
Abstract
Background
Neurogenetics investigations and diagnostic yield in patients with autism spectrum disorder (ASD) have significantly improved over the last few years. Yet, many patients still fail to be systematically investigated.
Methods
To improve access to services, an ambulatory team has been established since 1998, delivering on-site clinical genetics consultations and gradually upgrading services to 502 children and young adults with ASD in their standard environment across 26 day-care hospitals and specialized institutions within the Greater Paris region. The evaluation included a clinical genetics consultation, screening for fragile X syndrome, metabolic workup, chromosomal microarray analysis, and, in a proportion of patients, next-generation sequencing of genes reported in ASD and other neurodevelopmental disorders.
Results
Fragile X syndrome and pathogenic copy number variants (CNVs) accounted for the disease in 10% of cases, including 4/312 (1.3%) with fragile X syndrome and 34/388 (8.8%) with pathogenic CNVs (19 de novo and 4 inherited). Importantly, adding high-throughput resequencing of reported intellectual disability/ASD genes to the screening procedure had a major impact on diagnostic yield in the 141 patients examined most recently. Pathogenic or likely pathogenic sequence variants in 27 disease genes were identified in 33/141 patients (23.4%; 23 were de novo and 10 inherited, including five X-linked and five recessive compound heterozygous variants). Diagnosed cases presented atypical and/or syndromic ASD with moderate to severe intellectual disability. The diagnostic yield of fragile X syndrome and array CGH testing combined with next-generation sequencing was significantly higher than fragile X syndrome and array CGH alone (p value 0.009). No inborn errors of metabolism were detected with the metabolic screening.
Conclusion
Based on the diagnostic rate observed in this cohort, we suggest that a stepwise procedure be considered, first screening pathogenic CNVs and a limited number of disease genes in a much larger number of patients, especially those with syndromic ASD and intellectual disability.
Similar content being viewed by others

Background
Autism spectrum disorder (ASD) is a major health care issue, affecting 1/200 live births, with a male to female ratio of 4/1 [1, 2]. In the last few years, important advances in deciphering the neurogenetic bases of ASD have been achieved [1, 2]. However, many patients still fail to be offered systematic investigations. In order to improve patients’ access to services, disseminate knowledge, and counteract the loss of opportunity to diagnose a genetic condition, an ambulatory team was established and has visited day-care hospitals across the Greater Paris region since 1998. The team offered comprehensive clinical genetics consultations and gradually improved genetics services to ASD patients in their standard environment.
Here, we show that high-throughput resequencing of reported disease genes had a major impact on diagnostic yield. As cost and access to genomic facilities are common issues, we suggest that a stepwise procedure be considered, first screening a limited number of disease genes in a much larger number of individuals, especially those with syndromic ASD and intellectual disability. Moreover, owing to constraints imposed by the special needs of those patients, we suggest that this flexible method of on-site genetics services be considered, to implement improved standard of care, navigate referrals, and counteract the loss of opportunity to diagnose a genetic condition in patients with ASD and their relatives.
Patients and methods
The ambulatory team is based in the medical genetics clinic of the Necker-Enfants Malades University hospital, in Paris. Initially, it included one clinical geneticist (0.2 full-time equivalent, FTE), one case manager (0.5 FTE), and one clinical psychologist (0.5 FTE). The team has grown over time and now includes six members: two clinical geneticists (1.4 FTE), two FTE case managers, one FTE genetic counselor, and one FTE neuropsychologist. The institutions that were visited include 26 day-care hospitals and special schooling medical institutions, under the authority of the Greater Paris Regional Health Agency. Initiated in 1998, the program is still ongoing and will continue to serve the community with a multi-annual budget of the Greater Paris Regional Health Agency.
All institutions were visited upon request. They were founded in the mid-1960s at the initiative of parents and family support groups eager to prevent the psychiatric hospitalization of children. Parents’ consent, and when possible, patient’s consent, was obtained prior to the consultation in accordance with French legislation. All patients diagnosed with ASD based on Diagnostic and Statistical Manual of Mental Disorders criteria [3] were offered a consultation. Standardized clinical assessment supported multidimensional symptoms (Childhood Autism Rating Scale, Autism Diagnostic Observation Schedule, and/or Autism Diagnostic Interview-Revised). Global cognitive testing indicated cognitive dysfunctions (especially attention and/or visual-spatial impairments) and various degrees of intellectual disability in all patients. Local child psychiatrists attended the medical genetics consultation. For the sake of privacy, a confidential consultation with the clinical geneticist was offered to the family and seldom accepted. Consultations reviewed personal and family history, pedigree, and photo albums and included a complete clinical examination of the child in the presence of a local team member. The procedure addressed the following questions: (i) is ASD isolated or part of a recognizable syndrome? (ii) is the case sporadic or familial? (iii) with or without intellectual disability? and (iv) are there risk factors? (paternal age, in vitro fertilization, prematurity, drug intake during pregnancy). Owing to rapid changes in the field, a minority of medical genetics records were considered up-to-date. None of these patients has been reported previously.
Ambulatory workup included (i) screening for FMR1 expansion, (ii) metabolic workup (amino acid and organic acid chromatography, succinyl purines, sialo transferrin, creatine synthesis intermediates), and (iii) array comparative genomic hybridization (CGH), replacing high-resolution karyotype from 2005 onwards [4]. Agilent 60 K microarrays (Agilent Technologies, Santa Clara, CA) were used for genomic copy number analyses on blood samples. Chromosomal rearrangements were confirmed by fluorescence in situ hybridization (FISH). The pathogenicity of copy number variants (CNVs) was assessed according to the guidelines of the American College of Medical Genetics [5]. When this first series of tests were negative, they were followed by a brain magnetic resonance imaging (MRI) in 347/502 patients, with nuclear magnetic resonance (NMR) spectroscopy and a computerized tomography (CT) scan upon short sedation, and electroencephalography (EEG).
From 2014, high-throughput next-generation sequencing (NGS) of intellectual disability/ASD genes was performed in a proportion of families (both parents and the child) [6]. The NGS panel used in this study was an in-house, non-commercial service panel designed at the Imagine Institute in Paris, and was based on a sequence capture method (Agilent Technologies). It screened a total of 439 genes, known to be implicated in intellectual disability/ASD or candidate genes reported at least twice in two distinct studies (Additional file 1: Table S1). Genomic DNA was extracted from peripheral blood using standard procedures. Agilent Sure Select libraries were prepared from 2 μg of genomic DNA sheared with a Covaris S2 Ultrasonicator. Regions of interest were captured with the corresponding 120-bp cRNA baits using the SureSelectXT Target Enrichment Reagent (Agilent) and the Ovation® Target Capture Module (NuGen). The targeted region was sequenced on an Illumina HiSeq2500 (Illumina Inc., San Diego, CA) generating 2 × 130 paired-end reads. Paired-end sequence datasets from Illumina HiSeq2500 runs were treated following three main steps: alignment against human genome release hg19 (using Bwa), calling of single nucleotide variants and small indels (using SAMtools, GATK, and Varscan), and variant annotation based on Ensembl human database (GRCh37 release). Data was integrated in pipelines enabling a CNV analysis based on a double normalization of depth coverage.
A minority of patients benefited from different gene panels or whole exome sequencing as part of research projects. Sequence variants were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines [7]. Variants were confirmed by Sanger sequencing and segregation analysis was performed in the families for which parental DNA was available. Paternity and maternity were confirmed in all patients carrying a de novo variant. Results and conclusions were communicated to patients and families during subsequent on-site multi-disciplinary consultations. Variants of uncertain significance (VOUS) were not reported to the parents [8,9,10,11].
Statistical analyses were performed using the non-parametric Fisher’s exact test (null hypothesis: no percentage difference between the pre-NGS versus NGS tested patients; two-sided test) [12,13,14].
Results
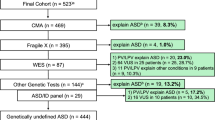
A total of 502 patients from 26 institutions were included in the program. There were 351 males and 151 females; most patients were unrelated, except for nine families with two affected siblings, and one family with three affected siblings. The distribution of patients by age categories was as follows: < 10 years, 34; 11–20 years, 194; 21–30 years, 211; > 30 years, 63. The majority of parents were positive about the on-site medical genetics consultations. Less than 1% of families declined the offer to participate, arguing that no immediate benefit would follow. Meeting with families and drawing the pedigree occasionally recognized X-linked or autosomal recessive forms of ASD. Frequently, significant and previously unreported information was first disclosed to the clinical geneticist (family history, affected relatives, serious medical events during pregnancy, in vitro fertilization, etc.). Brain MRI detected overt, yet nonspecific, isolated, or combined anomalies in 146/347 (42%) patients [15], including (i) punctuated white matter hyper intensities, (ii) impaired gray/white matter differentiation of temporal horns, and (iii) dilation of the Virchow-Robin spaces (Table 1).
On-site visits allowed the review of laboratory investigations including screening for fragile X syndrome, metabolic workup, and cytogenetic analyses. No screening for fragile X syndrome was available in 312/502 patients. The previously untested patients were screened and 1.3% (4/312) were found to be positive for fragile X syndrome. Systematic metabolic workup made a marginal contribution, especially as neonatal screening for phenylketonuria, and hypothyroidism is widespread in France.
Systematic array CGH replaced high-resolution karyotype from 2005 onwards and was performed in 388/502 patients. Pathogenic CNVs were found in 8.8% (34/388) of cases, including 19 de novo, 4 inherited, and 11 of unknown inheritance (adopted child, parent deceased, or unavailable) (Table 2). Most diagnosed patients presented atypical and/or syndromic ASD with moderate to severe intellectual disability.
Of our 502 patients, 193 were seen prior to and 309 after inception of NGS in 2014. Among the 309 patients seen from 2014 onwards, a pathogenic CNV was found in 18/309. Owing to funding restriction, only a fraction of patients without pathogenic CNVs (141/291) had NGS. NGS consisted in either panel resequencing of 439 known intellectual disability/ASD genes or candidate genes (Additional file 1: Table S1) or different gene panels in 116/141 patients, or whole exome sequencing in 25/141. A pathogenic or likely pathogenic variant was identified in 23.4% (33/141) of cases (Table 3). Most variants occurred de novo (23/33, 70%), while X-linked inheritance accounted for 5/33 (15%) cases and compound recessive heterozygosity for 5/33 (15%) cases. A total of 27 different disease genes were found to be mutated in our series. All diagnosed cases were syndromic forms of ASD, with moderate to severe intellectual disability. VOUS were identified in 23.4% (33/141) cases (data not shown) and were not reported to parents.
Overall, on-site medical genetics consultations in specialized institutions identified previously undiagnosed genetic conditions in 71 ASD children and young adults and the implementation of NGS significantly improved diagnostic yield. The difference in diagnostic yield of array CGH and fragile X syndrome testing either alone or combined with NGS was strongly significant (Fisher’s exact test, p value 0.00998).
Parents frequently mentioned that putting a name on the disease mechanism was not perceived as a “stigmatization,” but instead, they described it as a “relief” that helped them understand and overcome hardships and a connection to family support groups and other families facing similar situations.
Occasionally, couples reported that genetic counseling arrived too late, when they already had a second affected child (or relative) or had given up the idea of having another child. Local team members frequently considered identifying the disease mechanism as an opportunity to improve specific management and gain access to relevant literature and future clinical trials. When the procedure proved unsuccessful, on-site follow-up appointments were offered to families and possible inclusion in research programs was discussed (whole exome and whole genome sequencing).
Discussion
Taking advantage of on-site medical genetics consultations, we estimated the impact of systematic resequencing of reported disease genes on the diagnostic rate in day-care hospitals and special schooling institutions within the Greater Paris area. While array CGH and screening for fragile X syndrome detected pathogenic variants in 10% of patients, further implementation of high-throughput sequencing of intellectual disability/ASD genes identified pathogenic or likely pathogenic variants in 23.5% of investigated patients. Most variants occurred de novo and only 27 genes were found to be mutated in our series [7, 8]. All diagnosed cases were syndromic forms of ASD, with moderate to severe intellectual disability. Some patients had undiagnosed early-onset, transient epilepsy, later ascribed to a genetic condition when deferred behavioral problems occurred. Overall, on-site medical genetics consultations identified previously undiagnosed genetic conditions in 71 ASD children and young adults. This diagnostic yield may be an under-estimate, given that variants of uncertain significance and variants in strong candidate genes were not regarded as the cause of the disease. With the rapid pace of gene discovery in intellectual disability and ASD, some of these uncertain findings will likely be reclassified as pathogenic over time.
Based on this study, we suggest offering systematic array CGH and panel resequencing of known disease genes in syndromic/atypical ASD individuals with an associated intellectual disability. Moreover, we suggest that a stepwise procedure be considered, first screening a limited number of disease genes in a much larger number of patients, especially those with syndromic ASD and intellectual disability. In the future, current guidelines will hopefully mention genetics screening of the most frequent ASD genes as an explicit recommendation to professionals, which is not currently the case [17, 18].
While recognizing a genetic condition had no immediate impact on the case management, this information was often received by parents as a “relief” that helped them overcome hardship and alleviate the sense of guilt and self-blame of having given birth to a child with ASD. Relating to support groups and other families facing similar situations was also appreciated, as it fostered studies aimed at delineating natural history and the long-term outcome of ASD sub-types. On-site consultations also helped offering actionable recommendations and cognitive/behavioral interventions [19, 20].
Conversely, on-site medical genetics consultations had a significant impact on genetic counseling especially when de novo sequence variants or CNVs were identified, as they significantly reduced recurrence risk to parents and relatives (with the reservation of low-recurrence risk germline mosaicism). Omitting or postponing medical genetics consultations and failing to warn of potential genetic risks may have serious consequences in inherited forms of ASD.
The reason why so many patients failed to be systematically investigated before our on-site consultations remains unclear. Possible explanations include a limited number of clinical experts, the congestion or inadequacy of outpatient hospital consultations for patients with special needs, and a lack of funding for genetics services. Furthermore, while parents usually accept being referred to a child neurologist, the perception of a genetics consultation is a much more sensitive issue at the early stages of the disease, i.e., when parents first meet with a child psychiatrist. In contrast, the opportunity to conduct or update the etiological investigations was accepted more easily later on, when there is no doubt regarding the ASD diagnosis, but there are still many remaining mechanism-related questions.
This study shows that ASD children and young adults admitted in specialized institutions within the Greater Paris area (and probably other regions of France as well) had limited access to genetic advances. Deferred, optional on-site interventions may help by offering specialized consultations and counteracting the loss of opportunity to diagnose a genetic condition for both patients and relatives. The fact that genetics services are underused by affected families is not specific to France; it is a major challenge worldwide [21]. For instance, a Spanish study exploring access to genetics services and parental perception of genetic risk in children with ASD revealed striking underuse of genetics services, with only 30% of families visiting a genetics service and 13% of patients undergoing the recommended genetic tests [22]. Similarly, a recent Taiwanese study revealed that two-thirds of parents with children with ASD had never heard about genetic testing for ASD, while the majority (71%) expressed an interest in learning more about such testing [23]. This lack of service provision significantly impacted family planning in both studies.
Limitations
It is worth noting that our study has several limitations. First, owing to the number of patients reported here, no details on the clinical assessment (CARS, ADOS, ADI-R) or the level of intellectual disability could be individually provided for patients carrying pathogenic CNVs or sequence variants. Second, patients seen by our ambulatory services in specialized institutions possibly differ from those who visit regular clinics, as the most severe cases are selected over time. It is likely that the less severely affected ones are not referred to the institutions visited, and that in these patients, the etiologic yield of the genetic explorations performed here could be lower. Third, the eponym of ASD actually comprises a variety of conditions, including a significant amount of overlooked genetic conditions (i.e., early-onset transient epileptic encephalopathy). Owing to difficulties accessing long-term medical/medico-social facilities for disabled children, recognition of “autistic features” in a disabled child might have channeled many patients towards these high-quality institutions. Finally, an obvious limitation stems from public funding restriction, as only a fraction of patients without pathogenic CNVs had NGS.
Conclusions
We suggest that on-site clinical genetics consultations be considered in day-care hospitals and specialized institutions, to implement a standard of care, navigate referrals, and counteract the loss of opportunity to diagnose a genetic condition in ASD patients. Particular attention should be paid to a stepwise procedure, first screening for pathogenic CNVs and sequence variants in frequently mutated genes in a much larger number of children with syndromic ASD and intellectual disability.
Availability of data and materials
Clinical information, brain MRIs, laboratory results, and panel data are available by mail to nicole.guyot-berard-ext@aphp.fr.
Abbreviations
- Array CGH:
-
Array comparative genomic hybridization
- ASD:
-
Autism spectrum disorder
- CNV:
-
Copy number variant
- CT scan:
-
Computerized tomography scan
- EEG:
-
Electroencephalography
- FMR1 :
-
Fragile X mental retardation 1
- MRI:
-
Magnetic resonance imaging
- NGS:
-
Next-generation sequencing
- NMR spectroscopy:
-
Nuclear magnetic resonance spectroscopy
- VOUS:
-
Variant of uncertain significance
References
Charman T. The new genetics of autism: a translational opportunity? Lancet Psychiatry. 2015;10:856–7.
Lai MC, Lombardo MV, Baron-Cohen S. Autism. Lancet. 2014;383:896–910.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th Edn. Washington, DC: American Psychiatric Association; 2013.
Leroy C, Jacquemont ML, Doray B. Xq25 duplication: the crucial role of the STAG2 gene in this novel human cohesinopathy. Clin Genet. 2016;89:68–73.
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST. Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 2011; 13:680–685.
Redin C, Gerard B, Lauer J. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J Med Genet. 2014;51:724–36.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17:405–424.
Yin J, Schaaf CP. Autism genetics - an overview. Prenat Diagn. 2017;37:14–30.
Morgan A, Gandin I, Belcaro C. Target sequencing approach intended to discover new mutations in non-syndromic intellectual disability. Mutat Res. 2015;781:32–6.
Grozeva D, Carss K, Spasic-Boskovic O. Targeted next-generation sequencing analysis of 1,000 individuals with intellectual disability. Hum Mutat. 2015;36:1197–204.
Martínez F, Caro-Llopis A, Roselló M. High diagnostic yield of syndromic intellectual disability by targeted next-generation sequencing. J Med Genet. 2017;54:87–92.
Chisq.test function {stats library}. R Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2018. https://www.R-project.org/.oci.
Agresti A. Categorical data analysis. New York: Wiley; 1990. p. 59–66.
Mehta CR, Patel NR. A network algorithm for performing Fisher’s exact test in r * contingency tables. J Am Stat Assoc. 1983;78:427–34.
Boddaert N, Zilbovicius M, Philippe A, Robel L, Bourgeois M, Barthélemy C, et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS One. 2009;4:e4415.
Hoyer J, Ekici AB, Endele S, Popp B, Zweier C, Wiesener A, et al. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet. 2012;90:565–72.
Barton KS, Tabor HK, Starks H, Garrison NA, Laurino M, Burke W. Pathways from autism spectrum disorder diagnosis to genetic testing. Genet Med. 2018;20:737–44.
Trouble du spectre de l’autisme. Signes d’alerte, repérage, diagnostic et évaluation chez l’enfant et l’adolescent. 2018. https://www.has-sante.fr/portail/jcms/c_468812/fr/recommandations-pour-la-pratique-professionnelle-du-diagnostic-de-l-autisme.
Vorstman JAS, Parr JR, Moreno-De-Luca D, Anney RJL. Autism genetics: opportunities and challenges for clinical translation. Nat Rev Genet. 2017;18:362–76.
Poisson A, Nicolas A, Cochat P. Behavioral disturbance and treatment strategies in Smith-Magenis syndrome. Orphanet J Rare Dis. 2015;10:111.
Lord C, Elsabbagh M, Baird G, Veenstra-Vanderweele J. Autism spectrum disorder. Lancet. 2018;392:508–20.
Codina-Solà M, Pérez-Jurado LA, Cusco I, Serra-Juhé C. Provision of genetic services for autism and its impact on Spanish families. J Autism Dev Disord. 2017;47:2947–56.
Chen LS, Min J, Zhao S, Yeh YC, Huang TY. Information needs in genetic testing: a needs assessment survey among Taiwanese parents of children with autism spectrum disorders. Autism. 2019;23:902–9.
Acknowledgements
We would like to thank Fondation Electricité de France for their generosity; Anne-Marie Armanteras de Saxcé, Christophe Devys (ARS Ile-de-France), and Francois Géraud (Fondation Elan Retrouvé) for their support over the last 20 years; Cécile Glaunec, Nicole Guyot-Bérard, and Sophie Martin for their invaluable contribution to implementing the program; and Roland Goepel, Jérôme Jeannette, and Pierre-Alexis Messarovitch for their assistance to child sedation prior to MRI and blood sampling. The ambulatory medical genetics team visited 26 centers within the Greater Paris region. Among them, 20 day-care hospitals and institutions provide care to children and adolescents (Françoise Grémy, Antony, Chevilly Larue, Fontenay aux Roses, André Boulloche, La Colline, Serge Lebovici; Alternance 92, Alternance 75, Cour de Venise, Villa d’Avray, Adam Shelton, du Breuil, Amalthée, Jeunes Appedia, Cognacq Jay, l’Arche à Paris, Centre Raphael, les Petites Victoires, Notre Ecole à Paris) and six focus on adult populations (Le Cèdre Bleu, Alternat, Centre Georges et Lili Garel, Simone Veil, Alain Raoul Mossé, and Les Petites Victoires).
Funding
ARS Ile-de-France, Fondation Elan Retrouvé, and Fondation Electricité de France.
Author information
Authors and Affiliations
Contributions
AM and MA initiated on-site genetic consultations in France. AM wrote the draft of the manuscript, CD critically reviewed the draft and LF and CD contributed to technical and neuropsychological aspects. NB performed brain MRIs and the other authors were investigators in day-care hospitals. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All patients or their legal guardians gave their consent for this program according to French legislation and Hôpital Necker-Enfants Malades procedures.
Consent for publication
All patients and their parents or legal guardians gave their written or oral agreement for publication according to French legislation.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. List of 439 known intellectual disability/ASD genes or candidate genes tested by panel resequencing. (XLSX 196 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Munnich, A., Demily, C., Frugère, L. et al. Impact of on-site clinical genetics consultations on diagnostic rate in children and young adults with autism spectrum disorder. Molecular Autism 10, 33 (2019). https://doi.org/10.1186/s13229-019-0284-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13229-019-0284-2