
Abstract
The use of gene therapy (GT) for the treatment of primary immune deficiencies (PID) including severe combined immune deficiency (SCID) has progressed significantly in the recent years. In particular, long-term studies have shown that adenosine deaminase (ADA) gene delivery into ADA-deficient hematopoietic stem cells that are then transplanted into the patients corrects the abnormal function of the ADA enzyme, which leads to immune reconstitution. In contrast, the outcome was disappointing for patients with X-linked SCID, Wiskott–Aldrich syndrome and chronic granulomatous disease who received GT followed by autologous gene corrected transplantations, as many developed hematological malignancies. The malignancies were attributed to the predilection of the viruses used for gene delivery to integrated at oncogenic areas. The availability of safer and more efficient self-inactivating lentiviruses for gene delivery has reignited the interest in GT for many PID that are now in various stages of pre-clinical studies and clinical trials. Moreover, advances in early diagnosis of PID and gene editing technology coupled with enhanced abilities to generate and manipulate stem cells ex vivo are expected to further contribute to the benefit of GT for PID. Here we review the past, the present and the future of GT for PID, with particular emphasis on the Canadian perspective.
Similar content being viewed by others


Background
Primary immune deficiencies (PID) are a group of inherited immune disorders that can result in predisposition to infections, immune dysregulation, autoimmunity or malignancy. The introduction of newborn screening for severe immune defects as well as better diagnostic modalities and awareness have contributed to increase in identification of PID [1]. Early diagnosis, antibiotic prophylaxis and treatment, immunoglobulin replacement and immunosuppressive medications can help prevent or ameliorate many of the PID manifestations. However, such treatments often require life-long administration and are associated with significant emotional and financial burden to patients, families and society. Moreover, such treatments may lose their effectiveness over time and often do not prevent immune dysregulation disorders or malignancy. Hence, the ultimate cure for most PID requires correction of the defective gene responsible for the immune deficiency.
Hematopoietic stem cell transplantations for primary immune deficiency
Hematopoietic stem cell transplantations (HSCT) involve the infusion of stem cells typically obtained from bone marrow, peripheral blood or umbilical cord blood to re-establish the hematopoietic and/or immune function. Since the original description of allogeneic bone marrow transplantations for patients suffering from PID in 1968, HSCT have been performed across the world for many severe immune defects [2, 3]. These conditions range from severe combined immunodeficiency (SCID) encompassing all lymphoid lineages such as adenosine deaminase (ADA) deficiency, lymphoid subtypes such as “common” gamma chain (γc) defects to specific T cell defects such as immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Other PID that can be treated by HSCT include myeloid abnormalities such as Wiskott–Aldrich syndrome (WAS), leukocyte adhesion defect (LAD) or chronic granulomatous diseases (CGD). Throughout the years, HSCT using allogenic human leukocyte antigens (HLA) matched or mis-matched donors have cured thousands of patients with PID. However, allogeneic HSCT are associated with many complications. Chemotherapy is often required prior to HSCT to eliminate the recipient’s residual immune system, which helps prevent rejection of the donor cells. Moreover, many patients experience significant graft versus host (GvH) disease where the donor’s competent immune system recognizes the recipient HLA-expressing cells as foreign and attacks the recipient organs. When HLA-matched or mismatched unrelated donors are used for transplanting patients with PID, which occurs in the majority of HSCT, the risk for GvH disease increases to more than 70% [4]. Although earlier diagnosis, improved infections control, better HLA matching and lesser toxic conditioning regimens are expected to further improve the outcomes of HSCT, such procedures continue to carry significant complications.
Gene therapy
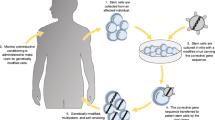
Gene therapy (GT), i.e. the use of genetic material to modify cells, has been investigated for numerous conditions since the development of recombinant DNA technology in the late 1970’s. Different forms of GT are being explored, including gene insertion into cells ex vivo that can then be transplanted into the recipient, expand and exert a desired biological effect (Fig. 1) or direct injection of the DNA into the body. GT using ex vivo modified autologous cells could avoid graft rejection and GvHD as the transplanted cells and the recipient have identical HLA. Therefore, it has long been proposed as an alternative treatment to allogeneic HSCT. Different gene delivery systems were developed, which provide either transient or stable gene transfer. Mechanical methods such as liposomes or electroporation can introduce nucleic acids into cells both in vitro and in vivo, albeit at low efficacy while damaging many of the cells. Biological systems, including transposons and viral vectors have been used at increasing frequency to insert genetic material into cells. Viruses now account for 67% of all delivery methods used in GT clinical trials worldwide, with adenovirus and retrovirus vectors representing the majority. Indeed, as data from the “Gene therapy Clinical Trials Worldwide” update from August 2016 (Fig. 2) indicate a steady increase in the use of adeno-associated virus and lentivirus vectors in clinical trials [5]. The viral vectors differ in the size of the genetic material they can harbor as well as their tropism to tissues and cells. Other differences include the virus’ ability to evade the recipient’s immune system, influencing the potential to trigger a neutralizing and possibly harmful immune response. Adenovirus associated virus has been commonly used for the correction of monogenetic disorders in post-mitotic tissues, while retroviral vectors can integrate into the host cell genome. Therefore retroviruses are preferred for the stable gene transfer into proliferating cells, such as hematopoietic stem cells. Indeed, most pre-clinical and clinical GT for PID used members of retroviridae family, including the Murine leukemia virus (MLV) and the human immunodeficiency virus (HIV) of the gamma-retrovirus and lentivirus (LV) genus, respectively. Inserting the gene of interest ex vivo into cells isolated from the patient, which are then given back to the patient, lowers the risk of unwanted effects associated with in vivo delivery, such as ectopic expression of the delivered gene in off-target organs. Furthermore, the therapeutic impact from ex vivo gene delivery is more robust since the gene-based correction is not subject to metabolic or renal clearance and is less likely to trigger immune responses. In some protocols, ex vivo GT may even allow for selection, expansion and quality control of the modified cells before reinfusion, thereby further improving safety and efficacy [6].

Ex vivo gene therapy. Patient’s cells are collected from bone marrow, peripheral blood or umbilical cord blood (1). A virus is altered ex vivo to increase safety and efficacy of gene delivery (2). A gene is inserted into the altered virus ex vivo (3). The altered virus containing the gene is added to the patient’s cells ex vivo (4). The cells are genetically altered ex vivo (5). The patient is treated with chemotherapy or radiotherapy (6). The genetically altered cells are transplanted into the conditioned patient (7). The genetically altered cells expand in the patient and exert their biological effects (8)

Viral vector use in gene therapy trials worldwide 2004–2016. Viral vectors account for 67% of the total vectors used for gene therapy clinical trials worldwide. The graph depicts the percentage (%) of AV adenovirus, RV retrovirus, AAV adeno-associated virus and LV lentivirus vector use of the total viral vectors
The use of LV vectors raised safety and ethical concerns about the possibility of transmitting or promoting HIV infection, including generation of replication competent LV during vector production or mobilisation of the vector by endogenous retroviruses in patients’ genomes. Some of the strategies developed to mitigate the risk for patients and the medical teams include limiting to the number of accessory viral genes, splitting the viral components to different plasmids, reducing the number of viral particles used for transduction and incorporating self-inactivating (SIN) vectors. Nevertheless, close monitoring of patients who receive LV GT is warranted. Importantly, despite the increasing use of LV, there have not been any reports of accidental development of HIV.
Gene therapy for primary immune deficiencies
GT has been a particularly attractive option for PID. The genes responsible for many PID have been identified, the diseases have been often fatal at a young age and current treatments have commonly been unsatisfactory. Moreover, the success of HSCT and spontaneous reverse mutations that corrected PID supported the hypothesis that autologous GT would be beneficial for such patients. Indeed, PID was the first human condition treated with GT and continues to be in the forefront of such attempts. Ideally, patients should be treated as early as possible before suffering significant infections and organ damage, and when the potential for immune reconstitution is maximal. Specific indications and inclusion criteria vary in accordance to the condition and the protocol. The subsequent discussion will detail the past current and future status of PID GT, with emphasis on Canadian experience and contributions.
Main text
Gene therapy for adenosine deaminase
Adenosine deaminase (ADA) is a ubiquitous enzyme that is crucial for the metabolism of adenosine and 2-deoxyadenosine. Impaired function of ADA leads to accumulation of purine metabolites that are particularly toxic to the rapidly proliferating bone marrow cells and thymocytes. Inherited ADA defects account for 15–20% of all causes of SCID, and some Canadian populations such as the Mennonite and Canadian First Nations seem to have increased frequency of ADA deficiency [7]. The implementation of newborn screening (NBS) for severe immune deficiency, and the inclusion of ADA among the conditions tested in some of the NBS algorithms, such as the one implemented in Ontario, are expected to reveal the true incidence of ADA deficiency. T, B and Natural Killer (NK) cells dysfunction is often present in ADA-deficient patients in infancy. In addition, patients may suffer from alveolar proteinosis [8], diverse neuro-developmental abnormalities [9] as well as bone and cartilage malformations [10]. Since the mid-1980, weekly injections of polyethylene glycol-modified bovine ADA (PEG-ADA) have been used to remove the toxic purine metabolites, improve T and B cell functions and correct for some of the non-immunologic abnormalities [11]. However, PEG-ADA does not cure ADA deficiency, as it is effective in only 80% of patients and the immune recovery often diminishes over time [12]. Moreover, the cost of PEG-ADA treatment (>$US 100,000/year) restricts its availability for many patients. Nevertheless, health Ministries of some Canadian provinces, such as Ontario and British Columbia have reluctantly sponsored PEG-ADA treatment. HSCT from an unaffected HLA identical sibling donor without any chemotherapy has been shown to restore immunity in ADA deficient patients, and is currently considered the treatment of choice [13]. However, HLA identical sibling donors are available for only a minority of patients while the use of HLA-mismatched related or unrelated donors is associated with significant morbidity and mortality [14]. These disappointing results of HSCT, together with the early availability of the ADA gene sequence, prompted the investigation of GT for ADA deficiency.
Initial GT trials for ADA deficiency, performed already in early 1990, delivered the ADA gene into T lymphocytes or umbilical cord blood/bone marrow progenitor cells using the murine gamma-retroviral vector. Similar to the protocols used for allogeneic HSCT, autologous GT were done in ADA-deficient patients without the use of cytoreductive conditioning. This was based on the assumption that ADA-proficient cells would have a “survival advantage” over the original ADA-deficient cells. Yet, for ethical reasons, patients enrolled in this trial were given PEG-ADA, which negated the survival advantage of the gene-corrected cells. Hence, despite detection of ADA-corrected cells in the host, inadequate amount of cells persisted to confer significant clinical benefit. It took almost a decade until the group in Italy, led by Drs. Aiuti and Naldini reintroduced non-myeloablative doses of busulfan or melphalan without PEG-ADA, into the GT trials for ADA deficiency [15]. Together with improved gene transduction techniques and the use of MLV-derived replication-deficient vector to deliver the ADA cDNA into cells, the Milan group was able to achieve lasting ADA expression in cells. This resulted in humoral and cellular immune reconstitution, decrease in susceptibility to infections [16] and correction of the bone abnormalities [17]. One Canadian patient who participated in this study is now almost 10 years after receiving GT and is clinically well. Subsequent studies at Great Ormond Street in the UK, at The National Human Genome Institute, the Children’s Hospital Los Angeles, and later the UCLA Mattel Children’s Hospital as well as Japan demonstrated the critical role of non-myeloablative pre-transplantation conditioning in gene therapy for ADA SCID [18–20]. Recently, long-term follow-up (range, 2.3–13.4 years) of the 18 ADA-deficient patients who received ADA GT in Milan revealed that all survived [21]. PEG-ADA was resumed in 3 patients, of which 2 later received HSCT from HLA identical sibling donors that were not available prior to GT. The relatively short follow-up of the ADA-deficient patients who received GT in England and North America precludes direct comparison with the Milan outcome, yet the overall results and safety of all these studies are encouraging. Indeed the success of the Milan ADA GT led to commercialization of the viral vector by GlaxoSmithKline (GSK) as Strimvelis™, which recently received marketing authorization in Europe. The impact of such move on ADA GT practicalities, including cost for patients and availability in North America are still not clear. Impressively, and in contrast to GT trials for other PID described below, all ADA-deficient patients who received GT in the USA and Europe survived, and none experienced abnormal clonal expansions or leuko/lympho-proliferative disorders. Although analyses of retroviral vector integrations in patients’ cells demonstrated insertion near proto-oncogenes sites (including LMO2) similar to those found in other PID trials, there was no skewing of the T cell repertoire or clonal selection/expansion in vivo. Despite the lack of insertional genotoxicity with gamma-retroviruses in ADA GT, concerns regarding leukemogenesis have led to the development of SIN LV vectors. Studies using these vectors for ADA deficiency are currently being completed in England (ClinicalTrials.gov Identifier: NCT01380990) and the USA (ClinicalTrials.gov Identifier: NCT01852071). Deletion of proteins from the vector packaging plasmids and the SIN mechanism have made their use safer. Moreover, as LV vectors can transduce non-dividing cells, such as quiescent hematopoietic stem cells, it is postulated that the efficacy of gene delivery into the very early stem cells will be improved. Interestingly, based on experiments in murine models, the current ADA SIN LV trials continue the administration of PEG-ADA for 30 days after the GT. More than 30 ADA-deficient patients have been treated with the SIN LV vector. Immune reconstitution has been achieved with no vector-related complications, although follow-up period for most patients is still short (<3 years). Several ADA-deficient patients from Quebec and Ontario, who lacked HLA-matched sibling donors, have already received GT under this protocol. Although GT is still very expensive (more than $ US 200,000/patient), the cost is less than the life-long continuation of PEG-ADA and possibly even less than an HLA-mismatched HSCT that is often associated with prolonged admissions and complications. Accordingly, the Ministry of Health in several Canadian provinces have approved the out-of-county expenses. After the control of infections and PEG-ADA administration, and coordination by the Canadian referring team with the centers performing the GT, patients typically spend 7–10 days at the GT center. During this period, the patients’ bone marrow cells are harvested, CD34 expressing cells are selected and transduced with the viral vector, busulfan is administered, and the gene-corrected cells are infused. Patients who are clinically well can return to the referral center prior to the development of chemotherapy-induced neutropenia. Close monitoring and frequent follow-ups are coordinated between the referring teams and the GT centers. In the future, shipping the patients’ bone marrow to designated centers might prevent the need for them to commute, further simplifying GT and reducing its costs. Indeed, researchers in the US and UK have began investigating the effects of cryopreservation of the cells on the success of LV GT for ADA-deficient patients (NCT02999984).
Gene therapy for common gamma chain
The interleukin-2 receptor gamma subunit (IL2R) gene on the X-chromosome encodes for the gamma chain (γc). The chain is a component for intracellular signaling of the IL-2, -4, -7, -9, -15, and -21 receptors, thus it is essential to the development and function of T, B and NK cells. Inherited defects in the γc are the most common cause of SCID in some medical centers [22], although not in others [23]. The male patients tend to present during infancy with recurrent and opportunistic infections such as Pneumocystis jiroveci pneumonia, unremitting candida and failure to thrive. The majority of patients lack T cells, yet expansion of B cells may prevent the characteristic lymphopenia. In recent years, with the introduction of NBS for SCID in most US states, as well as some Canadian provinces and European countries, IL2Rγ-deficient patients are being diagnosed earlier, prior to the development of infections.
Similar to other forms of SCID, HSCT can cure the immune defect caused by the impaired γc signaling. The best outcome, with >90% survival and excellent immune reconstitution can be achieved with the use of an HLA-identical sibling donor. Such transplants are typically done without any chemotherapy preparation. HSCT using HLA matched unrelated donors result in >80% survival and long-term immune reconstitution [4]. In recent years, improved outcome has also been reported with the use of HLA mismatched family donors, although immune reconstitution might be delayed and incomplete [24]. Hence, GT has been proposed as an alternative management option for patients without a suitable donor, particularly if patients also have active infections.
Gene therapy trials for X-linked SCID opened in 1999 and 2001 in Neckar, France and Great Ormond Street, UK, respectively. Both sites used autologous CD34+ cells that were transduced ex vivo with a murine gamma-retroviral vector. Gene-modified cells were returned to patients without cytoreductive conditioning. This led to improved cellular and humoral immunity, and patients were able to combat typical childhood infections and resume normal growth and development [25, 26]. However, 5 of the 20 patients treated at these centers developed T cell acute lymphoblastic leukemia 2.5–6 years after GT. The leukemic transformation was attributed to a predilection of the gamma-retroviral vector to integrate near oncogenes. The uncontrolled expression of a cytokine receptor important for the proliferation of T cells might also have contributed to the malignant transformation. While the overall outcome of these initial studies demonstrated that GT for the γc is possible, the significant concerns for safety halted clinical trials of GT for this condition (and others) for several years. Subsequently, modifications were made to the original gamma-retroviral vector to improve its safety, including creation of a SIN construct and replacement of the promoter. Results of GT with the modified construct used in 9 boys in parallel European and US trials, still without preparative conditioning, were recently reported [27]. One patient died from a preexisting adenoviral infection prior to immune reconstitution, while 7 of the 8 surviving patients had functional T cells and were free of infections. Four additional patients have received GT under this SIN gamma-retrovirus protocol (ClinicalTrials.gov Identifier: NCT01129544), which is close to completion. Integration analysis demonstrated no clonal skewing and none of the patients have developed malignancy, yet the follow-up period is still short. Similar to the trend in other PID, GT for X-linked SCID has recently shifted to the use SIN LV. A clinical trial using a codon-optimized SIN LV vector controlled by the ubiquitous elongation factor 1α promoter is being conducted in the US (ClinicalTrials.gov Identifier: NCT01306019). Interestingly, intermediate doses of busulfan were chosen for conditioning patients prior to GT. Initial results of this trial have already been published [28]. The 2 older subjects (aged 24 and 23 years respectively) cleared pre-existing viral infections and were able to stop immunoglobulin infusions. One patient died from pulmonary hemorrhage 27 months after GT while the other patient is clinically well 3 years after GT. Three younger patients (7–15 years old) were also treated, but conclusions regarding the safety and efficacy of this protocol cannot be drawn since their follow-up period is less than 1 year. In 2017, Boston Children’s and several collaborators are expected to open another trial with a SIN LV vector that will also involve administration of low dose busulfan in order to generate IL2Rγ-expressing B cells and to correct the humoral immunity. Future studies comparing the survival, complications and long-term immune reconstitution following HSCT from different donors and GT, with and without conditioning, will enable better assessment of the various treatment options for patients with X-linked SCID.
Using targeted genome editing by artificial nucleases is another interesting approach, although it is still in pre-clinical stages. These include the zinc finger nucleases (ZFNs), the transcription activator-like effector nucleases (TALENs) and the RNA-guided clustered regularly interspaced short palindromic repeats (CRISPR/Cas) nucleases that can efficiently and specifically cause a DNA break at a preselected site. Using ZFN and tailoring of delivery platforms and culture conditions, Naldini’s group were able to target a corrective cDNA into the IL2Rγ gene of stem cells from a patient with X-linked SCID, which led to normalization of hematopoiesis and generation of functional lymphoid cells [29].
Gene therapy for Wiskott Aldrich Syndrome
The WAS gene on the X chromosome encodes for the cytoplasmic WAS protein that affects actin polymerization in hematopoietic cells. WAS protein is important for leukocyte migration and formation of the immunologic synapse. WAS is characterized by increased susceptibility to infections, eczema, as well as the bleeding caused by the thrombocytopenia with platelets of low size and impaired function [30]. Patients also suffer from diverse autoimmune manifestations that may further contribute to the development of thrombocytopenia, vascular abnormalities and malignancies. Antibiotics and immunoglobulin prophylaxis as well as platelet transfusions and immune suppression may provide temporary relief for affected patients, yet most patients eventually develop life-threatening complications. Despite supportive care, the median life expectancy of patients is markedly reduced. Predicting outcome for specific patients based on the mutation, protein expression or clinical grading have been challenging, hence in most cases there should be an attempt to cure the disease. HSCT for WAS have been performed for almost 50 years, with excellent results, particularly if done early in life, with a well matched donor [31]. Indeed, the London group reported 100% survival rate in 34 patients treated between 1996 and 2016 using a variety of graft sources and tailored preparative regimens [32]. Nevertheless, GT is an attractive option for patients already harboring infections such as CMV, suffering from significant co-morbidities or lacking suitable HSCT donors.
The first WAS GT trial was performed between 2006 and 2009 in Munich, Germany and included 10 patients with severe phenotype. Patients received low doses of busulfan followed by transfusions of autologous CD34+ cells transduced with a WASP-expressing gamma-retroviral vector. GT reconstituted T cell function and antibody production. Platelets size normalized and their numbers increased, albeit often remaining below normal range, with resolution of hemorrhagic diatheses [33]. Yet, between 14 months and 5 years after gene therapy, 7 patients developed acute leukemia. Similar to the findings in X-linked SCID GT, the increased tendency of retroviruses to integrate near oncogenes, such as LMO2, was the probable reason. Subsequently, there have been 3 GT trials in Italy, the USA as well as France and England using a SIN LV and the endogenous WAS promoter [34–36]. As of April 2016, 8 patients received GT for WAS following reduced intensity conditioning in Milan, Italy (ClinicalTrials.gov Identifier: NCT01515462). At a median follow-up length of 3.8 years (range: 0.6–5.9), they are all alive and well. After immune reconstitution, marked reduction in severe infection rate was observed and 5 patients were able to stop immunoglobulin supplementation. There was a noticeable decrease in moderate-severe bleeding frequency and all patients became platelet transfusion independent, although platelet numbers have remained below normal. Importantly, no abnormal clonal proliferations were observed [37]. Seven patients suffering from WAS received GT in France (ClinicalTrials.gov Identifier: NCT01347346) and England (ClinicalTrials.gov Identifier: NCT01347242). At the time of the last reported follow-up, 6 were alive with no severe bleeding episodes, and were free of infections and leukemic events. One patient died 7 months after GT due to preexisting, refractory herpes virus infections. GT was also beneficial in a 30 year old patient with severe WAS manifesting with multiple inflammatory complications and lympho-proliferation who required long-term immunosuppressive treatment for disease control [38]. Another LV GT trial for WAS at Boston, USA (ClinicalTrials.gov Identifier: NCT01410825) has enrolled two patients who are reported to have improved immune and hematologic parameters without genotoxicity at early time points. The trials in France, UK and USA are currently recruiting patients. It is expected that the results from these autologous GT studies will enable better comparison with those of allogenic HSCT, including susceptibility to autoimmunity that has been frequently reported following partial correction of WAS [39].
Gene therapy for JAK3 deficiency
The JAK3 protein kinase delivers signalling into the cells following stimulation of the γc. Apart from the inheritance, which is autosomal recessive in JAK3 deficiency, patient often display a SCID phenotype similar to that seen in γc deficient patients. A single JAK3-deficient patient who failed HSCT received retroviral GT, however the results were only published in abstract form. Disappointingly, there was no evidence of immune reconstitution at 7 months posttreatment [40]. This clinical trial was stopped following the occurrence of leukemia in X-linked SCID GT.
Gene therapy for chronic granulomatous disease
CGD is caused by impaired function of the NADPH oxidase complex that is important for the production of reactive oxygen species in phagocytes. Consequently, patients are susceptible to infections by catalase-positive microorganisms such as Staphylococcus aureus, Nocardia spp, Serratia marcescens, Burkholderia cepacea and Salmonella spp as well as Aspergillus species. Infected areas typically include the lung, lymph nodes, liver, bones and skin. Dysregulated immune responses often result in granuloma formation and other inflammatory disorders involving the bowel. The most common form of CGD is caused by defects in the in the X-linked CYBB gene, which encodes gp91phox. Recognizing the poor long-term prognosis of patients with CGD, particularly those with markedly reduced neutrophil oxidative burst [41] and improvement in transplantation techniques have led to increasing numbers of patients who have benefited from HSCT [42]. Nevertheless, since patients often experience graft rejection or develop GvH disease and inflammatory exacerbations, GT for the X-linked CYBB gene defects has been explored. The first GT trial for CGD used a gamma-retroviral vectors to deliver human gp91phox. However, only few gene-corrected cells persisted, possibly because no pre-GT conditioning was given. Three patients who received GT at National Institute of Health (NIH) following reduced intensity conditioning showed slightly better engraftment and some clinical improvement; However 1 patient died 6 months after GT from a fungal infection [43]. GT performed in Germany involving 2 patients with CGD, using a similar vector, albeit with a different transcriptional control, led to the correction of 15% of the neutrophils shortly after treatment. Unfortunately, both patients developed fatal myelodysplasia secondary to insertional mutagenesis [44]. Similar complications were also noted in 2 additional children with CGD treated in Switzerland, and the patients were rescued with HSCT. Patients with CGD also received unsuccessful GT in London (4 patients) and Seoul (2 patients). To improve efficacy and safety of GT for CGD, SIN gamma-retroviral vector and a LV vector expressing gp91phox were developed with preparative conditioning regimens that are effective and well tolerated. Moreover, the newer constructs carry myeloid-specific promoters and/or allow for post-transcriptional down-regulation of expression in hematopoietic stem cells. Currently 3 US sites (NIH, Boston and Los Angeles) are recruiting patients with CGD who are 23 months or older for a trial with a 3rd generation SIN LV, which directs gp91phox expression from a codon-optimized form of the CYBB gene preferentially to myeloid cells (ClinicalTrials.gov Identifier: NCT02234934). Similar studies using LV are being conducted in Frankfurt, London and Zurich (ClinicalTrials.gov Identifier: NCT01855685), while the site in Paris is also accepting younger children (ClinicalTrials.gov Identifier: NCT02757911). So far, a single child with CGD and invasive liver, brain, abdominal, and pulmonary infections, and inflammatory complications received GT in Europe. He was reported to be stable for 3 months post GT, but then developed fatal respiratory complications [45].
Gene therapy for leukocyte adhesion defect
LAD is characterized by delayed separation of the cord, neutrophilia, severe gingivitis and periodontitis, and recurrent, cutaneous, non-healing wounds lacking puss formation. Patients commonly suffer from severe recurrent systemic bacterial infections. The classical form of LAD is caused by defects in the CD18 gene, also known as the beta-2 subunit of the leukocyte integrin family or ITGB2. Allogeneic HSCT are the only definitive cure for LAD, but complete donor engraftment has been difficult to achieve [46]. Two patients with severe LAD received RV mediated CD18 gene-corrected stem cells. After the infusion, only 0.1% of the patients’ neutrophils expressed CD18, and these cells disappeared within 2 months of GT. Subsequent GT for LAD has been restricted to animal models [47].
Gene therapy for other PID
GT for other PID are currently at various in vitro and in vivo pre-clinical stages (Table 1), often needing to address unique challenges associated with specific diseases.
The future of gene therapy
In recent years, advances in gene manipulation and vector design are expected to bring GT closer to clinical reality. A major breakthrough is the development of site-specific gene editing tools. By creating site-specific breaks in the DNA near the location of a known mutation, a cell’s natural repair mechanisms can be utilized to incorporate normal segments of DNA. This strategy positions genes in their endogenous locations under the control of normal regulatory elements, thereby decreasing the risk of insertional mutagenesis or ectopic protein expression. GT for PID uses HSC derived from patients’ bone marrow, mobilized peripheral mononuclear cells, or rarely from the recipient’s own cord blood. In the upcoming years, advances in reprograming blood, skin and other tissues into pluri-potent stem cells (iPSC) followed by ex vivo differentiation of these cells into hematopoietic and immune lineages are expected to limit the need for invasive procedures. Another important development is the ability to efficiently freeze, thaw and expand stem cells. This may circumvent the need to send Canadian patients, and families, out of the country to the few locations where gene delivery is currently being performed. Centralizing stem cells manipulation and gene delivery will enable resources and expertise to concentrate at specific GMP facilities, while allowing patients to continue receiving care at local Canadian centers experienced in transplantations. Centralization will also facilitate monitoring for long-term complications secondary to GT and conduction of novel GT trials.
Conclusions
The use of GT to cure PID is developing rapidly. While there are still significant challenges, the recently improved safety and efficacy measures in GT suggest that such treatment may soon become a standard of care for diverse PID, including many affected Canadian patients.
Abbreviations
- ADA:
-
adenosine deaminase
- CGD:
-
chronic granulomatous disease
- CRISPR:
-
clustered regularly interspaced short palindromic repeats
- γc:
-
gamma chain
- GT:
-
gene therapy
- GvH:
-
graft versus host
- HIV:
-
human immunodeficiency virus type 1
- HLA:
-
human leukocyte antigens
- HLH:
-
hemophagocytic lymphohistiocytosis
- HSC:
-
hematopoietic stem cell
- HSCT:
-
hematopoietic stem cell transplantation
- IL2Rγ:
-
interleukin-2 receptor gamma
- IPEX:
-
immune dysregulation, polyendocrinopathy, enteropathy, X-linked
- IPSCs:
-
induced pluripotent stem cells
- JAK:
-
Janus kinase
- LAD:
-
leukocyte adhesion deficiency
- LV:
-
lentivirus
- MLV:
-
Murine leukemia virus
- NIH:
-
National Institute of Health
- PEG-ADA:
-
polyethylene glycol-modified adenosine deaminase
- PID:
-
primary immunodeficiency diseases
- RAG:
-
recombination activating gene
- SCID:
-
severe combined immunodeficiency
- SCIDX-1:
-
X-linked severe combined immunodeficiency
- SIN:
-
self-inactivation
- TALEN:
-
transcription activator-like effector nucleases
- WAS:
-
Wiskott–Aldrich syndrome
- ZFNs:
-
zinc-finger nucleases
- NBS:
-
newborn screening
References
Meyts I, Bosch B, Bolze A, Boisson B, Itan Y, Belkadi A, Pedergnana V, Moens L, Picard C, Cobat A, Bossuyt X, Abel L, Casanova JL. Exome and genome sequencing for inborn errors of immunity. J Allergy Clin Immunol. 2016;138(4):957–69. doi:10.1016/j.jaci.2016.08.003.
Bach FH, Albertini RJ, Joo P, Anderson JL, Bortin MM. Bone-marrow transplantation in a patient with the Wiskott–Aldrich syndrome. Lancet. 1968;2(7583):1364–6.
Gatti RA, Meuwissen HJ, Allen HD, Hong R, Good RA. Immunological reconstitution of sex-linked lymphopenic immunological deficiency. Lancet. 1968;2(7583):1366–9.
Grunebaum E, Mazzolari E, Porta F, Dallera D, Atkinson A, Reid B, Notarangelo LD, Roifman CM. Bone marrow transplantation for severe combined immune deficiency. JAMA. 2006;295(5):508–18.
Edelstein M. Gene therapy clinical trials world wide. J Gene Med Website. 2016. http://www.abedia.com/wiley/vectors.php.
Kaufmann KB, Büning H, Galy A, Schambach A, Grez M. Gene therapy on the move. EMBO Mol Med. 2013;5(11):1642–61. doi:10.1002/emmm.201202287.
Grunebaum E, Cohen A, Roifman CM. Recent advances in understanding and managing adenosine deaminase and purine nucleoside phosphorylase deficiencies. Curr Opin Allergy Clin Immunol. 2013;13(6):630–8. doi:10.1097/ACI.0000000000000006.
Grunebaum E, Cutz E, Roifman CM. Pulmonary alveolar proteinosis in patients with adenosine deaminase deficiency. J Allergy Clin Immunol. 2012;129(6):1588–93. doi:10.1016/j.jaci.2012.02.003.
Rogers MH, Lwin R, Fairbanks L, Gerritsen B, Gaspar HB. Cognitive and behavioral abnormalities in adenosine deaminase deficient severe combined immunodeficiency. J Pediatr. 2001;139(1):44–50.
Manson D, Diamond L, Oudjhane K, Hussain FB, Roifman C, Grunebaum E. Characteristic scapular and rib changes on chest radiographs of children with ADA-deficiency SCIDS in the first year of life. Pediatr Radiol. 2013;43(5):589–92. doi:10.1007/s00247-012-2564-2.
Hershfield MS, Buckley RH, Greenberg ML, Melton AL, Schiff R, Hatem C, Kurtzberg J, Markert ML, Kobayashi RH, Kobayashi AL, et al. Treatment of adenosine deaminase deficiency with polyethylene glycol-modified adenosine deaminase. N Engl J Med. 1987;316(10):589–96.
Booth C, Hershfield M, Notarangelo L, Buckley R, Hoenig M, Mahlaoui N, Cavazzana-Calvo M, Aiuti A, Gaspar HB. Management options for adenosine deaminase deficiency; proceedings of the EBMT satellite workshop (Hamburg, March 2006). Clin Immunol. 2007;123(2):139–47.
Gaspar HB, Aiuti A, Porta F, Candotti F, Hershfield MS, Notarangelo LD. How i treat ADA deficiency. Blood. 2009;114(17):3524–32. doi:10.1182/blood-2009-06-189209.
Hassan A, Booth C, Brightwell A, Allwood Z, Veys P, Rao K, Hönig M, Friedrich W, Gennery A, Slatter M, Bredius R, Finocchi A, Cancrini C, Aiuti A, Porta F, Lanfranchi A, Ridella M, Steward C, Filipovich A, Marsh R, Bordon V, Al-Muhsen S, Al-Mousa H, Alsum Z, Al-Dhekri H, Al Ghonaium A, Speckmann C, Fischer A, Mahlaoui N, Nichols KE, Grunebaum E, Al Zahrani D, Roifman CM, Boelens J, Davies EG, Cavazzana-Calvo M, Notarangelo L, Gaspar HB, Inborn Errors Working Party of the European Group for Blood and Marrow Transplantation and European Society for Immunodeficiency. Outcome of hematopoietic stem cell transplantation for adenosine deaminase-deficient severe combined immunodeficiency. Blood. 2012;120(17):3615–24. doi:10.1182/blood-2011-12-396879 (quiz 3626).
Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, Morecki S, Andolfi G, Tabucchi A, Carlucci F, Marinello E, Cattaneo F, Vai S, Servida P, Miniero R, Roncarolo MG, Bordignon C. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296(5577):2410–3.
Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, Scaramuzza S, Andolfi G, Mirolo M, Brigida I, Tabucchi A, Carlucci F, Eibl M, Aker M, Slavin S, Al-Mousa H, Al Ghonaium A, Ferster A, Duppenthaler A, Notarangelo L, Wintergerst U, Buckley RH, Bregni M, Marktel S, Valsecchi MG, Rossi P, Ciceri F, Miniero R, Bordignon C, Roncarolo MG. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360(5):447–58. doi:10.1056/NEJMoa0805817.
Sauer AV, Mrak E, Hernandez RJ, Zacchi E, Cavani F, Casiraghi M, Grunebaum E, Roifman CM, Cervi MC, Ambrosi A, Carlucci F, Roncarolo MG, Villa A, Rubinacci A, Aiuti A. ADA-deficient SCID is associated with a specific microenvironment and bone phenotype characterized by RANKL/OPG imbalance and osteoblast insufficiency. Blood. 2009;114(15):3216–26. doi:10.1182/blood-2009-03-209221.
Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Zhang F, Adams S, Bjorkegren E, Bayford J, Brown L, Davies EG, Veys P, Fairbanks L, Bordon V, Petropoulou T, Kinnon C, Thrasher AJ. Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci Transl Med. 2011;3(97):97. doi:10.1126/scitranslmed.3002716.
Candotti F, Shaw KL, Muul L, Carbonaro D, Sokolic R, Choi C, Schurman SH, Garabedian E, Kesserwan C, Jagadeesh GJ, Fu PY, Gschweng E, Cooper A, Tisdale JF, Weinberg KI, Crooks GM, Kapoor N, Shah A, Abdel-Azim H, Yu XJ, Smogorzewska M, Wayne AS, Rosenblatt HM, Davis CM, Hanson C, Rishi RG, Wang X, Gjertson D, Yang OO, Balamurugan A, Bauer G, Ireland JA, Engel BC, Podsakoff GM, Hershfield MS, Blaese RM, Parkman R, Kohn DB. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood. 2012;120(18):3635–46. doi:10.1182/blood-2012-02-400937.
Otsu M, Yamada M, Nakajima S, Kida M, Maeyama Y, Hatano N, Toita N, Takezaki S, Okura Y, Kobayashi R, Matsumoto Y, Tatsuzawa O, Tsuchida F, Kato S, Kitagawa M, Mineno J, Hershfield MS, Bali P, Candotti F, Onodera M, Kawamura N, Sakiyama Y, Ariga T. Outcomes in two Japanese adenosine deaminase-deficiency patients treated by stem cell gene therapy with no cytoreductive conditioning. J Clin Immunol. 2015;35(4):384–98. doi:10.1007/s10875-015-0157-1.
Cicalese MP, Ferrua F, Castagnaro L, Pajno R, Barzaghi F, Giannelli S, Dionisio F, Brigida I, Bonopane M, Casiraghi M, Tabucchi A, Carlucci F, Grunebaum E, Adeli M, Bredius RG, Puck JM, Stepensky P, Tezcan I, Rolfe K, De Boever E, Reinhardt RR, Appleby J, Ciceri F, Roncarolo MG, Aiuti A. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood. 2016;128(1):45–54. doi:10.1182/blood-2016-01-688226.
Buckley RH, Schiff SE, Schiff RI, Markert L, Williams LW, Roberts JL, Myers LA, Ward FE. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl J Med. 1999;340(7):508–16.
Dalal I, Tasher D, Somech R, Etzioni A, Garti BZ, Lev D, Cohen S, Somekh E, Leshinsky-Silver E. Novel mutations in RAG1/2 and ADA genes in Israeli patients presenting with T-B-SCID or Omenn syndrome. Clin Immunol. 2011;140(3):284–90. doi:10.1016/j.clim.2011.04.011.
Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, Kapoor N, Hanson IC, Filipovich AH, Jyonouchi S, Sullivan KE, Small TN, Burroughs L, Skoda-Smith S, Haight AE, Grizzle A, Pulsipher MA, Chan KW, Fuleihan RL, Haddad E, Loechelt B, Aquino VM, Gillio A, Davis J, Knutsen A, Smith AR, Moore TB, Schroeder ML, Goldman FD, Connelly JA, Porteus MH, Xiang Q, Shearer WT, Fleisher TA, Kohn DB, Puck JM, Notarangelo LD, Cowan MJ, O’Reilly RJ. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med. 2014;371(5):434–46. doi:10.1056/NEJMoa1401177.
Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC, Martinache C, Rieux-Laucat F, Latour S, Belohradsky BH, Leiva L, Sorensen R, Debré M, Casanova JL, Blanche S, Durandy A, Bushman FD, Fischer A, Cavazzana-Calvo M. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2010;363(4):355–64. doi:10.1056/NEJMoa1000164.
Gaspar HB, Parsley KL, Howe S, King D, Gilmour KC, Sinclair J, Brouns G, Schmidt M, Von Kalle C, Barington T, Jakobsen MA, Christensen HO, Al Ghonaium A, White HN, Smith JL, Levinsky RJ, Ali RR, Kinnon C, Thrasher AJ. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet. 2004;364(9452):2181–7.
Hacein-Bey-Abina S, Pai SY, Gaspar HB, Armant M, Berry CC, Blanche S, Bleesing J, Blondeau J, de Boer H, Buckland KF, Caccavelli L, Cros G, De Oliveira S, Fernández KS, Guo D, Harris CE, Hopkins G, Lehmann LE, Lim A, London WB, van der Loo JC, Malani N, Male F, Malik P, Marinovic MA, McNicol AM, Moshous D, Neven B, Oleastro M, Picard C, Ritz J, Rivat C, Schambach A, Shaw KL, Sherman EA, Silberstein LE, Six E, Touzot F, Tsytsykova A, Xu-Bayford J, Baum C, Bushman FD, Fischer A, Kohn DB, Filipovich AH, Notarangelo LD, Cavazzana M, Williams DA, Thrasher AJ. A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med. 2014;371(15):1407–17. doi:10.1056/NEJMoa1404588.
De Ravin SS, Wu X, Moir S, Anaya-O’Brien S, Kwatemaa N, Littel P, Theobald N, Choi U, Su L, Marquesen M, Hilligoss D, Lee J, Buckner CM, Zarember KA, O’Connor G, McVicar D, Kuhns D, Throm RE, Zhou S, Notarangelo LD, Hanson IC, Cowan MJ, Kang E, Hadigan C, Meagher M, Gray JT, Sorrentino BP, Malech HL. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med. 2016;8(335):335ra57. doi:10.1126/scitranslmed.aad8856.
Genovese P, Schiroli G, Escobar G, Di Tomaso T, Firrito C, Calabria A, Moi D, Mazzieri R, Bonini C, Holmes MC, Gregory PD, van der Burg M, Gentner B, Montini E, Lombardo A, Naldini L. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510(7504):235–40. doi:10.1038/nature13420.
Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2006;117(4):725–38 quiz 739.
Moratto D, Giliani S, Bonfim C, Mazzolari E, Fischer A, Ochs HD, Cant AJ, Thrasher AJ, Cowan MJ, Albert MH, Small T, Pai SY, Haddad E, Lisa A, Hambleton S, Slatter M, Cavazzana-Calvo M, Mahlaoui N, Picard C, Torgerson TR, Burroughs L, Koliski A, Neto JZ, Porta F, Qasim W, Veys P, Kavanau K, Hönig M, Schulz A, Friedrich W, Notarangelo LD. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott–Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980–2009: an international collaborative study. Blood. 2011;118(6):1675–84. doi:10.1182/blood-2010-11-319376.
Elfeky R, Juliana S, Chiesa R, Rao K, Amrolia P, Lucchini G, Worth A, Thrasher A, Qasim W, Veys P. The London Wiskott–Aldrich syndrome cohort: 100% survival of 34 patients transplanted over a 20 year period. ESID6-0369. European Society of Immunodeficiency, Barcelona, Spain; 2016.
Braun CJ, Boztug K, Paruzynski A, Witzel M, Schwarzer A, Rothe M, Modlich U, Beier R, Göhring G, Steinemann D, Fronza R, Ball CR, Haemmerle R, Naundorf S, Kühlcke K, Rose M, Fraser C, Mathias L, Ferrari R, Abboud MR, Al-Herz W, Kondratenko I, Maródi L, Glimm H, Schlegelberger B, Schambach A, Albert MH, Schmidt M, von Kalle C, Klein C. Gene therapy for Wiskott–Aldrich syndrome—long-term efficacy and genotoxicity. Sci Transl Med. 2014;6(227):227ra33. doi:10.1126/scitranslmed.3007280.
Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, Dionisio F, Calabria A, Giannelli S, Castiello MC, Bosticardo M, Evangelio C, Assanelli A, Casiraghi M, Di Nunzio S, Callegaro L, Benati C, Rizzardi P, Pellin D, Di Serio C, Schmidt M, Von Kalle C, Gardner J, Mehta N, Neduva V, Dow DJ, Galy A, Miniero R, Finocchi A, Metin A, Banerjee PP, Orange JS, Galimberti S, Valsecchi MG, Biffi A, Montini E, Villa A, Ciceri F, Roncarolo MG, Naldini L. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott–Aldrich syndrome. Science. 2013;341(6148):1233151. doi:10.1126/science.1233151.
Hacein-Bey Abina S, Gaspar HB, Blondeau J, Caccavelli L, Charrier S, Buckland K, Picard C, Six E, Himoudi N, Gilmour K, McNicol AM, Hara H, Xu-Bayford J, Rivat C, Touzot F, Mavilio F, Lim A, Treluyer JM, Héritier S, Lefrère F, Magalon J, Pengue-Koyi I, Honnet G, Blanche S, Sherman EA, Male F, Berry C, Malani N, Bushman FD, Fischer A, Thrasher AJ, Galy A, Cavazzana M. Outcomes following gene therapy in patients with severe Wiskott–Aldrich syndrome. JAMA. 2015;313(15):1550–63. doi:10.1001/jama.2015.3253.
Candotti F. Advances of gene therapy for primary immunodeficiencies. F1000Res. 2016;5. pii: F1000 Faculty Rev-310. doi: 10.12688/f1000research.7512.1.
Ferrua F, Cicalese MP, Galimberti S, Giannelli S, F Dionisio, Bernardo ME, Migliavacca M, Barzaghi F, Assanelli A, Scaramuzza S, Brigda I, Salerio F, Panjo R, Castiello MC, Casiraghi M, Facchini M, Fossati C, Finocchi A, Metin A, Orange JS, Albert MH, Petrescu CAM, Xhafa M, Pesce F, Pignata C, Farah RA, Bosticardo A, Villa A, Dott C, van Rossem K, Valsecchia MG, Cicer F, Roncarolo MG, Naldini L, Aiuti A. Safety and clinical benefit of lentiviral hematopoietic stem cell gene therapy for Wiskott–Aldrich syndrome: the TIGIT-WAS clinical trial. ESID6-0935. European Society for Immunodeficiency. Barcelona, Spain; 2016.
Fox T, Morris E, Chakraverty R, Snell K, Rivat C, Gilmour K, Buckland K, Butler K, Salama A, Hara A, Burns S, Galy A, Gaspar B, Thrasher A. Gene therapy for Wiskott–Aldrich syndrome in a severely affected adult. ESID6-0325 European Society of Immunodeficiency, Barcelona, Spain; 2016.
Crestani E, Volpi S, Candotti F, Giliani S, Notarangelo LD, Chu J, Aldave Becerra JC, Buchbinder D, Chou J, Geha RS, Kanariou M, King A, Mazza C, Moratto D, Sokolic R, Garabedian E, Porta F, Putti MC, Wakim RH, Tsitsikov E, Pai SY, Notarangelo LD. Broad spectrum of autoantibodies in patients with Wiskott–Aldrich syndrome and X-linked thrombocytopenia. J Allergy Clin Immunol. 2015;136(5):1401–4. doi:10.1016/j.jaci.2015.08.010.
Sorrentino BP, Lu T, Ihle J. A clinical attempt to treat JAK3-deficient SCID using retroviral-mediated gene transfer to bone marrow CD34+ cells. Mol Ther. 2003;7(5S):S449.
Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, Uzel G, DeRavin SS, Priel DA, Soule BP, Zarember KA, Malech HL, Holland SM, Gallin JI. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363(27):2600–10. doi:10.1056/NEJMoa1007097.
Güngör T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, Vermont C, Ahmad I, Shaw PJ, Telles da Cunha JM, Schlegel PG, Hough R, Fasth A, Kentouche K, Gruhn B, Fernandes JF, Lachance S, Bredius R, Resnick IB, Belohradsky BH, Gennery A, Fischer A, Gaspar HB, Schanz U, Seger R, Rentsch K, Veys P, Haddad E, Albert MH, Hassan M, Inborn Errors Working Party of the European Society for Blood and Marrow Transplantation. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. 2014;383(9915):436–48. doi:10.1016/S0140-6736(13)62069-3.
Kang EM, Choi U, Theobald N, Linton G, Long Priel DA, Kuhns D, Malech HL. Retrovirus gene therapy for X-linked chronic granulomatous disease can achieve stable long-term correction of oxidase activity in peripheral blood neutrophils. Blood. 2010;115(4):783–91. doi:10.1182/blood-2009-05-222760.
Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, Schmidt M, Krämer A, Schwäble J, Glimm H, Koehl U, Preiss C, Ball C, Martin H, Göhring G, Schwarzwaelder K, Hofmann WK, Karakaya K, Tchatchou S, Yang R, Reinecke P, Kühlcke K, Schlegelberger B, Thrasher AJ, Hoelzer D, Seger R, von Kalle C, Grez M. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16(2):198–204. doi:10.1038/nm.2088.
Kuo CY, Kohn DB. Gene therapy for the treatment of primary immune deficiencies. Curr Allergy Asthma Rep. 2016;16(5):39. doi:10.1007/s11882-016-0615-8.
Qasim W, Cavazzana-Calvo M, Davies EG, Davis J, Duval M, Eames G, Farinha N, Filopovich A, Fischer A, Friedrich W, Gennery A, Heilmann C, Landais P, Horwitz M, Porta F, Sedlacek P, Seger R, Slatter M, Teague L, Eapen M, Veys P. Allogeneic hematopoietic stem-cell transplantation for leukocyte adhesion deficiency. Pediatrics. 2009 Mar;123(3):836–40. doi:10.1542/peds.2008-1191.
Bauer TR Jr, Gu YC, Creevy KE, Tuschong LM, Embree L, Holland SM, Sokolic RA, Hickstein DD. Leukocyte adhesion deficiency in children and Irish setter dogs. Pediatr Res. 2004;55(3):363–7.
Pike-Overzet K, Rodijk M, Ng YY, Baert MR, Lagresle-Peyrou C, Schambach A, Zhang F, Hoeben RC, Hacein-Bey-Abina S, Lankester AC, Bredius RG, Driessen GJ, Thrasher AJ, Baum C, Cavazzana-Calvo M, van Dongen JJ, Staal FJ. Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer. Leukemia. 2011;25(9):1471–83. doi:10.1038/leu.2011.106.
van Til NP, Sarwari R, Visser TP, Hauer J, Lagresle-Peyrou C, van der Velden G, Malshetty V, Cortes P, Jollet A, Danos O, Cassani B, Zhang F, Thrasher AJ, Fontana E, Poliani PL, Cavazzana M, Verstegen MM, Villa A, Wagemaker G. Recombination-activating gene 1 (Rag1)-deficient mice with severe combined immunodeficiency treated with lentiviral gene therapy demonstrate autoimmune Omenn-like syndrome. J Allergy Clin Immunol. 2014;133(4):1116–23. doi:10.1016/j.jaci.2013.10.009.
Punwani D, Kawahara M, Yu J, Sanford U, Roy S, Patel K, Carbonaro DA, Karlen AD, Khan S, Cornetta KG, Rothe M, Schambach A, Kohn DB, Malech HL, McIvor RS, Puck JM, Cowan MJ. Lentivirus mediated correction of artemis-deficient severe combined immunodeficiency. Hum Gene Ther. 2017;28(1):112–24. doi:10.1089/hum.2016.064.
Pacheco-Castro A, Martín-Fernández JM, Millán R, Sanal O, Allende L, Regueiro JR. Hum Gene Ther. 2004;14(17):1653–61. doi:10.1089/104303403322542293.
Lagresle-Peyrou C, Six EM, Picard C, Rieux-Laucat F, Michel V, Ditadi A, Demerens-de Chappedelaine C, Morillon E, Valensi F, Simon-Stoos KL, Mullikin JC, Noroski LM, Besse C, Wulffraat NM, Ferster A, Abecasis MM, Calvo F, Petit C, Candotti F, Abel L, Fischer A, Cavazzana-Calvo M. Human adenylate kinase 2 deficiency causes a profound hematopoietic defect associated with sensorineural deafness. Nat Genet. 2009;41(1):106–11. doi:10.1038/ng.278.
Liao P, Toro A, Min W, Lee S, Roifman CM, Grunebaum E. Lentivirus gene therapy for purine nucleoside phosphorylase deficiency. J Gene Med. 2008;10(12):1282–93. doi:10.1002/jgm.1261.
Adjali O, Marodon G, Steinberg M, Mongellaz C, Thomas-Vaslin V, Jacquet C, Taylor N, Klatzmann D. In vivo correction of ZAP-70 immunodeficiency by intrathymic gene transfer. J Clin Invest. 2005;115(8):2287–95.
Irla M, Saade M, Kissenpfennig A, Poulin LF, Leserman L, Marche PN, Jouvin-Marche E, Berger F, Nguyen C. ZAP-70 restoration in mice by in vivo thymic electroporation. PLoS ONE. 2008;3(4):e2059. doi:10.1371/journal.pone.0002059.
Bradley MB, Fernandez JM, Ungers G, Diaz-Barrientos T, Steimle V, Mach B, O’Reilly R, Lee JS. Correction of defective expression in MHC class II deficiency (bare lymphocyte syndrome) cells by retroviral transduction of CIITA. J Immunol. 1997;159(3):1086–95.
Kuo CY, Joglekar A, Kohn D. Targeted gene therapy in the treatment of X-linked hyper-IgM syndrome. JACI. 2014;133(2):AB162. doi:10.1016/j.jaci.2013.12.588.
Hubbard N, Hagin D, Sommer K, Song Y, Khan I, Clough C, Ochs HD, Rawlings DJ, Scharenberg AM, Torgerson TR. Targeted gene editing restores regulated CD40L function in X-linked hyper-IgM syndrome. Blood. 2016;127(21):2513–22. doi:10.1182/blood-2015-11-683235.
Passerini L, Rossi Mel E, Sartirana C, Fousteri G, Bondanza A, Naldini L, Roncarolo MG, Bacchetta R. CD4+ T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci Transl Med. 2013;5(215):215. doi:10.1126/scitranslmed.3007320.
Carmo M, Risma KA, Arumugam P, Tiwari S, Hontz AE, Montiel-Equihua CA, Alonso-Ferrero ME, Blundell MP, Schambach A, Baum C, Malik P, Thrasher AJ, Jordan MB, Gaspar HB. Perforin gene transfer into hematopoietic stem cells improves immune dysregulation in murine models of perforin deficiency. Mol Ther. 2015;23(4):737–45. doi:10.1038/mt.2014.242.
Tiwari S, Hontz A, Terrell CE, Arumugam P, Carmo M, Risma K, Jordan M, Malik P. High level of perforin expression is required for effective correction of hemophagocytic lymphohistiocytosis. Hum Gene Ther. 2016 [Epub ahead of print].
Rivat C, Booth C, Alonso-Ferrero M, et al. SAP gene transfer restores cellular and humoral immune function in a murine model of X-linked lymphoproliferative disease. Blood. 2013;121(7):1073–6.
Wang Y, Khan IF, Boissel S, Jarjour J, Pangallo J, Thyme S, Baker D, Scharenberg AM, Rawlings DJ. Progressive engineering of a homing endonuclease genome editing reagent for the murine X-linked immunodeficiency locus. Nucleic Acids Res. 2014;42(10):6463–75. doi:10.1093/nar/gku224.
Authors’ contributions
EG contributed to the conception, drafting and writing of the manuscript. XX and CST contributed to the revision and intellectual content of this manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was supported in part by the Audrey and Donald Campbell Chair for Immunology Research to EG. The authors would like to thank the patients and families affected by primary immunodeficiency who have contributed to the advancement of the field.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Consent for publication
All authors provided consent for publication.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xu, X., Tailor, C.S. & Grunebaum, E. Gene therapy for primary immune deficiencies: a Canadian perspective. Allergy Asthma Clin Immunol 13, 14 (2017). https://doi.org/10.1186/s13223-017-0184-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13223-017-0184-y