
Abstract
Background
The ABC model of flower development describes the molecular basis for specification of floral organ identity in model eudicots such as Arabidopsis and Antirrhinum. According to this model, expression of C-class genes is linked to stamen and gynoecium organ identity. The Zingiberales is an order of tropical monocots in which the evolution of floral morphology is characterized by a marked increase in petaloidy in the androecium. Petaloidy is a derived characteristic of the ginger families and seems to have arisen in the common ancestor of the ginger clade. We hypothesize that duplication of the C-class AGAMOUS (AG) gene followed by divergence of the duplicated AG copies during the diversification of the ginger clade lineages explains the evolution of petaloidy in the androecium. In order to address this hypothesis, we carried out phylogenetic analyses of the AG gene family across the Zingiberales and investigated patterns of gene expression within the androecium.
Results
Phylogenetic analysis supports a scenario in which Zingiberales-specific AG genes have undergone at least one round of duplication. Gene duplication was immediately followed by divergence of the retained copies. In particular, we detect positive selection in the third alpha-helix of the K domain of Zingiberales AGAMOUS copy 1 (ZinAG-1). A single fixed amino acid change is observed in ZinAG-1 within the ginger clade when compared to the banana grade. Expression analyses of AG and APETALA1/FRUITFULL (AP1/FUL) in Musa basjoo is similar to A- and C-class gene expressions in the Arabidopsis thaliana model, while Costus spicatus exhibits simultaneous expression of AG and AP1/FUL in most floral organs. We propose that this novel expression pattern could be correlated with the evolution of androecial petaloidy within the Zingiberales.
Conclusions
Our results present an intricate story in which duplication of the AG lineage has lead to the retention of at least two diverged Zingiberales-specific copies, ZinAG-1 and Zingiberales AGAMOUS copy 2 (ZinAG-2). Positive selection on ZinAG-1 residues suggests a mechanism by which AG gene divergence may explain observed morphological changes in Zingiberales flowers. Expression data provides preliminary support for the proposed mechanism, although further studies are required to fully test this hypothesis.
Similar content being viewed by others


Background
The genetic control of flower morphogenesis has long been studied in Arabidopsis thaliana and Antirrhinum majus [1]. Classically, floral organ specification has been described by combinatorial patterns of gene expression in what is well known as the ABC model of floral organ identity. In this model, specific domains of expression of A-, B-, and C-class MADS-box genes correlate with the position of the developing sepals (A-class genes only), petals (a combination of A- and B-class genes), stamens (a combination of B- and C-class genes), and gynoecium (C-class genes only): thus, gene expression is correlated with organ identity. In A. thaliana, there are two A-class genes (APETALA-1 (AP1), and APETALA-2 (AP2)), two B-class genes (APETALA-3 (AP3), PISTILLATA (PI)), and one C-class gene (AGAMOUS (AG)). With the exception of AP2, all other genetic components of the ABC model are type-II MIKCc MADS-box genes as determined by their arrangement of protein domains. Their proper function as transcriptional regulators is dependent on the protein-protein interactions that occur between the A-, B-, and C-class genes, as well as with the SEPALLATA genes [2], to form protein dimers and functional tetramers. The protein-level explanation for A-, B-, and C-class functions is known as the quartet model, and asserts that only in tetramers are the A-, B-, and C-class proteins capable of regulating downstream genes (for review, [3]).
Although the ABC model has proven fruitful in describing organ identity and floral organ development, it lacks a mechanism to explain how such robust gene expression patterns are established during development and how changes in expression, function, or copy number may correlate with evolutionary changes in organ morphology. In order to address this mechanism, the A-, B-, and C-class genes have been integrated into an elegant complex-system approach, capable of explaining the robustness of the ABC gene expression patterns during flower development [4,5]. By mapping the landscape of known gene interactions during floral development in A. thaliana, Mendoza and coworkers (1999) were able to recover the stable states (that is, attractors) that correspond to the gene expression patterns correlated to floral organ identity, as described by the ABC model. In doing so, the authors proposed a set of necessary and sufficient genes and genetic interactions that provide a dynamical and mechanistic explanation for the establishment of the ABC gene expression patterns [6,7].
Within the mapped gene interactions that constitute the floral organ identity gene regulatory network (FOS-GRN, Figure 1), AGAMOUS is one of the most highly interconnected genes, suggesting that alterations in this node are likely to constitute important changes in the stable states of the system, thereby functioning as a potential nexus for evolutionary change. In particular, AG may be a key regulator of androecial (stamen) morphology; stamen and petal stable gene expression patterns (also known as GRN “attractors”) in A. thaliana differ exclusively by opposite states of AP1 and AG expression in which AP1 is expressed in petals but not in stamens and AG is expressed in stamens but not in petals [8]. Thus, we propose that an understanding of the evolution of the AG family across Zingiberales, especially comparing the expression patterns of AG and determining its potential for interactions with AP1, can provide insight into the evolution of petaloidy in the stamen whorl.

Floral organ specification gene regulatory network (FOS-GRN), modified from Álvarez-Buylla, E.R et al. [6]). The circles (nodes) represent genes or proteins that are experimentally shown to participate in floral organ specification during flower morphogenesis in Arabidopsis thaliana. Arrows indicate positive interactions between nodes, while the T symbols indicate negative interactions between nodes. Fifteen genes are depicted: EMF1, LFY, AP2, WUS, AG, LUG, CLF, TFL1, PI, SEP, AP3, UFO, FUL, FT, and AP1. Direct interactions between AGAMOUS (AG) and other genes in the FOS-GRN are highlighted in black. Other interactions within the FOS-GRN not directly involving AG are depicted in gray, as well as genes that are not direct interactors with AG.
The AG gene was first isolated from A. thaliana over two decades ago [9], when fully penetrant mutations were shown to cause abnormalities in the development of the floral reproductive organs. AG has since been implicated in proper development of reproductive organ identity across flowering plants and is thought to play an additional role in ovule development and meristem determinacy in some lineages [10-12]. The evolution of the AG subfamily of transcription factors has been extensively studied across angiosperms. Phylogenetic analyses of the AG subfamily demonstrate that a duplication event occurring early during angiosperm diversification resulted in the origin of two major lineages: the AG and the AGAMOUS-like 11 (AGL11) lineages [13-16]. In the Arabidopsis canonical model, the AG and AGL11 lineages correspond functionally to C-class and D-class homeotic genes, respectively, in which C-class homeotic genes are involved in stamen and carpel identity and in floral meristem determinacy and D-class genes are more specifically involved in ovule and fruit development [14,12]. The AG lineage itself has undergone subsequent gene duplications, and parsing of function of duplicated copies is thought to have occurred independently in the major angiosperm lineages. Within the core eudicots, for example, the AG lineage is divided into the euAG and the PLENA (PLE) lineages [14,15] with various subclades within both lineages demonstrating neofunctionalization, subfunctionalization, redundancy, and loss of duplicated copies [17]. In Antirrhinum, the AG lineage genes PLE and FARINELLI (FAR) contribute unequally to specify male and female reproductive organs [18,19], while in Petunia FBP6 and PMADS3 act redundantly as C-function genes [20]. In Zea mays, however, the AG paralogs ZAG1 and ZMM2 appear to be expressed in spatially distinct domains of the developing flower, and may have subfunctionalized into carpel- and stamen-specific paralogs [21], while in Oryza sativa, AG paralogs OSMADS3 and OSMADS58 are essential for reproductive organ identity and together with AG11 lineage OSMADS13 are important for floral meristem determinacy [10,12,13].
In addition to its role in reproductive organ identity, differential expression of AG in A. thaliana has been shown to be involved in the development of petaloidy in the androecium. For instance, the ag-11 allele, bearing a single point mutation in the regulatory region of Arabidopsis AG, results in the transformation of stamens into petaloid organs [22]. Also, downregulation of AG by anti-sense RNA in A. thaliana leads to a variety of aberrant floral morphologies, including petaloid stamens [23]. Most recently, Tanaka et al. [24]) demonstrated the role of AG in regulating proper petal and stamen differentiation in cyclamen (Cyclamen persicum), where two paralogs (CpAG1 and CpAG2) are present and repression of CpAG2 leads to the formation of infertile and petaloid structures in the stamen whorl [24].
The molecular mechanisms underlying the evolution of androecial petaloidy in the angiosperms have not been studied in detail. It is likely that developmental processes underlying androecial petaloidy are homoplasious across flowering plants, as petal-like stamens have evolved independently in a variety of angiosperm lineages [25]. In male flowers of the early diverging Amborella trichopoda, stamen filaments are expanded into petal-like structures. In several other basal angiosperm and magnoliid lineages, flowers display a gradual transition between petal and stamen organs, with multiple degrees of androecial petaloidy present even within the same flower (for example, Nymphaea alba) [26]: A gradient of AG and B-class gene expression has been implicated in this gradual morphological transition between laminar petals and filamentous stamens [27].
In order to further explore the role of the AG gene in the evolution of androecial petaloidy across angiosperms, we focus our study on the Zingiberales, a group of monocots that exhibits extensive petaloidy in the androecial organs. Zingiberales are an order of tropical monocots comprising approximately 2,500 species. The order is divided into eight families, traditionally organized into the paraphyletic banana families (Musaceae, Lowiaceae, Strelitziaceae, and Heliconiaceae) and the derived ginger clade (Zingiberaceae, Costaceae, Cannaceae, and Marantaceae) ([28], Figure 2). In the Zingiberales, androecial petaloidy is an important component of floral morphological diversity: lineages of the ginger clade have a marked reduction in the number of fertile stamens. The staminodes (infertile stamens) develop as petal-like structures and usually constitute the bulk of floral display, with the petals of the same flower developing as relatively inconspicuous structures in comparison with the stamen whorl (Figure 2). In Costaceae and Zingiberaceae, 2 to 5 of the petaloid staminodes fuse together to form a novel organ, the labellum.

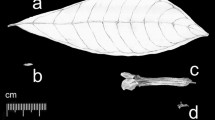
Phylogeny of Zingiberales with key events in androecial evolution. (A) Morphological character states of the androecium are mapped onto the most recent Zingiberales phylogeny [28]. The eight Zingiberales families are divided into two groups: the first diverging banana lineages (Heliconiaceae, Strelitziaceae, Musaceae, and Lowiaceae), and the derived ginger clade (Zingiberaceae, Costaceae, Marantaceae, and Cannaceae). Main changes in androecial morphology are depicted with numbers. (1) Reduction in the number of fertile stamens, from 5 to 6 fertile stamens in the banana lineages, to 1 fertile stamen in Zingiberaceae and Costaceae or ½ of a fertile stamen in Marantaceae and Cannaceae; (2) fusion of petaloid staminodes leading to the formation of the labellum. Five infertile stamens fuse in Costaceae, while 2 or 4 staminodes form the labellum in the Zingiberaceae; (3) laminar extension of the filament of the fertile stamen; (4) abortion of a theca of fertile stamen. (B) Costus sp. flowers. (La) labellum; Asterisk indicates the abaxial side of laminar connective of fertile stamen. (C) Canna indica one half fertile stamen. (Th) single theca; (Pa) petaloid appendage resulting from the laminar expansion of the filament [29].
Given the involvement of the AG gene lineage in reproductive organ development and its interconnectivity within the floral organ specification gene regulatory network (FOS-GRN), we hypothesize that gene duplication followed by potential functional divergence of the AG lineage in the ginger clade is correlated with the evolution of petaloidy in the androecium. In order to test our hypothesis, AG lineage genes were amplified from across the Zingiberales, and their expression was assessed during flower development in representative species. Tests of selection were carried out to investigate the role of selection on gene evolution and function. Our results suggest that positive selection has played a role in the evolution of AG across the Zingiberales order, particularly in protein divergence within the K domain. These protein modifications, together with comparative analyses of AG and AP1 expression across the order, suggest a mechanism by which androecial petaloidy may have evolved in the Zingiberales, and support the hypothesis that modifications in AG expression and function are correlated with androecial petaloidy.
Methods
Plant material, RNA extraction, and cDNA synthesis
Twenty species from seven of the eight Zingiberales families were sampled in order to represent the diversity of floral form observed in the order (Table 1). Fresh flowers were collected and stored in a homemade recipe equivalent of “RNA-later” for up to 2 weeks prior to RNA extraction. Total RNA was extracted from floral material using Plant RNA Extraction Reagent (Invitrogen, Carlsbad, CA, USA), according to Yockteng et al. [30]. RNA was stored at −80°C until further use. Prior to cDNA synthesis, RNA was treated with DNAse (Fermentas). cDNA synthesis was performed using iScript select cDNA synthesis Kit (Bio-Rad, Hercules, CA, USA) and polyT primers. Amplification of the ß-actin gene as a positive control for cDNA synthesis was performed using PCR primers (F: 5′ GGA CGA ACA ACT GGT ATC GTG CTG 3′ and R: 5′ GAT GGA TCC TCC AAT CCA GAC ACT GTA 3′) [31]. Reactions without reverse transcriptase (no-RT) were used as negative control.
Amplification of AGAMOUS genes in the Zingiberales
A multiple sequence alignment (MSA) for the AG gene lineage was generated from sequences downloaded from NCBI (Table 1). The MSA was used to design general primers for amplification of Zingiberales AG genes. Multiple primer combinations, with different degrees of degeneracy, were used in order to improve chances of assessing all copies of the AG gene lineage within the Zingiberales. Primer sequences were as follows: (i) forward primers: 5′ ACI AAY MGI CAR GTI ACI TTY TG 3′; 5′ ATG GSI MGI GGI AAR ATI SAR AT 3′; 5′ CAR GTK ACC TTC TGC AAG 3′; 5′ ATC CCA TGG AGC ATA AAG CA 3′; 5′ GRG GRA AGA TCG AGA TCA AG 3′; (ii) reverse primers: 5′ ACC CTA TCA GTC TCG GCG ATC TTG TTC C 3′; 5′ TCA TCG TTC AAC CAA AGT GG 3′; 5′ TTG MAK RAA GTT CCY TGA RTM RT 3′.
PCR reactions were carried out using Phire Hot Start II DNA Polymerase (Thermo Scientific) and 2 μl of 5X Phire buffer; 0.2 μl 10 mM dNTPs; 0.5 μl of each primer; 0.1 μl Phire Polymerase; 1 μl [1:10] cDNA; and ddH2O, for a total volume of 10 μl. Thermocycling conditions followed manufacturer’s recommendations. PCR products were visualized on a 1% agarose gel stained with GelRed™ (Phoenix Research Products) according to the manufacturer’s protocol. PCR products were cloned into Top10 cells and sequenced using Big Dye Terminator Kit v3.1 (Applied Biosystems) on a 3700 sequencer. At least 16 clones were sequenced for each of the species sampled. Over 40 clones were sequenced for Costus spicatus, in order to insure deep sampling of gene copy number in this species.
Phylogenetic analyses
A multiple sequence alignment was generated using MacClade (4.06 OS X) with all generated Zingiberales sequences aligned to outgroup sequences downloaded from NCBI (Table 1). Model selection for the final alignment was tested in jModeltest 0.1.1 [32] using the Bayesian information criterion (BIC). PartitionFinder [33] was used to test for the best partition scheme for the dataset and substitution model, also based on the BIC criterion. According to PartitionFinder as well as jModelTest, the best-fit model was K80 + G and no data partition was advised. The best-fit model was implemented in MrBayes 3.2 [34] and PhyML [35] in order to assess gene tree topology. MrBayes runs were implemented on the CIPRES Science Gateway (www.phylo.org) under the model specified above, as well as under variations of the best-fit model to ensure that topology was not influenced by model selection and ran for 1.5 M generations. Data were further analyzed for convergence with Tracer v1.5 (http://beast.bio.ed.ac.uk/). SumTrees v.3.0.0 using the DendroPy Phylogenetic Computing Library v3.7.1 [36] was used to calculate the burn-in and remove the appropriate trees saved prior to stationarity, and to assemble a 50% majority rule tree from the remaining trees. Maximum likelihood (ML) analyses were performed using PhyML implemented on the ATGC South of France bioinformatics platform (http://atgc.lirmm.fr/phyml/). Bootstrap support from 100 replicates and posterior probabilities (PP) were calculated for ML and MrBayes analyses, respectively, and are used as branch support in the gene tree (bootstrap/PP). In order to test the likelihood of different evolutionary scenarios for the AG gene tree, a Shimodaira-Hasegawa (SH) test [37] was performed by manually generating a constrained gene tree in which the two first branching lineages (all “ZinAG-1”) were forced to form a monophyletic group. The likelihood of the constrained tree was tested against the unconstrained gene tree obtained in our phylogenetic analysis using the likelihood ratio test (LRT). Likelihood tests for constrained topologies were obtained in PAUP* [38].
Selection tests
Both branch- and site-specific selection tests were performed in order to assess signals of selection across the AG subfamily, along branches leading to the major clades and at specific amino acid sites. Branch selection was assessed using PAML codeml branch models by setting the model = 2 in order to allow several omega (w) ratios compared to a fixed w, while site selection was evaluated using the site models M1a and M2a [39]. Site selection was also assessed using the fixed-effect likelihood (FEL) model in HyPhy 2.0 [40] under a stringent cut-off of 0.1, as suggested by the HyPhy program. A Bayesian 50% majority rule consensus tree was used for all selection analyses. For each node tested, a two-rate analysis was used to allow adjustment of the ratio of non-synonymous (dN) and synonymous (dS) substitutions across sites, and models determined by jModeltest were specified for the nucleotide model of evolution.
Gene expression of AGAMOUS and APETALA1 in Zingiberales developing flowers
RT-PCRs for ZinAG-1 and ZinAG-2 were carried out in all Musa basjoo floral organs. Primers were designed on intron-exon boundaries whenever possible, and sequences are as follows: MbAGcp1 forward 5′ TTG AAA GGT ATA AGA AAG CAT 3′; MbAGcp1 reverse 5′ TTA TTC TCG AGT TGC TTC ATG TCT 3′; MbAGcp2 forward 5′ TCG AGA GGT GGT ACA AGA AAG CAT GT 3′; MbAGcp2 reverse 5′ CGA GTC TCA AGC TGC TTC AG 3′. Reactions were carried out using Phire Hot Start II DNA Polymerase (Finnzymes, Finland) and the following protocol: 2 μl of 5X PHIRE buffer; 0.5 μl of each of the 10 mM primers; 0.1 μl of PHIRE polymerase; 0.2 μl of 50 mM dNTPs; 1 μl of [1:10] dilution of organ-specific cDNA; and water up to a 10 μl total volume, for 25 cycles. PCR reactions were performed on BioRad Thermocyclers and were visualized on 1% agarose gel, post-stained with Gel-Red™ (Biotium).
Expression of AGAMOUS and APETALA1 in C. spicatus and M. basjoo was assessed by generating organ-specific transcriptomes. cDNA libraries for sequencing on the Illumina platform were prepared using the TruSeq RNA sample prep kit v2. Two cDNA libraries each were prepared with 2.0 μg of RNA extracted from dissected tissue of the filament, theca, and free petal of M. basjoo and the petaloid filament, theca, petal, and labellum (fused petaloid staminodes) of C. spicatus. Libraries were multiplexed using barcoding set A. Samples were run on a HiSeq2000 at IIGB HT Sequencing Facility at the University of California, Riverside. Raw reads were trimmed to remove adapters and regions of poor quality with cutadapt [41]. Costus spicatus sequences were assembled into a reference transcriptome using Trinity [42] with minimum contig length of 300. All other parameters were used according to Trinity default settings. GMAP/GSNAP [43] was used to align M. basjoo trimmed reads to annotated CDS from the published Musa acuminata genome, while C. spicatus trimmed reads were aligned to the C. spicatus reference transcriptome. Expression of AG and AP1-like genes was estimated using eXpress [44] in units of FPKM (frequency per kilobase of exon per million aligned reads). Replicates were independently processed, and gene expression was compared between libraries for consistency. ACTIN1 expression was used to normalize targeted gene expression across transcriptome libraries. Error bars were calculated based on the standard deviation (SD) of the two normalized samples for each organ.
In order to confirm transcriptome expression data for C. spicatus AG and AP1-like genes, quantitative PCR was performed on each floral organ of C. spicatus flowers (sepals, petals, labellum, stamen, and gynoecium). A general C. spicatus AP1/FUL-like primer was designed, as at this point, we were not able to produce copy-specific primer pairs. qPCR was performed on C. spicatus floral organs using iQ™ SYBR® Green Supermix (Life Science Research), and following the manufacturer’s protocol. Clade-specific qPCR primers were designed based on the nucleotide differences between C. spicatus sequences from different clades on the AGAMOUS gene tree, and primer sequences are as follows: CsAGcp1 forward 5′ AAC AGC AGT GTG AGA GCG ACT 3′; CsAGcp1 reverse 5′ GGT CTC TAA GGC TCA TAG AAC CGA GA 3′; CsAGcp2 forward 5′ CCA ACA GTG TGA GAG CAA CAA 3′; CsAGcp2 reverse 5′ CTT CAT ATC GCG TAG GCT CA 3′; CsAP1/FUL-like forward 5′ ATA TCA GGT CAA GAA AGA ACC AAA TC 3′; CsAP1/FUL-like reverse 5′ GGG CTT GTT TGG ATT CGT T 3′. Two reactions were performed, one for ZinAG-1 and ZinAG-2, and another for AP1/FUL and ZinAG-1 with annealing temperatures of 60°C and 58°C, respectively. In both cases, three replicates per gene were performed and ACTIN1 was used as an internal control.
Results
Amplification and phylogenetic analyses
AG sequences were obtained for all families within the order, with the exception of Lowiaceae (Orchidantha), a monogeneric lineage. A multiple sequence alignment (MSA) of 558 bp was generated and encompasses all protein domains, with the exception of the first nine codons of the MADS domain and the end of the C-terminal domain, for which alignment to outgroup sequences became increasingly challenging. The final MSA comprises a total of 37 ingroup and 13 outgroup sequences. This MSA was used as the input to jModelTest, and determined the best-fit model as the K80 + G model. The best-fit model, as well as other more parameterized models (GTR, GTR + I, and GTR + I + G), was implemented in both MrBayes and PhyML.
Tree topology across methods and models was largely congruent (Figure 3). All AG sequences from Zingiberales form a monophyletic group with high support (76% bootstrap and posterior probability of 1). According to the species distribution on the gene tree, there are at least two copies of the AG gene in the Zingiberales, herein called ZinAG-1 and ZinAG-2 (Figure 3B). ZinAG-2 sequences form a monophyletic group, suggesting an orthologous relationship between copies found in the banana and ginger lineages, while ZinAG-1 sequences formed a grade at the base of the ZinAG-2 clade.

AGAMOUS ( AG ) gene tree for the Zingiberales. (A) Bayesian topology generated in MrBayes with 1.5 M generations, under the K80 + G model (best fit model according to jModelTest). The general tree topology agrees with results generated by maximum likelihood (ML) analysis (PhyML), as well as under different models for both Bayesian and ML analyses. Partition of the data set according to codon position rendered an unresolved tree with poor likelihood (data not shown). Only bootstrap support >50% are presented, followed by posterior probabilities (PP). At least two copies of the gene AG can be identified, according to the distribution of ginger clade and banana grade species in the gene tree. (B) Schematic representation of the AG gene tree. Black circles represent sequences from the ginger clade, while gray circles represent sequences from the banana grade. Each circle indicates the position of the corresponding sequences in the gene tree. This schematic tree depicts one clade (ZinAG-2) comprising ginger clade and banana grade sequences, and two early branching lineages comprising banana grade and ginger clade sequences (ZinAG-1).
According to the AG gene tree, several different evolutionary scenarios could account for the recovered topology for Zingiberales AG phylogeny (Figure 4A). Although ZinAG-1 sequences appear paraphyletic based on the recovered topologies, these sequences could result from a single duplication event predating the divergence of the Zingiberales that ultimately resulted in two clades: ZinAG-1 and ZinAG-2 (Figure 4A, scenario 1). Differential sequence divergence resulting from distinct evolutionary pressures on ZinAG-1 could result in an unresolved clade of copy 1 sequences, with phylogenetic analyses resolving a paraphyletic grade. In this case, the phylogenetic analysis would recover incongruence between the gene tree (placing the sequences as paraphyletic rather than within a single clade) and the organismal tree. Alternatively (Figure 4A, scenarios 2 and 3), a second duplication event may have occurred only in the Zingiberaceae lineage after it diverged from Costaceae, leading to a third lineage-specific AG copy (ZinAG-3) in the Zingiberaceae. This copy was retained while the paralogous duplicate was subsequently lost from the Zingiberaceae, yielding only two copies but with less clear orthology to the two copies found in the remaining Zingiberales lineages.

Evolution of the AGAMOUS gene lineage. (A) Potential AGAMOUS (AG) gene copy histories within the Zingiberales. Scenario 1 assumes one single duplication event at the base of the Zingiberales order, leading to two distinct orthologous AG lineages (ZinAG-1 and ZinAG-2). Scenarios 2 and 3 depict alternative histories of duplications and losses of the AG copies, particularly in the Zingiberaceae lineage. In both cases, orthologous relationships would be complicated by the existence of subsequent duplication events, unique to the Zingiberaceae lineage, leading to the evolution of yet another copy of AG, ZinAG-3. (B) Shimodaira-Hasewaga test (Shimodaira & Hasewaga [37]), SH test) was performed using PAUP* on a constrained topology, where the two first paraphyletic lineages were forced to form a monophyletic clade. The likelihood score for the constrained topology was compared to the likelihood score of the unconstrained gene tree, as obtained on Bayesian and maximum likelihood phylogenetic analysis (Figure 3). The constrained topology shows a better likelihood score than the one calculated for the gene tree topology presented here (although not significantly different), supporting the idea that the first two paraphyletic lineages are actually derived from a single duplication event. This result supports the evolutionary history depicted by Scenario 1, in which ZinAG-1 and ZinAG-2 are orthologous lineages.
The SH test [37] was performed using a constrained gene tree in which the two first branching lineages (all “ZinAG-1”) were forced to form a monophyletic group (Figure 4B), and compared for likelihood score against the unconstrained gene tree obtained in our analyses (Figure 3). Our results indicate that the constrained gene tree has a likelihood score that is not significantly different from the unconstrained analysis, suggesting that scenario 1 (Figure 4A) is equally as likely as scenario 2 in describing the evolutionary history for ZinAG. The most parsimonious explanation, then, is described by scenario 1, in which a single duplication event happened at the base of the Zingiberales order prior to lineage diversification.
Selection tests
Branch selection was detected using PAML codeml branch models. Branch-selection test show significant positive selection (omega (ω) = 1.2059; LRT = 1,622.95153, P = 0.000) at the base of the Zingiberales clade, suggesting that functional divergence between lineages might have happened soon after the duplication event (Figure 5A).

Selection test results. (A) PAML branch selection test. Omega (w) values are depicted for each branch in the gene tree. A likelihood ratio test (LTR) for branch models (M1a and M2a) was performed. PAML detects a strong selection signal at the base of the Zingiberales sequences, but nowhere else in the gene tree. (B) HyPhy (package FEL) site selection test. Tree shows nodes (in red) tested for positive selection. Balancing and positive selected residues are marked along the AGAMOUS protein domains. As expected, FEL detected various sites under balancing selection, while three sites were detected to be under positive selection, particularly in the I and K domains. Table shows selection test statistics for all positive selected sites observed in the analysis.
Sites under selection were identified using the FEL package of HyPhy as well as site selection models of PAML codeml. As expected, most sites are under balancing selection, while three sites show signs of positive selection (Figure 5B). Codon position 75 in the I domain, and codon positions 124 and 142 in the K domain show signs of positive selection (Figure 5B). Comparing these sites between species of the banana grade (for example, M. acuminata) and the ginger group (for example, C. spicatus and Canna sp.), most of the changes, although fixed between the two groups, do not result in changes to the chemical properties of the amino acids in these positions with the single exception being the change observed at codon position 142 of ZinAG-1 (Figure 6). In M. acuminata, position 142 is occupied by amino acids with charged polar side chains, such as asparagine (N) and histidine (H), while in Canna sp. and C. spicatus this position is occupied by tyrosine (Y), an amino acid with an uncharged side chain. Codon position 142 is part of the third alpha-helix of the K domain, also known as K3.

Amino acid changes within positive selected sites for the two copies of the AGAMOUS ( AG ) gene across Zingiberales species. The asterisk depicts the evolution of androecial petaloidy within the Zingiberales order. Note that it also corresponds to the base of the ginger clade (in blue). Marked in yellow are the paraphyletic lineages of the banana grade. For amino acid comparisons, Musa acuminata (Musaceae), Costus spicatus (Costaceae), and Canna indica (Cannaceae) AG sequences were used. Logos for the specific codons of the banana grade (bottom) and ginger clade (top) are shown. Single-letter amino acid traditional names were used (colored boxes). On the far right, images of Canna indica fertile stamen (top; Th-theca; Pa-petaloid appendage of the stamen); Costus sp. labellum (La, middle image); and Musa basjoo flower (bottom; Fp free petal) are shown. Also, note that Musa acuminata has four AG sequences due to a subsequent whole genome duplication event after the divergence of the Musaceae lineage [45].
Gene expression
AG expression in M. basjoo was initially assessed using RT-PCR (Figure 7). ZinAG-1 and ZinAG-2 were present in all M. basjoo floral organs examined.

Musa basjoo RT-PCR for ZinAG-1 and ZinAG-2 . RT-PCR was carried out for all M. basjoo floral organs, as well as for total flower cDNA as a positive control. RT-PCR results show expression of both copies of the gene AG in all floral organs studied.
Expression of AG was further investigated through organ-specific transcriptome data on M. acuminata and C. spicatus floral organs. ZinAG-1 and ZinAG-2 are expressed in filaments and theca of Musa, and in very low levels in the free petal (Figure 8A).

Musa basjoo and Costus spicatus gene expression based on transcriptomes of developing floral organs. (A) Musa acuminata and Costus spicatus AG expression based on normalized FPKMs. AG has four copies in Musa, due to an independent duplication event. Musa copies are distinguished by the letters ‘a’ and ‘b,’ while ‘ZinAG-1’ and ‘ZinAG-2’ relate to the Zingiberales broad duplication event. (B) Musa acuminata and Costus spicatus AP1/FUL-like gene expression based on normalized FPKMs. Error bars correspond to standard deviation of two replicates.
In the organ-specific transcriptome data, C. spicatus AG expression is dominated by ZinAG-1, and extremely low levels of ZinAG-2 are only observed in the petal (Figure 8A), although ZinAG-2 can be amplified by RT-PCR in these organs (data not shown). In M. basjoo, AP1/FUL-like expression largely agrees with that anticipated based on a hypothesis of mutual exclusion [46]: APETALA1/FRUITFULL-like (AP1/FUL-like) genes are mostly expressed in petals where there is very low expression of AG, while in stamens, AP1/FUL-like expression is almost abolished and AG is highly expressed (Figure 8B). In C. spicatus, AP1/FUL-like and AG gene expressions show a different pattern to that observed in Musa. Although AP1/FUL-like show very low expression values across Costus floral organs in comparison to AG expression, AG and AP1/FUL-like are simultaneously expressed in the androecial organs (labellum, stamen filament, and stamen theca), suggesting that ZinAG-1 is not capable of fully suppressing AP1/FUL-like expression in these organs.
In order to confirm transcriptome expression data, qPCR was performed in all organs of C. spicatus flower (sepals, petals, labellum, stamen, and gynoecium). In general, AG and AP1/FUL-like expression patterns largely agree with transcriptome data. In contrast with transcriptome data, however, ZinAG-2 is expressed in stamen and gynoecium, with low levels of expression in labellum, petals, and sepals. The expression pattern of ZinAG-2 agrees, in this case, with the classical expression pattern of AG found in model species (Figure 9A): This may indicate that ZinAG-2 maintained the AGAMOUS functionality within the ginger clade. ZinAG-1 exhibits a consistent pattern of expression between the qPCR data and the transcriptome data, showing higher levels of expression in the labellum, stamen, and gynoecium (Figure 9A). Despite its low levels when compared to ZinAG-1, AP1/FUL-like gene expression in C. spicatus floral organs agrees with transcriptome data (Figure 9B). In general, AP1/FUL-like expression can be detected in all floral organs, including stamen and gynoecium, potentially due to the inability of AG to fully suppress its expression in inner floral whorls (Figure 9B). It is important to notice, however, that there might be other copies of AP1/FUL-like genes in C. spicatus (as suggested by the transcriptome), and a thorough analysis of this gene family should be carried out in order to better understand the role of this gene family in flower development and morphological evolution in the Zingiberales.

qPCR results for Costus spicatus AG and AP1/FUL -like genes. (A) C. spicatus ZinAG-1 and ZinAG-2 mean expression in all floral organs. (B) C. spicatus ZinAP1 and ZinAG-1 expression in all floral organs. Results are based on three replicates, normalized by ACTIN1. Error bars depict standard deviation for the three replicates.
Discussion
The AG gene subfamily has been extensively implicated in the development of reproductive organs (carpels and stamens) and meristem determinacy in angiosperms. In both monocots and eudicots, the conservation of these functions by AG lineage genes is remarkable considering multiple gene duplication and subfunctionalization events [12,16,21], even though AGL11 lineage genes might act redundantly in some lineages [10,20]. In the Zingiberales, at least one lineage-specific duplication event is observed within the AG lineage. Sequence divergence between the two copies (ZinAG-1 and ZinAG-2), as well as their expression patterns, suggests the involvement of Zingiberales AG genes in the evolution of reproductive organ development and the evolution of petaloidy in the order.
Based on the branch selection patterns observed in the Zingiberales AG gene tree, it is likely that functional divergence between lineages happened early after the duplication event, at the base of the ginger clade. As expected for functionally important and highly interconnected genes, most of the observed site selection is due to balancing selection, suggesting functional conservation. However, three residues in the Zingiberales show signs of positive selection, and fixed differences among members of the ginger clade indicate that these modifications might be implicated in the morphological changes observed in the androecium of the Zingiberales.
In particular, the positive selected amino acid change observed at position 142 of the K domain is of particular relevance. The role of subdomains of the K domain in MADS-box protein-protein interactions has been studied, especially in the formation of dimers between B-class genes and SEP genes [47,48]. The K domain of the MADS proteins are involved in the formation of protein complexes for DNA binding. In particular, K1 and K2 helices are involved in dimer formation, while K3 is involved in the formation of tetramers [48-51]. Also, in Antirrhinum, a single amino acid change has been implicated in differences in the establishment of male and female identity between AG lineage genes PLE and FAR. A single glutamine insert in the K3 domain of FAR leads to a limited protein-protein interaction between AG and SEPALLATA (SEP) proteins, underlying the functional differences observed between FAR and PLE genes in determining reproductive organ identity [18].
In Zingiberales, it is possible that the amino acid change at the K3 domain observed in ZinAG-1 between the banana grade and the ginger group might change AG protein ability to form higher level complexes while maintaining the capacity to form protein dimers. This suggests an interesting mechanism in which ZinAG-1 from the ginger group could act as a negative regulator of tetramer formation: while binding to AG interacting proteins to form dimers, this complex would be less likely involved in the formation of quartets, resulting in a post-transcriptional downregulation of AG downstream targets.
If one assumes that ZinAG-1 in the ginger clade (exemplified by Costus) inhibits quartet formation, and thus its expression leads to the downregulation of downstream targets in C. spicatus (as suggested by the amino acid change; Figure 6), we expect that high levels of Costus ZinAG-1 would lead to a stronger suppression of downstream genes, and a more petal-like phenotype in the stamen whorl. The correlation between higher levels of ZinAG-1 in Costus labellum and filament and a petaloid phenotype of these organs is consistent with increased levels of ZinAG-1 in the labellum and filament and decreasing levels of ZinAG-1 expression towards the fertile theca.
This interpretation is also supported by changes in the expression profile of AP1/FUL-like genes across floral organs of the Zingiberales. In A. thaliana, relatively high levels of AP1/FUL were detected in petaloid stamens and sepaloid carpels of flowers with reduced levels of AG due to anti-sense (RNAi) knockdown [23]. Accordingly, petaloid organs in the androecium such as those observed in C. spicatus are characterized by simultaneous expression of AG and AP1/FUL-like, indicating a lack of negative interaction between these two gene families. Morphologically, this expression profile corresponds to a ‘hybrid’ organ (petaloid staminode) and could potentially represent a ‘mix-attractor’ between stamen and petal in the A. thaliana FOS-GRN ([8], modified in Figure 1). However, this ‘hybrid’ attractor has not been observed as a stable state of the A. thaliana FOS-GRN, potentially due to the presumed fixed mutual negative regulation between AG and AP1/FUL. It is possible that duplication of many of the FOS-GRN genes observed in the Zingiberales could lead to stable states that are not observed in Arabidopsis, as different lineage-specific duplication events and subsequent differential retention/loss of duplicated copies as well as sequence divergence would provide the opportunity for novel protein interactions leading to novel stable states.
Such novel interactions are suggested by data from other monocot lineages, where an expanded AP1/FUL-like expression pattern has been observed in various grass lineages [52,53]. Interestingly, in Z. mays, constitutive expression of one of the AP1/FUL-like copies (ZmFUL2a) leads to the development of undifferentiated floral organs in the male spikelet. The authors propose an ‘interference hypothesis’ where interference of AP1/FUL-like proteins in the formation of proper protein-protein interactions during particular stages of development could result in the observed phenotypes [54]. Although the precise function of A. thaliana AP1/FUL gene might be specific to Arabidopsis and closely related species, studies in grasses support the idea that AP1/FUL-like genes do play a role in transition to flowering, meristem and perianth identity, or even in determining the identity of all floral organs ([53] and references therein).
It is important to note that the results on AP1/FUL-like genes presented here are preliminary. Although there is an unexpected expansion of AP1/FUL-like gene expression towards the inner flower whorls, a more in-depth analysis of this gene family within the Zingiberales, as well as a comprehensive survey of the expression patterns of AG downstream genes, is required to fully test our hypothesized scenario. Also, protein-protein interaction studies are critical to test the functions of the described AG protein modifications observed across the Zingiberales.
In transgenic Arabidopsis plants carrying AG anti-sense RNA, a range of floral organ phenotypes is observed including the occurrence of petaloid stamens [23]. Likewise, mutations in the regulatory site of AG in Arabidopsis can lead to the development of petaloidy in the androecium [22]. Here, we show that androecial petaloidy in the Zingiberales is likewise associated with evolution of the AG lineage, and may result from a single amino acid change in the K domain of ZinAG-1 after the divergence of the banana lineages and the ginger clade.
Conclusions
The results presented here suggest a scenario in which positive selection acting upon AG genes in the Zingiberales has resulted in a fixed change in the K3 domain that can potentially explain the evolution of androecial petaloidy and infertility observed in the order. Selected amino acid changes in the K3 domain might result in differential abilities to form higher level protein-protein complexes between ZinAG-1 and its interaction partners, resulting in a post-translational downregulation of downstream genes. While further studies are needed to fully test this hypothesis, our expression data are consistent with this model. If it is the case that the changes in the AG genes are responsible for the observed changes in floral morphology across Zingiberales, a clear trade-off between production of fertile stamens and increased petaloidy has been fixed by positive selection in this group. Although androecial petaloidy is a remarkable feature of Zingiberales floral evolution, no changes have been observed in meristem determinacy. This might be explained, at least in part, by the potential functional redundancy between AG and AGL11 lineage genes, as already reported for in rice and petunia [10,20]. Further studies of the AG subfamily genes in the Zingiberales will help understand the complete role of the AG subfamily in floral development and evolution across the Zingiberales.
Abbreviations
- AG :
-
AGAMOUS
- AGL11 :
-
AGAMOUS-like 11
- AP1/FUL :
-
APETALA1/FRUITFULL
- AP2 :
-
APETALA2
- AP3 :
-
APETALA3
- BS:
-
bootstrap support
- CsAGcp :
-
Costus spicatus AGAMOUS copy 1
- CsAGcp2 :
-
Costus spicatus AGAMOUS copy 2
- CsAP1/FUL-like :
-
Costus spicatus APETALA1/FRUITFULL-like gene
- CpAG1 :
-
Cyclamen persicum AGAMOUS copy 1
- CpAG2 :
-
Cyclamen persicum AGAMOUS copy 2
- DN:
-
ratio of non-synonymous substitution rates
- DS:
-
ratio of synonymous substitution rates
- euAG :
-
eudicot AGAMOUS
- FAR :
-
FARINELLI
- FBP6 :
-
Floral binding protein gene 6
- FEL:
-
fixed effect likelihood model
- FOS-GRN:
-
floral organ specification gene regulatory network
- FPKM:
-
frequency per kilobase of exon per million aligned reads
- H:
-
histidine
- K domain:
-
keratin domain
- ML:
-
maximum likelihood
- MbAGcp1 :
-
Musa basjoo AGAMOUS copy 1
- MbAGcp2 :
-
Musa basjoo AGAMOUS copy 2
- MSA:
-
multiple sequence alignment
- N:
-
asparagine
- OSMADS3 :
-
Oryza sativa MADS-box gene 3
- OSMADS13 :
-
Oryza sativa MADS-box gene 13
- OSMADS58 :
-
Oryza sativa MADS-box gene 58
- PI :
-
PISTALLATA
- PLE :
-
PLENA
- PMADS3 :
-
Petunia MADS-box gene 3
- PP:
-
posterior probability support
- SE :
-
SEPALLATA
- SD:
-
standard deviation
- SH:
-
Shimodaira-Hasegawa test
- Y:
-
tyrosine
- ZAG1 :
-
Zea mays AGAMOUS 1
- ZinAG-1 :
-
Zingiberales AGAMOUS copy 1
- ZinAG-2 :
-
Zingiberales AGAMOUS copy 2
- ZMM2 :
-
Zea mays MADS-box gene 2
References
Coen ES, Meyerowitz EM. The war of the whorls: genetic interactions controlling flower development. Nature. 1991;353:31–7.
Pelaz S, Ditta GS, Baumann E, Wisman E, Yanofsky MF. B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature. 2000;405:200–3.
Melzer R, Theissen G. Reconstitution of ‘floral quartets’ in vitro involving class B and class E floral homeotic proteins. Nucleic Acids Res. 2009;37:2723–36.
Mendoza L, Alvarez-Buylla E. Dynamics of the genetic regulatory network for Arabidopsis flower morphogenesis. J Theor Biol. 1998;193:307–19.
Mendoza L, Thieffry D, Alvarez-Buylla E. Genetic control of flower morphogenesis in Arabidopsis thaliana: a logical analysis. Bioinformatics. 1999;15:593–606.
Alvarez-Buylla ER, Benítez M, Corvera-Poiré A, Chaos Cador A, de Folter S, Gamboa de Buen A, et al. Flower development. In: American Society of Plant Biologists, editor. The Arabidopsis book. 2010. doi: 10.1199/tab.0127.
Álvarez-Buylla ER, Azpeitia E, Barrio R, Benítez M, Padilla-Longoria P. From ABC genes to regulatory networks, epigenetic landscapes and flower morphogenesis: making biological sense of theoretical approaches. Semin Cell Dev Biol. 2010;21:108–17.
Barrio RÁ, Hernández-Machado A, Varea C, Romero-Arias JR, Álvarez-Buylla ER. Flower development as an interplay between dynamical physical fields and genetic networks. PLoS One. 2010;5:e13523.
Yanofsky MF, Ma H, Bowman JL, Drews GN, Feldman KA, Meyerowitz EM. The protein encoded by the Arabidopsis homeotic gene agamous resembles transcription factors. Nature. 1990;346:35–9.
Dreni L, Pilatone A, Yun D, Erreni S, Pajoro A, Caporali I, et al. Functional analysis of all AGAMOUS subfamily members in rice reveals their roles in reproductive organ identity determination and meristem determinacy. Plant Cell. 2011;23:2850–63.
Fourquin C, Ferrándiz C. Functional analyses of AGAMOUS subfamily members in Nicotiana benthamiana clarify the evolution of early and late roles of C-function genes in eudicots. Plant J. 2012;71:990–1001.
Yamaguchi T, Lee DY, Miyao A, Hirochika H, An G, Hirano HY. Functional diversification of the two C-class MADS box genes OSMADS3 and OSMADS58 in Oryza sativa. Plant Cell. 2006;18:15–28.
Dreni L, Osnato M, Kater MM. The ins and outs of the rice AGAMOUS subfamily. Mol Plant. 2013;6:650–64.
Dreni L, Kater MM. MADS reloaded: evolution of AGAMOUS subfamily genes. New Phytol. 2014;201:717–32.
Kramer EM, Jaramillo MA, Di Stilio VS. Patterns of gene duplication and functional evolution during the diversification of the AGAMOUS subfamily of MADS box genes in Angiosperms. Genetics. 2004;166:1011–23.
Zahn LM, Leebens-Mack JH, Arrington JM, Hu Y, Landherr LL, DePamphilis CW, et al. Conservation and divergence in the AGAMOUS subfamily of MADS-box genes: evidence of independent sub- and neofunctionalization events. Evol Dev. 2006;8:30–45.
Airoldi CA, Davies B. Gene duplication and the evolution of plant MADS-box transcription factors. J Genet Genomics. 2012;13:157–65.
Airoldi CA, Bergonzi S, Davies B. Single amino acid change alters the ability to specify male or female organ identity. Proc Natl Acad Sci U S A. 2010;107:18898–902.
Davies B, Motte P, Keck E, Saedler H, Sommer H, Schwarz-Sommer Z. PLENA and FARINELLI: redundancy and regulatory interactions between two Antirrhinum MADS-box factors controlling flower development. EMBO J. 1999;18:4023–34.
Heijmans K, Ament K, Rijpkema AS, Zethof J, Wolters-Arts M, Gerats T, et al. Redefining C and D in petunia ABC. Plant Cell. 2012;24:2305–17.
Mena M, Ambrose BA, Meeley RB, Briggs SP, Yanofsky MF, Schmidt RJ. Diversification of C-function activity in maize flower development. Science. 1996;274:1537–40.
Hong RL, Hamaguchi L, Busch MA, Weigel D. Regulatory elements of the floral homeotic gene AGAMOUS identified by phylogenetic footprinting and shadowing. Plant Cell. 2003;15:1296–309.
Mizukami Y, Ma H. Separation of AG function in floral meristem determinacy from that in reproductive organ identity by expressing antisense AG RNA. Plant Mol Biol. 1995;28:767–84.
Tanaka Y, Oshima Y, Yamamura T, Sugiyama M, Mitsuda N, Ohtsubo N, et al. Multi-petal cyclamen flowers produced by AGAMOUS chimeric repressor expression. Scientific Rep. 2013; 3: doi: 10.1038/srep02641
Walker-Larsen J, Harder LD. The evolution of staminodes in angiosperms: patterns of stamen reduction, loss, and functional re-invention. Am J Bot. 2000;87:1367–84.
Volkova PA, Choob VV, Shipunov AB. The flower organ transition in water lily (Nymphaea alba s.l., Nymphaeaceae) under cross-examination with different morphological approaches. Belg J Bot. 2007;140:60–72.
Soltis D, Chanderbali A, Kim S, Buzgo M, Soltis PS. The ABC model and its applicability to basal angiosperms. Ann Bot. 2007;100:155–63.
Kress WJ, Prince LM, Hahn WJ, Zimmer EA. Unraveling the evolutionary radiation of the families of the Zingiberales using morphological and molecular evidence. Syst Biol. 2001;50:926–44.
Almeida AMR, Brown A, Specht CD. Tracking the development of the petaloid fertile stamen in Canna indica: insights into the origin of androecial petaloidy in the Zingiberales. AoB PLANTS. 2013;5:plt009.
Yockteng R, Almeida AMR, Yee S, Andre T, Hill C, Specht CD. A method for extracting high-quality RNA from diverse plants for next-generation sequencing and gene expression analyses. Appl Plant Sci. 2013;1:1300070.
Bartlett ME, Specht CD. Evidence for the involvement of GLOBOSA-like gene duplications and expression divergence in the evolution of floral morphology in the Zingiberales. New Phytol. 2010;187:521–41.
Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253–6.
Lanfear R, Calcott B, Ho SYW, Guindon S. Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29:1695–701.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.
Sukumaran J, Holder MT. DendroPy: a Python library for phylogenetic computing. Bioinformatics. 2010;26:1569–71.
Shimodaira H, Hasewaga M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol. 1999;16:1114–6.
Swofford DL. PAUP*: Phylogenetic analysis using parsimony (and other methods) 4.0 Beta. Sinauer Associates. 2002.
Yang Z. PAML 4: a program package for phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91.
Pond SLK, Frost SDW, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–9.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet Journal. 2011;17:10–2.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–75.
Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12:R22.
D’Hont A, Denoeud F, Aury J-M, Baurens F-C, Carreel F, Garsmeur O, et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature. 2012;488:213–9.
Gustafson-Brown C, Savidge B, Yanofsky MF. Regulation of the Arabidopsis floral homeotic gene APETALA1. Cell. 1994;76:131–43.
Yang Y, Fanning L, Jack T. The K domain mediates heterodimerization of the Arabidopsis floral organ identity proteins, APETALA3 and PISTILLATA. Plant J. 2003;33:47–59.
Yang Y, Jack T. Defining subdomains of the K domain important for protein–protein interactions of plant MADS proteins. Plant Mol Biol. 2004;55:45–59.
Gramzow L, Theissen G. A hitchhicker’s guide to the MADS world of plants. Genome Biol. 2010;11:214. http://genomebiology.com/2010/11/6/214.
Immink RGH, Tonaco IAN, de Folter S, Shchennikova A, van Dijk ADJ, Busscher-Lange J, et al. SEPALLATA3: the ‘glue’ for MADS box transcription factor complex formation. Genome Biol. 2009;10:R24.
Kaufmann K, Melzer R, Theissen G. MIKC-type MADS-domain proteins: structural modularity, protein interactions and network evolution in land plants. Gene. 2005;347:183–98.
Gocal GF, King RW, Blundell CA, Schwartz OM, Andersen CH, Weigel D. Evolution of floral meristem identity genes. Analyses of Lolium temulentum genes related to APETALA1 and LEAFY of Arabidopsis. Plant Physiol. 2001;125:1788–801.
Preston JC, Kellogg EA. Conservation and divergence of APETALA1/FRUITFULL-like gene function in grasses: evidence from gene expression analyses. Plant J. 2007;52:69–81.
Heuer S, Hansen S, Bantin J, Brettschneider R, Kranz E, Lörz H, et al. The maize MADS box gene ZmMADS3 affects node number and spikelet development and is co-expressed with ZmMADS1 during flower development in egg cells, and early embryogenesis. Plant Physiol. 2001;127:33–45.
Acknowledgements
Research contributing to this manuscript was supported by NSF Doctoral Dissertation Improvement Grant (DEB 1110461; AMRA and CDS), an NSF CAREER award to CDS (IOS 0845641), and research support from UC Berkeley Committee on Research (COR) and AES. AMRA was supported by a CAPES/Fulbright Fellowship. The authors thank M. E. Maldaner, A. Brown, and C. Sangster for their help in gathering Zingiberales AGAMOUS sequences, and E. Alvarez-Buylla for insightful discussion during the preparation of this manuscript. The authors thank H. Forbes from the UC Botanical Garden for support with fresh plant material. Publication made possible in part by support from the Berkeley Research Impact Initative (BRII) sponsored by the UC Berkeley Library.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AMRA contributed with the conceptual and experimental design, data collection and analysis, and drafted the manuscript. RY and WCO participated in the data collection and analysis, and manuscript editing. CDS provided the conceptual advice and experimental design, edited the manuscript, and provided financial support. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Almeida, A.M.R., Yockteng, R., Otoni, W.C. et al. Positive selection on the K domain of the AGAMOUS protein in the Zingiberales suggests a mechanism for the evolution of androecial morphology. EvoDevo 6, 7 (2015). https://doi.org/10.1186/s13227-015-0002-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13227-015-0002-x