ability suggests that modulation of this pathway may be an effective intervention for neurobehavioral impairments. In fact, the elevation of P-S6 would implicate mTORC1, hence, the potential efficacy of mTOR inhibition. The ongoing multisite randomized controlled trial of RAD001 (Everolimus) for PTEN-associated neurodevelopmental disorders will be an important test of this hypothesis (NCT02991807). However, it is also important to note that, in this study, the effects of PTEN and AKT were only minimally mediated by EIF2A and not mediated by other downstream molecules. Thus, it will be important to further explore the exact molecular and neural mechanisms by which PTEN and AKT protein levels influence cognitive ability and language and the developmental timing of these mechanisms.
The variability in PTEN downstream molecules both in human peripheral blood and in the PTEN-murine heterozygote model brain, in spite of a highly conserved genetic background and consistent environment, suggests that other non-PTEN influences may interact with the PTEN mutation to ultimately affect phenotype. We have recently shown that CNV load may interact with germline PTEN mutations to favor ASD over cancer phenotypes [53]. Similarly, the metabolic milieu may also crosstalk with the PTEN mutation, in a phenotype-specific manner [54]. We already appreciate the complexity of ASD even with the contribution of the genetic underpinnings. Modifier effects, as they crosstalk with Mendelian genes, will become germane in human ASD predisposed by germline mutations in Mendelian genes [36].
Decreases in MnSOD and elevations in P-S6 in ASD cases, irrespective of genotype, are consistent with prior molecular findings in idiopathic ASD [55,56,57,58,59]. Specifically, MnSOD is a redox catalyst, located in the mitochondrial matrix, and plays a role in many biological processes to reduce O2 and producing an influx of O2˙radicals. Decreased levels of MnSOD have been associated with increased oxidative stress in idiopathic ASD [59]. Prior research has suggested that subsets of idiopathic ASD cases show oxidative stress [55, 56] and mitochondrial dysfunction [57, 58] and metabolomic algorithms have shown promise in differentiating ASD and healthy control cases [60]. Furthermore, increased oxidative stress has been associated with muscle function [61] and, in the current investigation, both ASD groups had substantially decreased motor functioning, a well-replicated finding in idiopathic ASD [62,63,64]. The potential to impact at least the motor portion of the PTEN mutation neurobehavioral phenotype via improving the functioning of oxidative stress pathways should be explored in future research and could represent an augmentative strategy for future clinical trials.
The increases in P-S6 observed in ASD groups within this study are congruent with the notion of increased protein translation as a converging signaling mechanism within at least some ASD cases [36, 65, 66]. Altered protein translation also provides a molecular mechanism for excessive early life head growth [67, 68], often resulting in macrocephaly [69, 70], within a subset of ASD. Alternatively, or possibly simultaneously, increased P-S6 may contribute to dysfunctional energy metabolism associated with abnormal mTOR [52, 71] or insulin signaling pathway activation [72]. Based on these findings, it is plausible that MnSOD and P-S6 could serve as early peripheral markers of ASD within PTEN mutation cases and potentially in children with excessive early life head growth and/or macrocephaly who are at higher risk for ASD, while preliminary, the observation of upregulation of phosphorylated AKT and S6 from both peripheral blood protein lysate and PTEN model murine brains and livers, lends hope that at least some PTEN signaling molecules in the periphery do mirror those in the brain.
As a group, canonical PTEN pathway proteins significantly predicted several neurobehavioral domains, including cognitive ability, language, frontal sub-cortical function, and externalizing behavior. Larger samples and future waves of longitudinal data collection will be useful for replicating these effects and determining which proteins are playing important roles. Regardless, these findings imply that canonical PTEN pathways are an important part of understanding and predicting the variability in neurobehavioral outcomes in PTEN mutation patients. Beyond replication and clarification of these findings, future studies should search for other non-canonical mechanisms, as there is substantial variance remaining to be explained. If replicated, and additional factors are identified with useful predictive validity, an aggregate index of peripheral molecular markers could be a useful advance for early prediction of neurobehavioral outcome and would allow for better planning and tailoring of early intervention strategies. This index might also have value for longitudinal tracking, including evaluating and understanding target engagement and efficacy within clinical trials.
Limitations
While reasonable for a rare genetic syndrome, sample sizes in the present study were small and precluded analysis of cross-level relationships within each patient group. This step is critical before final conclusions are made since relationships observed might be driven by the specific pathophysiology observed in one group and not the other. A key task of future longitudinal waves will be to retain existing patients while adding new patients to the cohort. These waves would also be wise to add protein and other molecular assays for non-canonical PTEN effects. This will increase the power and explanatory value of future molecular and cross-level analyses and could improve the validity of an aggregate molecular index in predicting neurobehavioral outcomes.
An additional limitation was the lack of additional time points to provide both replication and evaluation of stability versus change in protein levels, neurobehavioral functions, and their inter-relationship. Furthermore, lack of neural systems measures leaves an important gap for evaluating how protein measures might influence neural structure and function to produce neurobehavioral effects. Adding intermediate neural system measures that can be readily acquired in all PTEN patients, such as resting electroencephalogram or event-related potential paradigms, would provide key information between molecular assays and neurobehavioral outcomes and could clarify the full picture of how PTEN mutations result in widely disparate phenotypic outcomes.
Samples sizes were also insufficient to evaluate the functional impact of missense mutations in PTEN patients [51]. Future work in larger samples, computing fitness [48] and abundance [49] scores and identifying candidate dominant negative mutations, will be crucial to understanding whether different mutations influence peripheral protein levels and/or neurobehavioral outcomes.
Conclusions
The present study identified several protein-level group differences and relationships between PTEN pathway proteins and neurobehavioral functions that clarify how germline heterozygous PTEN mutations may impact neurobehavioral function. If replicated, these protein changes could not only improve understanding of individual differences in neurodevelopment within PTEN mutation cases but could also serve as peripheral markers of key neurobehavioral outcomes.
Availability of data and materials
The data generated and/or analyzed for the current study are not publicly available due to an embargo period, but, when this period has terminated, data will be made available by the RDCRN upon reasonable request.
Abbreviations
- ASD:
-
Autism spectrum disorder
- DSM-5:
-
Diagnostic and Statistical Manual of Mental Disorders—Fifth Edition
- IQ:
-
Intelligence quotient/global cognitive ability
- Macro-ASD:
-
Patients with macrocephalic autism spectrum disorder without PTEN mutations
- PTEN-ASD:
-
Patients with PTEN mutations and autism spectrum disorder
- PTEN no-ASD:
-
Patients with PTEN mutations without autism spectrum disorder
- PVDF:
-
Polyvinylidene difluoride
References
Yehia L, Ngeow J, Eng C. PTEN-opathies: from biological insights to evidence-based precision medicine. J Clin Invest. 2019;129(2):452–64.
van Diepen MT, Eickholt BJ. Function of PTEN during the formation and maintenance of neuronal circuits in the brain. Dev Neurosci. 2008;30(1–3):59–64.
Delatycki MB, Danks A, Churchyard A, Zhou X-P, Eng C. De novo germline PTEN mutation in a man with Lhermitte–Duclos disease which arose on the paternal chromosome and was transmitted to his child with polydactyly and Wormian bones. J Med Genet. 2003;40:e92.
Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22(3):183–98.
Buxbaum JDC, Cai G, Chaste P, Nygren G, Goldsmith J, Reichert J, Anckarsäter H, et al. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet Part B N(europsychiatric Genet). 2007;144:484–91.
Butler MG, Dasouki MJ, Zhou X-P, Talebizadeh Z, Brown M, Takahashi TN, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–21.
Klein S, Sharifi-Hannauer P, Martinez-Agosto JA. Macrocephaly as a clinical indicator of genetic subtypes in autism. Autism Res. 2013;6(1):51–6.
McBride KL, Varga EA, Pastore MT, Prior TW, Manickam K, Atkin JF, et al. Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 2010;3(3):137–41.
Orrico A, Galli L, Buoni S, Orsi A, Vonella G, Sorrentino V. Novel PTEN mutations in neurodevelopmental disorders and macrocephaly. Clin Genet. 2009;75(2):195–8.
Varga EA, Pastore M, Prior T, Herman GE, McBride KL. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet Med. 2009;11(2):111–7.
Hobert JA, Embacher R, Mester JL, Frazier TW, Eng C. Biochemical screening and PTEN mutation analysis in individuals with autism spectrum disorders and macrocephaly. Eur J Hum Genet. 2014;22(2):273–6.
Herman GE, Butter E, Enrile B, Pastore M, Prior TW, Sommer A. Increasing knowledge of PTEN germline mutations: two additional patients with autism and macrocephaly. Am J Med Genet. 2007;143A:589–93.
Tilot AK, Frazier TW 2nd, Eng C. Balancing proliferation and connectivity in PTEN-associated autism spectrum disorder. Neurotherapeutics. 2015;12(3):609–19.
Yuen RK, Merico D, Bookman M, Howe JL, Thiruvahindrapuram B, Patel RV, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. 2017;20(4):602–11.
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–21.
O’Roak BJ, Stessman HA, Boyle EA, Witherspoon KT, Martin B, Lee C, et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun. 2014;5:5595.
Trost B, Engchuan W, Nguyen CM, Thiruvahindrapuram B, Dolzhenko E, Backstrom I, et al. Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature. 2020;586(7827):80–6.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485(7397):237–41.
Tammimies K, Marshall CR, Walker S, Kaur G, Thiruvahindrapuram B, Lionel AC, et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA. 2015;314(9):895–903.
Schaaf CP, Betancur C, Yuen RKC, Parr JR, Skuse DH, Gallagher L, et al. A framework for an evidence-based gene list relevant to autism spectrum disorder. Nat Rev Genet. 2020;21(6):367–76.
Hansen-Kiss E, Beinkampen S, Adler B, Frazier T, Prior T, Erdman S, et al. A retrospective chart review of the features of PTEN hamartoma tumour syndrome in children. J Med Genet. 2017;54(7):471–8.
Ciaccio C, Saletti V, D’Arrigo S, Esposito S, Alfei E, Moroni I, et al. Clinical spectrum of PTEN mutation in pediatric patients. A bicenter experience. Eur J Med Genet. 2018;62:103596.
Amiri A, Cho W, Zhou J, Birnbaum SG, Sinton CM, McKay RM, et al. Pten deletion in adult hippocampal neural stem/progenitor cells causes cellular abnormalities and alters neurogenesis. J Neurosci. 2012;32(17):5880–90.
Fraser MM, Zhu X, Kwon CH, Uhlmann EJ, Gutmann DH, Baker SJ. Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res. 2004;64(21):7773–9.
Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, et al. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–88.
Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, et al. Pten regulates neuronal soma size: a mouse model of Lhermitte–Duclos disease. Nat Genet. 2001;29(4):404–11.
Page DT, Kuti OJ, Prestia C, Sur M. Haploinsufficiency for Pten and Serotonin transporter cooperatively influences brain size and social behavior. Proc Natl Acad Sci USA. 2009;106(6):1989–94.
Tilot AK, Bebek G, Niazi F, Altemus JB, Romigh T, Frazier TW, et al. Neural transcriptome of constitutional Pten dysfunction in mice and its relevance to human idiopathic autism spectrum disorder. Mol Psychiatry. 2016;21:118–25.
Tilot AK, Gaugler MK, Yu Q, Romigh T, Yu W, Miller RH, et al. Germline disruption of Pten localization causes enhanced sex-dependent social motivation and increased glial production. Hum Mol Genet. 2014;23(12):3212–27.
Frazier TW, Hardan AY. A meta-analysis of the corpus callosum in autism. Biol Psychiatry. 2009;66(10):935–41.
Just MA, Cherkassky VL, Keller TA, Kana RK, Minshew NJ. Functional and anatomical cortical underconnectivity in autism: evidence from an FMRI study of an executive function task and corpus callosum morphometry. Cereb Cortex. 2007;17(4):951–61.
Fishman I, Keown CL, Lincoln AJ, Pineda JA, Muller RA. Atypical cross talk between mentalizing and mirror neuron networks in autism spectrum disorder. JAMA Psychiatry. 2014;71(7):751–60.
Frazier TW, Embacher R, Tilot AK, Koenig K, Mester J, Eng C. Molecular and phenotypic abnormalities in individuals with germline heterozygous PTEN mutations and autism. Mol Psychiatry. 2015;20(9):1132–8.
Busch RM, Chapin JS, Mester J, Ferguson L, Haut JS, Frazier TW, et al. Cognitive characteristics of PTEN hamartoma tumor syndromes. Genet Med. 2013;15(7):548–53.
Busch RM, Srivastava S, Hogue O, Frazier TW, Klaas P, Hardan A, et al. Neurobehavioral phenotype of autism spectrum disorder associated with germline heterozygous mutations in PTEN. Transl Psychiatry. 2019;9(1):253.
Muhle RA, Reed HE, Stratigos KA, Veenstra-VanderWeele J. The emerging clinical neuroscience of autism spectrum disorder: a review. JAMA Psychiatry. 2018;75(5):514–23.
Hoang N, Cytrynbaum C, Scherer SW. Communicating complex genomic information: a counselling approach derived from research experience with autism spectrum disorder. Patient Educ Couns. 2018;101(2):352–61.
Tan MH, Mester J, Peterson C, Yang Y, Chen JL, Rybicki LA, et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet. 2012;88(1):42–56.
Jaini R, Loya MG, King AT, Thacker S, Sarn NB, Yu Q, et al. Germline PTEN mutations are associated with a skewed peripheral immune repertoire in humans and mice. Hum Mol Genet. 2020;29(14):2353–64.
Sarn N, Jaini R, Thacker S, Lee H, Dutta R, Eng C. Cytoplasmic-predominant Pten increases microglial activation and synaptic pruning in a murine model with autism-like phenotype. Mol Psychiatry. 2020. https://doi.org/10.1038/s41380-020-0681-0.
Little RJ, Rubin DB. Statistical analysis with missing data. 2nd ed. New York: Wiley; 2002.
Schafer JL, Graham JW. Missing data: our view of the state of the art. Psychol Methods. 2002;7(2):147–77.
Bhaskaran K, Smeeth L. What is the difference between missing completely at random and missing at random? Int J Epidemiol. 2014;43(4):1336–9.
Muthén BO, Muthén LK, Asparouhov T. Regression and mediation analysis using MPlus. Los Angeles: Muthén & Muthén; 2016.
Corp IBM. IBM SPSS Statistics for Windows. 260th ed. Armonk: IBM Corp; 2018.
Muthén LK, Muthén BO. Mplus User’s Guide. 7th ed. Los Angeles: Muthén & Muthén; 1998.
Zablotsky B, Black LI, Maenner MJ, Schieve LA, Danielson ML, Bitsko RH, et al. Prevalence and trends of developmental disabilities among children in the united states: 2009–2017. Pediatrics. 2019;144(4):e20190811.
Mighell TL, Evans-Dutson S, O’Roak BJ. A saturation mutagenesis approach to understanding PTEN lipid phosphatase activity and genotype-phenotype relationships. Am J Hum Genet. 2018;102(5):943–55.
Matreyek KA, Starita LM, Stephany JJ, Martin B, Chiasson MA, Gray VE, et al. Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat Genet. 2018;50(6):874–82.
Papa A, Wan L, Bonora M, Salmena L, Song MS, Hobbs RM, et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell. 2014;157(3):595–610.
Post KL, Belmadani M, Ganguly P, Meili F, Dingwall R, McDiarmid TA, et al. Multi-model functionalization of disease-associated PTEN missense mutations identifies multiple molecular mechanisms underlying protein dysfunction. Nat Commun. 2020;11(1):2073.
Ganesan H, Balasubramanian V, Iyer M, Venugopal A, Subramaniam MD, Cho SG, et al. mTOR signalling pathway—a root cause for idiopathic autism? BMB Rep. 2019;52(7):424–33.
Yehia L, Seyfi M, Niestroj LM, Padmanabhan R, Ni Y, Frazier TW, et al. Copy number variation and clinical outcomes in patients with germline PTEN mutations. JAMA Netw Open. 2020;3(1):e1920415.
Yehia L, Ni Y, Feng F, Seyfi M, Sadler T, Frazier TW, et al. Distinct alterations in tricarboxylic acid cycle metabolites associate with cancer and autism phenotypes in cowden syndrome and Bannayan–Riley–Ruvalcaba syndrome. Am J Hum Genet. 2019;105(4):813–21.
Kern JK, Jones AM. Evidence of toxicity, oxidative stress, and neuronal insult in autism. J Toxicol Environ Health Part B. 2006;9(6):485–99.
Abruzzo PM, Matte A, Bolotta A, Federti E, Ghezzo A, Guarnieri T, et al. Plasma peroxiredoxin changes and inflammatory cytokines support the involvement of neuro-inflammation and oxidative stress in Autism Spectrum Disorder. J Transl Med. 2019;17(1):332.
Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry. 2012;17(3):290–314.
Weissman JR, Kelley RI, Bauman ML, Cohen BH, Murray KF, Mitchell RL, et al. Mitochondrial disease in autism spectrum disorder patients: a cohort analysis. PLoS ONE. 2008;3(11):e3815.
Yenkoyan K, Harutyunyan H, Harutyunyan A. A certain role of SOD/CAT imbalance in pathogenesis of autism spectrum disorders. Free Radic Biol Med. 2018;123:85–95.
Smith AM, Natowicz MR, Braas D, Ludwig MA, Ney DM, Donley ELR, et al. A metabolomics approach to screening for autism risk in the children’s autism metabolome project. Autism Res. 2020;13(8):1270–85.
Lustgarten MS, Jang YC, Liu Y, Qi W, Qin Y, Dahia PL, et al. MnSOD deficiency results in elevated oxidative stress and decreased mitochondrial function but does not lead to muscle atrophy during aging. Aging Cell. 2011;10(3):493–505.
Larson JC, Mostofsky SH. Motor deficits in autism. In: Tuchman R, Rapin I, editors. Autism: a neurological disorder of early brain development. London: Mac Keith Press; 2006.
Dewey D, Cantell M, Crawford SG. Motor and gestural performance in children with autism spectrum disorders, developmental coordination disorder, and/or attention deficit hyperactivity disorder. J Int Neuropsychol Soc. 2007;13(2):246–56.
Licari MK, Alvares GA, Varcin K, Evans KL, Cleary D, Reid SL, et al. Prevalence of motor difficulties in autism spectrum disorder: analysis of a population-based cohort. Autism Res. 2020;13(2):298–306.
Kelleher RJ 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135(3):401–6.
Hooshmandi M, Wong C, Khoutorsky A. Dysregulation of translational control signaling in autism spectrum disorders. Cell Signal. 2020;75:109746.
Hazlett HC, Gu H, Munsell BC, Kim SH, Styner M, Wolff JJ, et al. Early brain development in infants at high risk for autism spectrum disorder. Nature. 2017;542(7641):348–51.
Piven J, Elison JT, Zylka MJ. Toward a conceptual framework for early brain and behavior development in autism. Mol Psychiatry. 2018;23(1):165.
Lainhart JE. Increased rate of head growth during infancy in autism. J Am Med Assoc. 2003;290:393–4.
Lainhart JE, Bigler ED, Bocian M, Coon H, Dinh E, Dawson G, et al. Head circumference and height in autism: a study by the collaborative program of excellence in autism. Am J Med Genet A. 2006;140(21):2257–74.
Winden KD, Ebrahimi-Fakhari D, Sahin M. Abnormal mTOR activation in autism. Annu Rev Neurosci. 2018;41:1–23.
Zhang J, Gao Z, Ye J. Phosphorylation and degradation of S6K1 (p70S6K1) in response to persistent JNK1 activation. Biochim Biophys Acta. 2013;1832(12):1980–8.
Acknowledgements
We are sincerely indebted to the generosity of the families and patients in PTEN clinics across the USA who contributed their time and effort to this study. We would also like to thank the PTEN Hamartoma Syndrome Foundation and the PTEN Research Foundation for their continued support of PTEN research.
We would also like to acknowledge the other members of the Developmental Synaptopathies Consortium supporting Project 2, including: Simon K. Warfield, PhD, Department of Radiology, Boston Children’s Hospital; Benoit Scherrer, PhD, Department of Radiology, Boston Children’s Hospital; Kira Dies, ScM, CGC, Department of Neurology, Boston Children’s Hospital; Rajna Filip-Dhima, MS, Department of Neurology, Boston Children’s Hospital; Amanda Gulsrud, PhD, UCLA Semel Institute for Neuroscience & Human Behavior, David Geffen School of Medicine at UCLA; Ellen Hanson, PhD, Department of Developmental Medicine, Boston Children’s Hospital; and Jennifer M. Phillips, PhD, Department of Psychiatry, Stanford University.
Funding
This study was funded, in part, by the National Institute of Neurological Disorders and Stroke (U54NS092090; PI/Network Director: Sahin; Project 2 Leaders/PIs: Eng & Busch) and the Ambrose Monell Foundation (to CE). The Developmental Synaptopathies Consortium (U54NS092090) is part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS). Research reported in this publication was also supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD) and the National Institute of Mental Health (NIMH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health (NIH). CE is the Sondra J. and Stephen R. Hardis Endowed Chair of Cancer Genomic Medicine at the Cleveland Clinic and an ACS Clinical Research Professor. MS is the Rosamund Stone Zander Chair at Boston Children’s Hospital. TF’s participation in this project was funded, in part, by Autism Speaks and the Hartwell Foundation.
Author information
Authors and Affiliations
Consortia
Contributions
TWF, RMB, and CE conceptualized and designed the study. All authors participated in data collection. TWF conducted the statistical analyses. RJ, MW, and CE interpreted the experimental data. TWF, RMB, AYH, JAMA, MS, and CE made substantial contributions to the interpretation of the overall data. TWF drafted the manuscript, CE critically revised the manuscript, and TWF finalized the manuscript. All authors substantively contributed. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the local institutional review boards of the participating centers, and written informed consent was obtained from all participants or their legal guardians with assent obtained from participants age < 18 years who were deemed able to provide assent.
Consent for publication
Not applicable.
Competing interests
TWF has received funding or research support from, acted as a consultant to, received travel support from, and/or received a speaker’s honorarium from Quadrant Biosciences, Impel NeuroPharma, F. Hoffmann-La Roche AG Pharmaceuticals, the Cole Family Research Fund, Simons Foundation, Ingalls Foundation, Forest Laboratories, Ecoeos, IntegraGen, Kugona LLC, Shire Development, Bristol-Myers Squibb, Roche Pharma, National Institutes of Health, and the Brain and Behavior Research Foundation and has an investor stake in Autism EYES LLC. JAMA has no competing interests. MS reports grant support from Novartis, Roche, Pfizer, Ipsen, LAM Therapeutics, Astellas, Aucta, Bridgebio, and Quadrant Biosciences. He has served on Scientific Advisory Boards for Sage, Roche, Celgene, Aeovian, Regenxbio, and Takeda.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Frazier, T.W., Jaini, R., Busch, R.M. et al. Cross-level analysis of molecular and neurobehavioral function in a prospective series of patients with germline heterozygous PTEN mutations with and without autism. Molecular Autism 12, 5 (2021). https://doi.org/10.1186/s13229-020-00406-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13229-020-00406-6