
Abstract
Purpose
d-psicose-3-epimerase (DPEase) catalyses the isomerisation of d-fructose to d-psicose, a rare sugar in nature with unique nutritional and biological functions. An effective industrial-scale method is needed for d-psicose production. Herein, the expression of a neutral and a slightly acidic pH DPEase in Bacillus subtilis was evaluated.
Methods
Two DPEase genes from Clostridium bolteae and Dorea sp. were separately expressed in B. subtilis via plasmid pSTOP1622, and an extra P43 promoter was employed to the expression cassette. The fermentation conditions of the engineered B. subtilis strains were also optimised, to facilitate both cell growth and enzyme production.
Result
The introduction of P43 promoter to the two DPEase genes increased enzyme production by about 20%. Optimisation of fermentation conditions increased DPEase production to 21.90 U/g at 55 °C and 24.01 U/g at 70 °C in B. subtilis expressing C. bolteae or Dorea sp. DPEase, equating to a 94.67% and 369.94% increase, respectively, relative to controls.
Conclusion
Enhanced DPEase production was achieved in B. subtilis expressing C. bolteae or Dorea sp. DPEase genes.
Similar content being viewed by others

Introduction
d-psicose, the C-3 epimer of d-fructose, is rarely found in nature. It has 70% relative sweetness but 0.3% energy of sucrose and is suggested as an ideal substitute for sucrose in foodstuffs due to its low energy and safety (generally recognised as safe (GRAS)) (Zhang et al. 2017b). Recently, d-psicose has attracted increasing attention for its ability reduce blood glucose levels and intra-abdominal fat accumulation, protect against atherosclerosis, and scavenge reactive oxygen species (ROS) (Chen et al. 2019; Shintani et al. 2017). Also, some d-psicose derivatives exhibit anti-cancer and anti-viral activities (Lim and Oh 2011; Yadav et al. 2018). Currently, d-psicose is only found in limited quantities in some natural plants such as cane and wheat (Mu et al. 2012). Therefore, it is necessary to develop a method for d-psicose production to meet the increasing market demand.
In the Izumoring strategy, biological production of d-psicose from d-fructose is catalysed by d-psicose-3-epimerase (DPEase) (Granstrom et al. 2004). Heterologous expression of microbial DPEases have been achieved in Escherichia coli, with d-psicose productivity of 20 to 30% (Jia et al. 2014; Tseng et al. 2018; Zhang et al. 2015). Nevertheless, products from recombinant E. coil can be controversial for food-grade production (Chen et al. 2016a). As a promising host, the Gram-positive bacterium Bacillus subtilis is attractive because it is non-pathogenic and has acquired GRAS status (Tännler et al. 2008). Moreover, B. subtilis exhibits no codon preferences and does not produce inclusion bodies, both of which may dramatically enhance the production of enzymes (Cui et al. 2017). B. subtilis is now one of the most commonly used hosts in industrial enzyme production processes.
During industrial monosaccharide bioconversion processes, maintaining a slightly acidic pH can effectively reduce non-enzymatic reactions and the accumulation of unnecessary by-products (Sang-Jae et al. 2005). For all DPEases identified to date, the most suitable working conditions are weak alkalinity, except for two enzymes from Clostridium bolteae and Dorea sp. (Jia et al. 2014; Zhang et al. 2015). Heterologous expression of DPEase from Ruminococcus sp. DPEase, Clostridium scindens, and Agrobacterium tumefaciens in B. subtilis cells or spores has been accomplished (Chen et al. 2016a; He et al. 2016a; He et al. 2016b). However, the expression of DPEases from C. bolteae and Dorea sp. has received minimal attention. In industrial-scale processes, whole-cell systems can minimise damage from external environmental factors, maintain stability under high-temperature conditions (Zhang et al. 2017a), and facilitate the application of desired enzymes.
In the present study, production of d-psicose was achieved via a whole-cell reaction by expressing DPEase from C. bolteae and Dorea sp. in B. subtilis. Additionally, the strength of the promoter, induction conditions, and optimal fermentation conditions were investigated to maximise overproduction of DPEase.
Materials and methods
Bacterial strains, plasmids, and DNA manipulation
DPEase-encoding genes from C. bolteae ATCC BAA-613 (Genbank: CP022464.2) and Dorea sp. CAG317 (Genbank: FR892665.1) were artificially synthesised with codon optimisation by Sangon (Shanghai China), and the gene sequences were available in the Additional file 1. During the experimental process, most molecular biology manipulation methods used for plasmid construction and gene amplification by PCR were performed using standard reagents and procedures. CaCl2-mediated transformation of E. coli competent cells and two-step transformation of B. subtilis were performed as described previously (Kim 2017).
Culture media and growth condition
Strains used in this study are listed in Table 1. E. coli DH5α (TaKaRa, Dalian, China) served as a host for cloning manipulation and plasmid amplification. B. subtilis WB800 was used as a host for DPEase expression. The inducible shuttle vector pSTOP1622 was used for DPEase gene expression. E. coli and B. subtilis WB800 transformants were incubated in Luria-Bertani (LB) medium containing peptone (1%), yeast extract (0.5%), and NaCl (1%), supplemented with ampicillin (100 μg/mL) and agar (1%) as appropriate. Engineered B. subtilis cells were fermented in basal medium containing glucose (10 g/L), yeast extract (15 g/L), Na2HPO4 (1 g/L), MgSO4·7H2O (0.5 g/L), and NaCl (8 g/L), supplemented with tetracycline (20 μg/mL) as appropriate. E. coli and B. subtilis strains were incubated at 37 °C with shaking at 200 r/min. All experiments were repeated at least three times, and mean values were used for comparison.
Construction and transformation of recombinant plasmids
Primers used in this study are listed in Table 2. The endogenous P43 promoter was amplified from the B. subtilis WB800 chromosome using primer pairs P1/P2 and P1/P5. DPEase-encoding genes from C. bolteae (cb-dpe) and Dorea sp. (ds-dpe) were amplified via PCR using PUC-cb-dpe and PUC-ds-dpe templates and P3/P4 and P6/P7 primer pairs, respectively. Primer P2 is the reverse complement of primer P3, and primer P5 is the reverse complement of P7. The P43 promoter was fused with cb-dpe or ds-dpe to generate an expression cassette via splicing overlap extension PCR (SOE-PCR) using P1 and P4, or P1 and P7 primers, respectively. Finally, P43-cb-dpe and P43-ds-dpe expression cassettes were incorporated into the pMD-19T vector.
During the experimental process, a facile method was applied to construct the dpe gene expression plasmid, involving a sequence-independent approach without restriction enzymes or ligases. The procedure consists of three steps: (1) the expression vector pSTOP1622 was digested with BamH I; (2) DNA multimers were subsequently generated based on the vector fragments, and cb-dpe, ds-dpe, P43-cb-dpe, and P43-ds-dpe fragments were amplified by prolonged overlap extension PCR (POE-PCR) without primers. The resulting chimeric pSTOP1622-cb-dpe, pSTOP1622-ds-dpe, pSTOP1622-P43-cb-dpe, and pSTOP1622-P43-ds-dpe plasmids were transformed into E. coli DH5α, and transformants were grown on LB agar plates supplemented with ampicillin; and (3) DNA multimers were transformed into B. subtilis WB800 competent cells, and transformants were selected on LB agar plates containing tetracycline.
Preparation of crude enzymes and sodium dodecyl sulphate-polyacrylamide gel electrophoresis
B. subtilis cells harbouring C. bolteae or Dorea sp. DPEase were harvested by centrifugation at 8000 r/min for 5 min. After washing three times, recombinant cells were resuspended in 50 mM Tris-HCl (pH 7.5) buffer containing 20 mg/mL lysozyme and 500 mM NaCl. The cell suspension was incubated at 37 °C for 30 min, and cells were disrupted by sonication for 10 min on ice (300 W, 2 s pulses, 3 s pauses). Unbroken cells and cell debris were removed by centrifugation.
The crude enzyme was analysed by SDS-PAGE and Coomassie Brilliant Blue R-250 staining as described in a previous report (Wanmeng et al. 2011). Electrophoresis was performed using a 12% polyacrylamide gel at room temperature, and premixed protein markers were obtained from TaKaRa.
B. subtilis whole-cell reaction
To evaluate the B. subtilis whole-cell reaction, 500 μL of 100 g/L d-fructose in 50 mM Tris-HCl (pH 7.5) served as a substrate, and 500 μL of washed wet engineered B. subtilis WB800 strains resuspended in 50 mM phosphate-buffered saline (PBS) buffer (pH 7.5) and served as an enzyme solution for measuring DPEase activity. Reactions were performed at 55 °C for 10 min and terminated by boiling at 100 °C for 5 min (He et al. 2016c). The resulting d-psicose in the mixture was filtered with 0.22 μm membrane before being measured via high-performance liquid chromatography (HPLC) using a Sugar-Pak1 column (300 mm × 6.5 mm; Waters, Wexford, Ireland) and a 2414 refractive index detector (Waters, Wexford, Ireland). The column was eluted with fresh ultra-pure water at a constant flow rate of 0.4 mL/min at 85 °C (Kai et al. 2018). One unit of DPEase activity was defined as the amount of enzyme that catalysed the production of 1 μmol d-psicose per minute (Chen et al. 2017).
Optimisation of fermentation conditions
We examined the efficiency of DPEase assays using whole-cell reactions to determine optimal carbon and nitrogen sources. Glucose (1% w/v) in the basal medium was replaced separately with sucrose, glycerol, fructose, lactose, or starch. Yeast extract (2% w/v) in the basal medium was substituted by beef extract, soy peptone, tryptone, casein, or peptone.
To determine the optimal culture induction time, we varied the xylose inducing time from 0 to 12 h and calculated the recombinant DPEase enzyme activity, as described above. The optimum inducer concentration was also investigated by testing xylose concentrations from 0 to 25 g/L.
To explore the effect of pH on DPEase activity, substrate pH ranges from 5.0 to 9.0 were tested using three buffer systems (50 mM): acetate buffer (pH 4.5 to 6.0), phosphate buffer (pH 6.0 to 7.0), and Tris-HCl buffer (pH 7.0 to 9.0). Again, DPEase activity was measured using the whole-cell reaction method.
To address the effect of temperature on DPEase activity, we set the reaction temperature between 45 and 85 °C, and the activity of DPEase was measured according to the above method.
Results
Heterologous expression of DPEase in B. subtilis
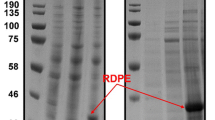
In this work, DPEase genes from C. bolteae and Dorea sp. strains were expressed in B. subtilis WB800 using the inducible episome plasmid pSTOP1622, resulting in engineered B. subtilis strains WB800-cb and WB800-ds. The theoretical molecular mass of recombinant Cb-DPEase and Ds-DPEase is 34.2 kDa and 32.7 kDa, respectively, based on the respective 302 and 290 amino acid sequences. Expression of DPEases in B. subtilis was induced by xylose, and recombinant enzymes were verified by SDS-PAGE analysis (Fig. 1). After incubation for 24 h in the basal medium at 37 °C with shaking at 200 r/min, the activities of recombinant DPEases from C. bolteae and Dorea sp. reached 11.25 U/g and 6.22 U/g, respectively.

SDS-PAGE analysis of recombinant DPEase expression in B. subtilis. Lane M, protein molecular weight markers; lane 1, recombinant strain WB800-P43-Cb; lane 2, recombinant strain WB800-Cb; lane 3, recombinant strain WB800-P43-Ds; lane 4, recombinant strain WB800-Ds; lane 5, strain WB800 (negative control)
Exploring the regulation effect of a dual-promoter system on DPEase expression
Promoters are important regulatory elements in expression systems, and dual promoter induced various expression effects in previous reports (Liu et al. 2019; Yang et al. 2013). To address the strength of promoter on DPEase expression, a constitutive promoter P43 was employed to the expression cassette (Fig. 2). The SDS-PAGE results indicated an increase in the protein expression level under the promoter of P43, compared to control (Fig. 1). Also, the introduction of P43 increased the DPEase activity to 13.62 U/g and 7.36 U/g in engineered B. subtilis strains harbouring C. bolteae and Dorea sp. DPEase genes, 21.07% and 18.32% higher than that of the control, respectively.

Construction of a P43-DPE tandem promoter expression cassette
Optimisation of xylose induction conditions
The PxylA promoter in vector pSTOP1622 is strictly regulated by xylose (Bhavsar et al. 2001). To optimise the DPEase production, a more detailed analysis of B. subtilis WB800-cb and WB800-ds cultivation was performed. With 0.5% xylose as an inducer, the activity of Cb-DPEase and Ds-DPEase in WB800 was monitored from 0 to 12 h at intervals of 2 h, and the highest activity was 13.55 U/g and 6.54 U/g, respectively, at 6 h (Fig. 3a, b). These results revealed that dpe genes could be induced by xylose, and the expression appeared to correspond with DPEase catalysis. In addition, enzyme assays confirmed that DPEase production increased gradually within the first 6 h then decreased as the induction time was extended for both B. subtilis WB800-cb and WB800-ds. Therefore, 6 h was considered the optimal induction time for DPEase production.

Effect of xylose utilisation and induction time in the xylose-inducible expression system. a Effect of induction time on the production of DPEase in WB800-Cb. b Effect of induction time on the production of DPEase in WB800-Ds. c Effect of xylose concentration on the production of DPEase in WB800-Cb. d Effect of xylose concentration on the production of DPEase in WB800-Ds. All tests were performed in triplicate, and the results are presented as the means ± standard deviation
Xylose can also be consumed as a carbon source by B. subtilis. To ensure that the xylose concentration was sufficient to regulate the expression of the DPEase genes, we evaluated the effect of xylose concentration (0, 2, 6, 10, 15, 20, and 25 g/L) on the enzyme activity. The results showed that Cb-DPEase and Ds-DPEase achieved maximum enzyme activities of 17.50 U/g and 18.01 U/g, respectively, at 15 g/L xylose (Fig. 3c, d). Additionally, DPEase activities tended to increase with increasing xylose concentration up to 15 g/L, then gradually declined. Thus, a xylose induction concentration of 15 g/L was considered optimal.
Optimisation of fermentation conditions
Carbon and nitrogen sources are vital components for the synthesis of microbial cell proteins and nucleic acids (Kand et al. 2018). To maximise DPEase production, the effects of different carbon and nitrogen sources on recombinant DPEase expression were evaluated by measuring the enzyme activity. As shown in Fig. 4a, b, among the five carbon sources tested, maximum Cb-DPEase and Ds-DPEase production was achieved using lactose, which yielded enzyme activity of 17.30 U/g and 8.88 U/g, respectively. Supplementation with the monosaccharides lactose or fructose stimulated expression of DPEases. Under the same conditions, glycerol and starch increased cell growth but decreased DPEase activity relative to glucose. Specifically, glycerol doubled the growth rate compared to lactose, but only trace amounts of DPEase were produced, possibly because it enhanced the competitive growth ability of the strains (Espinoza-Molina et al. 2016).

Effects of carbon and nitrogen source on DPEase production and cell growth. a Effect of different carbon sources on the production of DPEase in WB800-Cb. b Effect of different nitrogen sources on the production of DPEase in WB800-Ds. c Effect of different carbon sources on the production of DPEase in WB800-Cb. d Effect of different nitrogen sources on the production of DPEase in WB800-Ds. All tests were performed in triplicate, and the results are presented as the means ± standard deviation
Different nitrogen sources also impacted the yield of recombinant DPEases. Beef extract was the best nitrogen source for strain WB800-cb, while soy peptone was the best for WB800-ds, with relative enzyme activities of 14.22 U/g and 8.33 U/g, respectively. The comparison of nitrogen sources for WB800-cb showed that soy peptone achieved more vigorous cell growth, but enzyme production was only half of that achieved by casein peptone as a nitrogen source (Fig. 4c, d). These results could be because casein peptone contains a wide range of amino acids, peptides, and proteins, as well as small amounts of sugars and growth factors required by the organism (Liu et al. 2018).
Optimisation of pH and temperature conditions for whole-cell biocatalyst
The effect of pH on the enzymatic activity of DPEases from C. bolteae and Dorea sp. was also evaluated. The optimal pH for both enzymes was pH 7.0 and 6.0, and Cb-DPEase exhibited a high relative activity at pH values ranging from 6.5 to 7.5 (Fig. 5a), with an enzyme activity reaching 11.35 U/g. Meanwhile, Ds-DPEase exhibited high relative activity at pH values ranging from 5.5 to 6.5, with an enzyme activity reaching 6.82 U/g (Fig. 5b).

Effects of pH on DPEase production. a Effect of pH on DPEase production in WB800-Cb. b Effect of pH on DPEase production in WB800-Ds
Temperature plays an important role in the bioproduction of engineered enzymes; thus, we investigated the yield of DPEase under temperature between 45 and 85 °C with an interval of every 5 °C. The enzyme activity in engineered B. subtilis harbouring C. bolteae DPEase-encoding gene increased rapidly as incubation temperature increases from 45 °C, and reached to the maximum at 55 °C of 11.57 U/g, then followed with a gradual decrease as the temperature increased (Fig. 6a). In contrast, the activity of DPEase from Dorea sp. in engineered B. subtilis strains increased from 45 °C and reached to the maximum at 70 °C of 9.33 U/g (Fig. 6b).

Effects of temperature on DPEase activity. a Effect of temperature on DPEase activity in WB800-Cb. b Effect of temperature on DPEase activity in WB800-Ds
Under the above optimal fermentation conditions, DPEase production increased to 21.90 U/g and 24.01 U/g in B. subtilis expressing C. bolteae or Dorea sp. DPEase, a 94.67% and 369.94% higher than that of control, respectively.
Discussion
Given the bright prospects of rare sugars such as d-psicose in the food and medical industries, the production of d-psicose via DPEase catalysis has attracted wide attention. Although overproduction of DPEase in engineered strains has been achieved in previous studies, these DPEase enzymes preferred alkaline conditions. In the present study, neutral and slightly acidic pH DPEases from C. bolteae and Dorea sp. were respectively expressed in B. subtilis, and the production of d-psicose by whole-cell reactions was evaluated.
B. subtilis strains are widely applied for enzymatic production due to their GRAS status. Induced production of target enzymes can be more effective than the expression under the control of a constitutive promoter (Chen et al. 2016b). We therefore evaluated the production of DPEases in B. subtilis under the xylose-inducible promoter PxylA, encoded by the XylR gene (Fig. 7). In the absence of xylose, XylR binds to the two tandem overlapping operator sequences located in PxylA and prevents transcription of the dpe gene. However, in the presence of xylose, the sugar binds to the repressor XylR, resulting in a conformational change in XylR that causes it to dissociate from the promoter (Biedendieck et al. 2010). This system is rigorously regulated and has a high induction ratio.

Schematic diagram of the PxylA promoter
In addition to acting as an inducer, xylose may be consumed as a carbon source by B. subtilis strains, which lowers xylose levels in the culture medium. If the xylose concentration drops below a threshold value, the gene expression could and DPEase production may decrease. To moderate competition between the synthesis of DPEase and cell growth, an effective induction strategy was explored (Fig. 3). Promoters are important cis-regulatory elements for regulating gene expression and would impact on protein production (Cui et al. 2018). However, previous reports showed that enhancing the strength of the promoter increased enzyme production at different levels, from 7.62 to 79.9% of increase under different dual-promoter strategies (Liu et al. 2019; Okegawa and Motohashi 2016). The results obtained in the present study indicated that the introduction of P43 to improve the promoter strength was beneficial to DPEase production (Fig. 2).
Optimising medium components and culture conditions can enhance the yield of target products. Since carbon and nitrogen sources play vital roles in both cell growth and product synthesis, optimising these parameters can maximise target protein production (Su et al. 2018). During fermentative production of DPEases, the highest enzyme activity occurred with lactose as a carbon source and casein peptone or soy peptone as a nitrogen source. In addition, the two different dpe genes exhibited different preferences for nitrogen sources during heterologous expression. Furthermore, controlling pH and temperature proved advantageous for strengthening the catalytic ability of DPEases.
This work established a production strategy for the expression of dpe genes from C. bolteae and Dorea sp. in B. subtilis for DPEase production. Production of engineered DPEase enzymes was increased using the non-toxic and harmless xylose as an inducer. Evaluation of industry-scale production is required in future studies.
References
Bhavsar AP, Zhao X, Brown ED (2001) Development and characterization of a xylose-dependent system for expression of cloned genes in Bacillus subtilis: conditional complementation of a teichoic acid mutant. Appl Environ Micro 67:403–410
Biedendieck R, Bunk B, Fürch T, Franco-Lara E, Jahn M, Jahn D (2010) Systems biology of recombinant protein production in Bacillus megaterium. Adv Biotechm Eng Biotechnol. 120:133–161
Chen J, Zhao L, Fu G, Zhou W, Sun Y, Zheng P, Sun J, Zhang D (2016a) A novel strategy for protein production using non-classical secretion pathway in Bacillus subtilis. Microb Cell Fact. 15:69
Chen J, Zhu Y, Fu G, Song Y, Jin Z, Sun Y, Zhang D (2016b) High-level intra- and extra-cellular production of D-psicose 3-epimerase via a modified xylose-inducible expression system in Bacillus subtilis. J Ind Microbiol Biot. 43:1577–1591
Chen JJ, Huang WL, Zhang T, Lu M, Jiang B (2019) Anti-obesity potential of rare sugar D-psicose by regulating lipid metabolism in rats. Food Funct. 10:2417–2425
Chen X et al (2017) Production of D-psicose from D-glucose by co-expression of D-psicose 3-epimerase and xylose isomerase. Enzyme Microb Tech. 105:18–23
Cui W, Han L, Suo F, Liu Z, Zhou L, Zhou Z (2018) Exploitation of Bacillus subtilis as a robust workhorse for production of heterologous proteins and beyond. World J Microb Bio. 34:145
Cui Y, Meng Y, Zhang J, Cheng B, Yin H, Chao G, Ping X, Yang C (2017) Efficient secretory expression of recombinant proteins in Escherichia coli with a novel actinomycete signal peptide. Protein Expres Pur 129:69–74
Espinoza-Molina JA, Acosta-Muniz CH, Sepulveda DR, Zamudio-Flores PB, Rios-Velasco C (2016) Codon optimization of the “Bos Taurus Chymosin” gene for the production of recombinant chymosin in Pichia pastoris. Mol Biotechnol. 58:657–664
Granstrom TB, Takata G, Tokuda M, Izumori, K. Izumoring (2004) A novel and complete strategy for bioproduction of rare sugars. J Biosci Bioeng 97: 89–94.
He W, Jiang B, Mu W, Zhang T (2016a) Production of D-allulose with D-psicose 3-epimerase expressed and displayed on the surface of Bacillus subtilis spores. J Agr Food Chem 64:7201–7207
He W, Mu W, Jiang B, Yan X, Zhang T (2016b) Construction of a food grade recombinant Bacillus subtilis based on replicative plasmids with an auxotrophic marker for biotransformation of D-fructose to D-psic. J Agr Food Chem. 64:3243–3250
He W, Mu W, Jiang B, Yan X, Zhang T (2016c) Food-grade expression of D-psicose 3-epimerase with tandem repeat genes in Bacillus subtilis. J Agr Food Chem. 64:5701–5707
Jia M, Mu W, Chu F, Zhang X, Jiang B, Zhou LL, Zhang T (2014) A D-psicose 3-epimerase with neutral pH optimum from Clostridium bolteae for D-psicose production: cloning, expression, purification, and characterization. Appl Microbiol Biot 98:717–725
Kai H, Xiao G, Bo J, Sen L (2018) Construction of a food-grade arginase expression system and its application in L-ornithine production with whole cell biocatalyst. Process Biochem 73:94–101
Kand D, Raharjo IB, Castro-Montoya J, Dickhoefer U (2018) The effects of rumen nitrogen balance on in vitro rumen fermentation and microbial protein synthesis vary with dietary carbohydrate and nitrogen sources. Anim Feed Sci Tech 241:184–197
Kim J (2017) Surface display of lipolytic enzyme, Lipase A and Lipase B of Bacillus subtilis on the Bacillus subtilis spore. Biotechnol Bioproc E 22:462
Lim YR, Oh DK (2011) Microbial metabolism and biotechnological production of D-allose. Appl Microbiol Biot 91:229–235
Liu X, Wang H, Wang B, Pan L (2018) High-level extracellular protein expression in Bacillus subtilis by optimizing strong promoters based on the transcriptome of Bacillus subtilis and Bacillus megaterium. Protein Expres Purif 151:72–77
Liu Z, Zheng W, Ge C, Cui W, Zhou L, Zhou Z (2019) High-level extracellular production of recombinant nattokinase in Bacillus subtilis WB800 by multiple tandem promoters. BMC Microbiol 19:89
Mu W, Zhang W, Feng Y, Jiang B, Zhou L (2012) Recent advances on applications and biotechnological production of D-psicose. Appl Microbiol Biot 94:1461–1467
Okegawa Y, Motohashi K (2016) Expression of spinach ferredoxin-thioredoxin reductase using tandem T7 promoters and application of the purified protein for in vitro light-dependent thioredoxin-reduction system. Protein Expr Purif 121:46–51
Sang-Jae L, Dong-Woo L, Eun-Ah C, Young-Ho H, Seong-Bo K, Byoung-Chan K, Yu-Ryang P (2005) Characterization of a thermoacidophilic L-arabinose isomerase from Alicyclobacillus acidocaldarius: role of Lys-269 in pH optimum. Appl Environ Micro 71:7888–7896
Shintani T, Yamada T, Hayashi N, Iida T, Nagata Y, Ozaki N, Toyoda Y (2017) Rare sugar syrup containing D-allulose but not high-fructose corn syrup maintains glucose tolerance and insulin sensitivity partly via hepatic glucokinase translocation in Wistar rats. J Agr Food Chem 65:2888–2894
Su L, Sun F, Liu Z, Zhang K, Wu J (2018) Highly efficient production of Clostridium cellulolyticum H10 D-psicose 3-epimerase in Bacillus subtilis and use of these cells to produce D-psicose. Microb Cell Fact 17:188
Tännler S, Zamboni N, Kiraly C, Aymerich S, Sauer U (2008) Screening of Bacillus subtilis transposon mutants with altered riboflavin production. Metab Eng 10:216–226
Tseng WC, Chen CN, Hsu CT, Lee HC, Fang HY, Wang MJ, Wu YH, Fang TY (2018) Characterization of a recombinant D-allulose 3-epimerase from Agrobacterium sp. ATCC 31749 and identification of an important interfacial residue. Inter J Biol Macromol 112:767–774
Wanmeng M, Feifei C, Qingchao X, Shuhuai Y, Leon Z, Bo J (2011) Cloning, expression, and characterization of a D-psicose 3-epimerase from Clostridium cellulolyticum H10. J Agri Food Chem 59:7785–7792
Yadav D, Kim SJ, Bae MA, Kim JR, Cho KH (2018) The ability of different ketohexoses to alter Apo-A-I structure and function in vitro and to induce hepatosteatosis, oxidative stress, and impaired plasma lipid profile in hyperlipidemic zebrafish. Oxid Med Cell Long:1–12
Yang M, Zhang W, Ji S, Cao P, Chen Y, Zhao X (2013) Generation of an artificial double promoter for protein expression in Bacillus subtilis through a promoter trap system. Plos One 8:e56321
Zhang W, Li H, Jiang B, Zhang T, Mu W (2017a) Production of D-allulose from D-glucose by Escherichia coli transformant cells co-expressing D-glucose isomerase and D-psicose 3-epimerase genes. J Sci Food Agr 97:3420–3426
Zhang W, Li H, Zhang T, Jiang B, Zhou L, Mu W (2015) Characterization of a D-psicose 3-epimerase from Dorea sp. CAG317 with an acidic pH optimum and a high specific activity. J Mol Catal B-Enzyme 120:68–74
Zhang WL, Zhang T, Jiang B, Mu WM (2017b) Enzymatic approaches to rare sugar production. Biotechnol Adv 35:267–274
Funding
This work was supported by the National Natural Science Foundation of China (31460026), the National Natural Science Foundation of Guangxi Province (2018GXNSFAA050126), and the Bosch Young Teachers Innovation Training Project (BRP180215).
Author information
Authors and Affiliations
Contributions
Hongbei Wei participated in the design of the study, performed research, analyzed data, and drafted the paper. Ruoxuan Zhang performed SDS-PAGE experiments and analyzed the data. Leyi Wang performed the enzyme activity test and analyzed the data. Donglong Li performed the fermentation experiments and analyzed the data. Fangxue Hang performed the enzyme properties and analyzed the data. Jidong Liu conceived the study, designed and coordinated the experiments, and critically revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent for publication
Informed consent was obtained from all individual participants included in the study.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wei, H., Zhang, R., Wang, L. et al. Expression of d-psicose-3-epimerase from Clostridium bolteae and Dorea sp. and whole-cell production of d-psicose in Bacillus subtilis. Ann Microbiol 70, 9 (2020). https://doi.org/10.1186/s13213-020-01548-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13213-020-01548-x