
Abstract
Purpose
Microbial community composition is crucial for the flavor and quality of fermented foods. However, the microbiota of Chinese traditional dough starter (Jiaozi) from different origins has scarcely been studied. The aim of this study was to determine the composition of bacterial and fungal communities in six Jiaozi collected from two provinces in northwest China.
Methods
Our study determined the composition of bacterial and fungal communities in six Jiaozi through Illumina MiSeq sequencing of the 16S rRNA gene and the ITS regions.
Result
A total of 234 operational taxonomic units (OTUs) for bacteria and 490 OTUs for fungi were identified. Furthermore, Lactobacillus, Weissella, Acetobacter, Sphingomonas, and Serratia were identified as the predominant bacterial genera in Jiaozi samples, while Saccharomyces, Candida, Alternaria, unclassified Filobasidiales, and Mycosphaerella were the most abundant fungal genera. The results revealed that the six samples could be grouped into two groups based on their province of origin. In the results of PCA and HCA analysis, the first three principal components which were chosen could explain 99.93% and 90.99% of the total bacterial and fungal communities, respectively.
Conclusion
The results indicated high levels of bacteria and fungi in traditional Jiaozi and highlighted the possible influence of geographic areas on microbial diversity.
Similar content being viewed by others

Introduction
Chinese traditional dough fermentation starter (locally called “Jiaozi”) has a long history in the preparation of steamed bread (Mantou). Mantou has been one of the most important fermented staple foods in northwest China for over two millennia. Jiaozi fermentation has a remarkable influence on the overall quality of Mantou due to an abundant metabolic repertoire of its stable symbiotic culture of bacteria, yeast, and mold. Traditional Jiaozi is a Chinese handcrafted product made from wheat flour or maize flour, which is obtained through repeated multi-culture natural fermentation without prior sterilization. Jiaozi mixtures contain abundant bacteria and fungi, which can increase desirable volatile constituents and aromatic precursors of Mantou (Hu et al. 2015; Li et al. 2016; Li et al. 2017).
Traditional microbiological tests, PCR-DGGE, amplicon sequencing, and metagenomics analysis methods, have been widely used to study the microbial composition of sourdough and Chinese traditional Jiaozi (Iacumin et al. 2009; Li et al. 2016). With the development of next-generation sequencing and advanced analytical methods, there are increasing studies exploring the microbial community of traditional fermented foods from different countries (Kergourlay et al. 2015; Chen et al. 2017). Recently, Li et al. (2017) performed a comparative study of the bacterial diversity in traditional Jiaozi and sourdough by high-throughput sequencing of 16S rRNA amplicons and found that the bacterial communities of Jiaozi are more complex than those of sourdough because of the different production methods. At the same time, fungi are also essential microbes active in Jiaozi, which are responsible for dough leavening and aromatic compound formation (De Vuyst et al. 2016).
Extensive research reports have revealed that traditional fermented foods are influenced by geographical environmental conditions, which determine their microbial ecosystem (Sun et al. 2014; Kergourlay et al. 2015; Liu et al. 2018b). However, the influence of geographical regions on the microbial community of Jiaozi is yet to be thoroughly investigated. Therefore, it is necessary to form a deeper and more accurate understanding of the microbial diversity of Jiaozi derived from various geographic areas. Hence, the main objective of this study was to characterize the bacterial and fungal communities of six traditional Jiaozi samples collected from different regions in northwest China using high-throughput sequencing. The results improve our understanding of the differences in bacterial communities between regions, thereby lending new insights into the microorganism community of traditional Jiaozi and provide a theoretical basis for the production of distinctive, safe, high-quality Jiaozi.
Materials and methods
Sample collection and determination of pH and total titratable acidity (TTA)
Samples were aseptically collected from two provinces in northwest China, which included Jiaozi manufactured by the traditional hand-made process in six private households, following good hygiene practices. All of the Jiaozi samples were made of wheat flour as a raw material, and Chinese Xiaoqu (a traditional starter for making Chinese rice wine) was used as microbial inoculum. It is obtained through air-dried after repeated three fermentations in local natural environment. These households belong to the following municipalities: KEL (Xinjiang Province), WS (Xinjiang province), KS (Xinjiang province), YA (Shaanxi Province), XA (Shaanxi Province), and YL (Shaanxi Province) (Table 1). All samples were stored in labeled sterile resealable plastic bags at − 20 °C. The pH values of the samples were measured using a pH meter and the TTA values of the samples were defined as the amount (mL) of a 0.1 M NaOH required for 10 g of Jiaozi sample to reach a pH value of 8.5 according to the method of Li et al. (2017).
Total DNA extraction
Total DNA was extracted using a Rapid Soil DNA Isolation Kit (Sangon Biotech Co., Ltd., Shanghai, China), based on the manufacturer’s instructions. DNA integrity was evaluated by 1% agarose gel electrophoresis under ultraviolet light. DNA concentrations were measured using a Qubit 2.0 Fluorometer (Life Technologies, CA, USA). DNA purity was determined by A260/A280. Lastly, DNA of sufficient quantity and quality was stored at − 20 °C pending sequence analysis.
High-throughput sequencing
For each sample, the barcoded primers341F-806R (341F: 5′-CCTAYGGGRBGCASCAG-3′; 806R: 5′-GGACTACNNGGGTATCTAAT-3′) for bacteria (Liu et al. 2018c) and ITS1F-ITS2R (ITS1F: 5′-CTTGGTCATTTAGAGGAAGTAA-3′; ITS2R: 5′-GCTGCGTTCTTCATCGATGC-3′) for fungi (Wang et al. 2017) were synthesized and used to amplify specific regions by PCR on an ABI GeneAmp® 9700 system. Reactions consisted of 4 μL 5× FastPfu buffer, 2 μL 2.5 mM dNTPs, 0.8 μL each of 5 μM forward and reverse primer, 0.4 μL FastPfu Polymerase (TransGen catalog no. AP221–02), and 10 ng template DNA in a total volume of 20 μL. Samples were denatured at 95 °C for 3 min and amplified over 27 (bacteria) or 32 cycles (fungi) at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 45 s, followed by final extension at 72 °C, for 10 min and rest at 10 °C. Samples were amplified in triplicate. Then, the PCR products were used to verify amplification success with 2% agarose gel electrophoresis, followed by purification with a AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) and elution with Tris-HCl. Purified PCR products were assessed a second time on 2% agarose gels, and then quantified with a blue fluorescence quantitative system (Quanti FluorTM-ST, Promega Corporation, Madison, WI, USA). Finally, high-throughput sequencing was performed using the Illumina MiSeq PE250 platform (San Diego, CA, USA), according to the manufacturer’s instructions.
Bioinformatic analyses
After Illumina MiSeq PE250 sequencing, the raw 16S rRNA and ITS sequencing files were demultiplexed and quality-filtered using the Usearch software, version v10. Furthermore, operational taxonomic units (OTUs) were clustered using UPARSE (version 7.1) with a cutoff of 97% similarity, and UCHIME was used to identify and remove chimeric sequences. The phylogenetic affiliation of each 16S rRNA gene sequence was analyzed by UCLUST Classifier against the SILVA 16S rRNA database (Release119 http://www.arb-silva.de). The fungal ITS sequencing data were classified by using the UNITE ITS database (Release 6.0 http://unite.ut.ee/index.php). All sequences obtained were submitted to the NCBI to identify the approximate species.
Alpha diversity was performed using Mothur v.1.21.1 (Schloss et al. 2009) to generate the rarefaction curves, ACE, Chao1, Good’s coverage, and Shannon and Simpson indices. The beta diversity analysis was evaluated with UniFrac (Lozupone et al. 2011) to compare the microbial composition and complexity between Jiaozi samples. PCA analyses were performed in R software using the vegan package. Moreover, we performed clustering on genera obtained from the RDP Classifier by means of the complete linkage hierarchical clustering technique using the R package HCLUST and VEGDIST. Statistical analysis was performed through subjection of the data to analysis of variance (ANOVA) using IBM SPSS Statistics 21 (NY, USA). Means were subject to Duncan’s test, and p < 0.05 was considered statistical significance.
Results
Acidity analysis of Jiaozi
The pH and TTA of each of the Jiaozi samples collected from the different regions were shown in Table 1. It can be seen that the lowest pH value was 3.63 for the sample KEL and having the highest TTA value 11.12 mL. These results implied that the acidity value of sample KEL was the highest among all the samples. For the samples of XA and YL, the pH values respectively were 4.64 and 4.62, as well as not statistically significant. However, it was interesting that the TTA values of them were 7.31 and 9.42, which had a significant difference. For the samples of WS, KS, and YA, the pH value and TTA value were 5.13 and 6.32, 4.82 and 5.80, and 3.74 and 8.53, respectively.
Characteristics of the sequencing results and alpha diversity
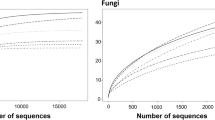
In this study, after quality merging, filtering, and trimming, Illumina MiSeq PE250 sequencing-based analysis of the six Jiaozi samples generated 219,221 16S rRNA gene sequences and 215,999 fungal ITS sequences. On average, 36,537 ± 1768 sequences per Jiaozi sample were detected for bacterial community and 36,000 ± 3613 sequences per Jiaozi sample were detected for fungal community. Based on 97% nucleotide sequence identity between reads, a total of 234 OTUs for bacteria and 490 OTUs for fungi were identified (Table 2). Rarefaction curve analysis at 97% similarity levels for the sampling showed that the curves reached a plateau, implying a sufficient and reasonable sampling of the bacterial and fungal communities (Fig. 1). This conclusion is further supported by the results of Good’s coverage estimator, which returned values higher than 0.999 in all cases (Table 2).

Rarefaction analysis at 97% similarity levels for Jiaozi samples collected from the six geographical regions. a Bacterial community; b fungal community
Alpha diversity, as measured by statistical analysis and various indices, captures the abundance and diversity of microbial communities. We estimated the ACE and Chao 1 indices for community richness and the indices of Shannon and Simpson for community diversity (Table 2). ACE, Chao1, and Shannon values increase with community richness and diversity, while Simpson values are inversely correlated with microbial community diversity. The results showed that the alpha diversity indices for fungal communities in samples from KEL, WS, XA, and YL were significantly higher than those of samples from KS and YA. However, for the bacterial communities, these indices showed conflicting results.
Bacterial community diversity
At the phylum level, the bacteria present in all six Jiaozi samples correspond to six phyla (Fig. 2a). Firmicutes was the dominant phyla in Jiaozi samples, except for the sample from KEL in which Proteobacteria was the dominant phyla with 59.08% presence. In samples from WS, KS, YA, XA, and YL, Firmicutes levels were 97.19%, 98.57%, 53.62%, 97.61%, and 95.89%, respectively. The Proteobacteria phylum was the second most common phylum in Jiaozi. The bacterial community of Jiaozi shows a great level of diversity at genus level. A total of 34 bacterial genera were identified in Jiaozi, with different levels of each genus within different samples (Fig. 2b). Members of the genus Lactobacillus were the dominant bacteria in samples from KEL, KS, YA, XA, and YL, with 40.41%, 98.55%, 53.61%, 97.51%, and 91.62% respective relative abundances.

Relative abundance of bacterial community proportions of different Jiaozi samples, all the six samples of two province at the level of phyla (a) and the genus (b) respectively
Fungal community diversity
At the phylum level, a total of five fungal phyla were identified in Jiaozi from six geographic locations. The main fungal phylum was Ascomycota, which comprised 68.88%, 80.77%, 58.22%, 99.80%, 70.28%, and 98.99% of the fungal communities of samples from KEL, WS, KS, YA, XA, and YL, respectively (Fig. 3a). In addition to this dominant phylum, the Basidiomycota and Anthophyta phyla were identified in Jiaozi samples at different levels, with Basidiomycota being present at higher levels than Anthophyta. Additionally, low levels of Zygomycota and others were also identified in all six Jiaozi samples. At genus level, a total of 110 fungal genera were identified at different levels within different samples. The richness of fungal genera in the samples of Xinjiang province was higher than in samples from Shaanxi province (Fig. 3b). Clear differences in fungal diversity of Jiaozi at genus level can be observed from Fig. 3b. Samples from KS, WS, and KEL were similar in fungal communities. For the KEL sample, with the composition including unclassified unclassified Filobasidiales (20.44%), Saccharomyces (15.36%), Gibberella (11.29%), Alternaria (9.93%), and Aspergillus (8.78%). Similarly, Alternaria (17.75%), Mycosphaerella (11.07%), unclassified Filobasidiales (10.70%), and Cryptococcus (10.03%) were the dominant genera in the KS sample. In addition, the predominant genera of fungi in the WS sample were Saccharomyces (17.32%), Aspergillus (9.42%), Alternaria (8.97%), Mycosphaerella (8.18%), and Gibberella (7.57%).

Relative abundance of fungal community proportions of different Jiaozi samples, all the six samples of two provinces at the level of phyla (a) and the genus (b) respectively
Community comparisons
PCA and hierarchical clustering analysis (HCA) were used to compare the community structures of bacteria and fungi collected from six Jiaozi samples (Fig. 4). Of the total variance in the data set, the first three principal components were chosen to define the PCA results, because they altogether explained 99.93% and 90.99% of the total bacterial and fungal communities, respectively.

Principal component analysis (PCA) of bacterial community and fungal community for Jiaozi. Each color dot represents data from same province (n = 3 per group). a Bacterial community; b fungal community. Hierarchical cluster tree constructed based on a distance matrix calculated using the unweighted UniFrac algorithm for six Jiaozi samples collected from two province in china. c Bacterial community; d fungal community
The community structure may be largely responsible for these differences. As shown in Fig. 4a, the bacterial communities of the KEL and WS samples were distinct from those of samples from Shaanxi province (XA, YA, and YL) and the KS sample based on PC1, PC2, and PC3 (69.3%, 27.41%, and 3.22% variance, respectively). The analysis also revealed that the bacterial communities of samples collected in Shaanxi province were similar, whereas more variety was observed between the three samples of Xinjiang province. HCA also indicated that the KEL and WS samples were clearly distinct from each other (Fig. 4c), whereas the other four groups were separated as two clades, with a much smaller within-the-clade distance between the YL and XA samples compared with the YA sample. PCA analysis also revealed that the fungal communities of Shaanxi province samples were distinct from those of Xinjiang province samples, based on the first three principal components (PC1, PC2, and PC3 accounted for 65.99, 23.29, and 1.71% of variance, respectively) (Fig. 4b). This was confirmed by HCA, which revealed the differences in fungal communities of Jiaozi samples from the two different provinces (Fig. 4d). The WS, KEL, and KS samples had very similar fungal communities, while the three samples from Shaanxi showed more variety.
Discussion
In the present study, we collected six traditional Jiaozi samples from two provinces in northwestern China, and the values of pH and TTA were analyzed. The pH values can reflect the amount of strong acid produced via bacteria in the sourdough, whereas the TTA values can give an indication on the total acidity found in the sourdough (Zhang and He 2013). Interestingly, there is no significant difference in the pH value of XA and YL samples, but the TTA values of the two samples were significantly different (p < 0.05) (Table 1). Liu et al. (2016) hypothesized that this result may be due to the variable acid producing microbial community held by the samples and the different buffering capacity of the flour used.
Illumina MiSeq sequencing (16S rRNA gene and the ITS regions) was used to explore the bacterial and fungal diversity. We obtained sufficient sequences, as Good’s coverage values were above 90% for bacterial and fungal sequences (Table 2). Results revealed high levels of accuracy in the sequencing data of microbial communities, indicating that they were reliable for further study. Interestingly, the observation of community richness and diversity revealed numerous microbial communities among Jiaozi samples collected from different regions.
Bacterial community composition and structural analysis showed that the predominant phyla found in Jiaozi were Firmicutes and Proteobacteria. These results are in line with the main taxonomy of sourdough and Jiaozi from different geographical regions (Li et al. 2017). High relative levels of Lactobacillus, Weissella, Acetobacter, Sphingomonas, Serratia, Halomonas, and Leuconostoc were also identified in Jiaozi. Our findings are consistent with previous observations. Lactobacillus sanfranciscensis belongs to the Lactobacillus genus, is a predominant key LAB, has been widely isolated from traditionally fermented sourdoughs, and is shown to influence the quality of fermented foods by affecting their texture and flavor, as well as by conferring a longer shelf life (Gobbetti and Corsetti 1997). Yang et al. (2017) reported the genotypic characterization of L. sanfranciscensis strains isolated from Chinese traditional sourdoughs in different regions and found that the geographical origin of the strains was not related to their genotypic diversity and population evolution.
Previously, several studies showed that Acetobacter, Weissella, and Leuconostoc are also genera commonly found in traditional sourdough (Iacumin et al. 2009; Zhang et al. 2011; Zhang and He 2013). The fermentation of Jiaozi is a complex process, and the bacterial communities involved are very diverse. As shown in Fig. 2b, there were numerous differences in bacterial community structures among the six Jiaozi samples collected from different provinces. Similar results have been reported by Li et al. (2017), who showed that Acetobacter and Pediococcus were the primary bacteria genera in the studied Jiaozi samples. They also found that the bacterial community structures of Jiaozi are different from those of sourdough, which were characterized by the presence of Lactobacillus, Weissella, and Lactobacter. The bacterial diversity of Jiaozi is influenced by many factors, which include not only the types of raw materials, manufacture methods, and the propagation times, but also environmental factors, which play an important role in species distribution. This effect has also been observed in sourdoughs collected from different geographical regions (Scheirlinck et al. 2007; Zhang and He 2013; Li et al. 2016). In short, the bacterial communities of Jiaozi are complex, diverse, variable, and may depend upon several environmental factors, thus requiring further investigation.
For fungal communities, the sequences of ITS regions obtained from all studied samples mainly correspond to two phyla: Ascomycota and Basidiomycota. The most abundant fungal genera observed in all six Jiaozi samples were Saccharomyces, Candida, Alternaria, unclassified Filobasidiales, Mycosphaerella, Gibberella, Aspergillus, etc. Previously, fungal communities of Jiaozi were analyzed by culture-dependent and DGGE methods, and little diversity in Jiaozi fungal communities was reported, mainly including Saccharomyces cerevisiae (S. cerevisiae), Wickerhamomyces anomalus, Candida tropicalis, Saccharomycopsis fibuligera, and some molds (Li et al. 2016). In this study, Saccharomyces was identified in samples from KS, YA, and XA with low incidence, being most abundant in the YL sample and absent in the sample from KEL and WS. Zhang et al. (2011) reported similar results regarding the diversity of yeasts in Jiaozi by DGGE method and revealed that S. cerevisiae was the dominant yeast species in traditional sourdoughs. Multiple previous studies have reported that S. cerevisiae is the most widely used baker’s yeast, and Mantou made with S. cerevisiae featured a relatively high content of volatile compounds (Liu et al. 2018a). Candida genera were only detected in the YA sample at high levels. Gullo (2003) previously reported that Candida humilis was the dominant species in sourdoughs used for the production of durum wheat bran flour bread. Rhodotorula mucilaginosa (R. mucilaginosa), another dominant yeast species, has been isolated from traditional sourdough collected from Alashan League of Inner Mongolia by pure culture method (Zhang et al. 2011). A recent finding by Ma et al. (2018) indicated that an exopolysaccharide REPS2-A extracted from R. mucilaginosa could be quickly produced and collected from liquids and can significantly inhibit cancer cell growth. Hence, R. mucilaginosa has excellent potential as a probiotic and microorganism-based therapeutic agent. Apart from yeasts, some molds also play an important role in the fermentation of sourdough, but this has scarcely been reported (Zhang et al. 2015; Li et al. 2016). In our sequencing results, some molds with research value were identified and can be further studied in the future. In summary, our Jiaozi samples have rich and various fungi which are mainly dominated by yeast as previously shown for other traditional sourdough collected from different regions of the world detected by both culture-dependent and culture-independent methods.
The microbial communities of fermented foods have typical regional characteristics (Kergourlay et al. 2015; Chen et al. 2017). Sun et al. (2014) reported that bacterial and fungal communities in different tarag (a fermented milk product of cows in Mongolia and the northwest of China) samples were stratified by geographical region. Many factors may influence the microbial communities of sourdough, including the geographical region, environmental conditions, materials, and production methods (Minervini et al. 2014). Previous studies have demonstrated the geographical origin significantly influences the LAB community structure of Belgian sourdoughs, irrespective of the type of flour used to prepare the sourdough (Scheirlinck et al. 2007). Moreover, an analysis of the microflora of traditional sourdoughs collected from the western region of Inner Mongolia found large differences among different samples (Zhang et al. 2011). Liu et al. (2018b) investigated the bacterial diversity of traditional sourdoughs collected from three regions of China and found that the diversity of bacterial flora in Chinese western samples was different from that of northern and southern samples due to the genera Lactobacillus and Pediococcus.
Further study of the function of bacterial and fungal communities in Jiaozi using metagenomics, metatranscriptomics, and metaproteomics might help elucidate their interactions, as well as the metabolic network of substances involved in the process of multi-strain fermentation in Jiaozi.
Conclusions
In the present study it was found that the traditional Jiaozi has a rich microbial community diversity and varied with the geographic location. Six traditional dough samples (Jiaozi) used for manufacturing fermented staple foods collected from the Xinjiang and Shaanxi provinces in northwestern China were analyzed using high-throughput sequencing. Bacterial communities of the three samples XA, YA, and YL in the Shaanxi province were similar and could be differentiated from the communities of the Xinjiang province. Fungal communities WS, KEL, and KS from Xinjian were very similar while samples from Shaanxi province showed more variability and were distinct from those of Xinjiang province samples. This study provides a basis for further research on bacteria and fungi, naturally occurring in Jiaozi, with excellent fermentation and probiotic properties.
References
Chen G, Chen C, Lei Z (2017) Meta-omics insights in the microbial community profiling and functional characterization of fermented foods. Trends Food Sci Technol 65:23–31
De Vuyst L, Harth H, Van Kerrebroeck S, Leroy F (2016) Yeast diversity of sourdoughs and associated metabolic properties and functionalities. Int J Food Microbiol 239:26–34
Gobbetti M, Corsetti A (1997) Lactobacillus sanfrancisco a key sourdough lactic acid bacterium: a review. Food Microbiol 14:175–187
Gullo M (2003) Candida humilis—dominant species in sourdoughs for the production of durum wheat bran flour bread. Int J Food Microbiol 80:55–59
Hu X, Sheng X, Liu L, Ma Z, Li X, Zhao W (2015) Food system advances towards more nutritious and sustainable mantou production in China. Asia Pac J Clin Nutr 24:199–205
Iacumin L, Cecchini F, Manzano M, Osualdini M, Boscolo D, Orlic S, Comi G (2009) Description of the microflora of sourdoughs by culture-dependent and culture-independent methods. Food Microbiol 26:128–135
Kergourlay G, Taminiau B, Daube G, Champomier Verges MC (2015) Metagenomic insights into the dynamics of microbial communities in food. Int J Food Microbiol 213:31–39
Li H, Li Z, Qu J, Wang J (2017) Bacterial diversity in traditional Jiaozi and sourdough revealed by high-throughput sequencing of 16S rRNA amplicons. LWT Food Sci Technol 81:319–325
Li Z, Li H, Bian K (2016) Microbiological characterization of traditional dough fermentation starter (Jiaozi) for steamed bread making by culture-dependent and culture-independent methods. Int J Food Microbiol 234:9–14
Liu T, Li Y, Chen J, Sadiq FA, Zhang G, Li Y, He G (2016) Prevalence and diversity of lactic acid bacteria in Chinese traditional sourdough revealed by culture dependent and pyrosequencing approaches. LWT Food Sci Technol 68:91–97
Liu T, Li Y, Sadiq FA, Yang H, Gu J, Yuan L, Lee YK, He G (2018a) Predominant yeasts in Chinese traditional sourdough and their influence on aroma formation in Chinese steamed bread. Food Chem 242:404–411
Liu X, Zhou M, Jiaxin C, Luo Y, Ye F, Jiao S, Hu X, Zhang J, Lü X (2018b) Bacterial diversity in traditional sourdough from different regions in China. LWT Food Sci Technol 96:251–259
Liu Z, Wang Z, Lv X, Zhu X, Chen L, Ni L (2018c) Comparison study of the volatile profiles and microbial communities of Wuyi Qu and Gutian Qu, two major types of traditional fermentation starters of Hong Qu glutinous rice wine. Food Microbiol 69:105–115
Lozupone C, Lladser M. E, Knights D, Stombaugh J, Knight R (2011) UniFrac: an effective distance metric for microbial community comparison. ISME J 5:169-172.
Ma W, Chen X, Wang B, Lou W, Chen X, Hua J, Sun Y. J, Zhao Y, Peng T (2018) Characterization, antioxidativity, and anti-carcinoma activity of exopolysaccharide extract from Rhodotorula mucilaginosa CICC 33013. Carbohydr Polym 181:768-777.
Minervini F, De Angelis M, Di Cagno R, Gobbetti M (2014) Ecological parameters influencing microbial diversity and stability of traditional sourdough. Int J Food Microbiol 171:136–146
Scheirlinck I, Van der Meulen R, Van Schoor A, Vancanneyt M, De Vuyst L, Vandamme P, Huys G (2007) Influence of geographical origin and flour type on diversity of lactic acid bacteria in traditional Belgian sourdoughs. Appl Environ Microbiol 73:6262–6269
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Sun Z, Liu W, Bao Q, Zhang J, Hou Q, Kwok L, Sun T, Zhang H (2014) Investigation of bacterial and fungal diversity in tarag using high-throughput sequencing. J Dairy Sci 97:6085–6096
Wang X, Du H, Zhang Y, Xu Y. J. A (2017) Environmental microbiota drives microbial succession and metabolic profiles during Chinese liquor fermentation. Appl Environ Microbiol 84: AEM.02369-17.
Yang H, Liu T, Zhang G, Chen J, Gu J, Yuan L, He G (2017) Genotyping of Lactobacillus sanfranciscensis isolates from Chinese traditional sourdoughs by multilocus sequence typing and multiplex RAPD-PCR. Int J Food Microbiol 258:50–57
Zhang G, He G (2013) Predominant bacteria diversity in Chinese traditional sourdough. J Food Sci 78:M1218–M1223
Zhang G, Sadiq FA, Zhu L, Liu T, Yang H, Wang X, He G (2015) Investigation of microbial communities of Chinese sourdoughs using culture-dependent and DGGE approaches. J Food Sci 80:M2535–M2542
Zhang J, Liu W, Sun Z, Bao Q, Wang F, Yu J, Chen W, Zhang H (2011) Diversity of lactic acid bacteria and yeasts in traditional sourdoughs collected from western region in Inner Mongolia of China. Food Control 22:767–774
Funding
This work was financially supported by the National Natural Science Foundations of China (Grant No. 31671889) and Shaanxi key research and development program (2018TSCXL-NY-03-02). In addition, we especially wish to thank Xi’an Xiding Biotechnology Co., Ltd. for its assistance in sequencing and bioinformatics analysis.
Author information
Authors and Affiliations
Contributions
Ting Liu & Jiamu Kang: Formal analysis, Investigation, Writing Original Draft; Xiaolong Wang & Xiaoping Li: Formal analysis, Writing - Review & Editing; Liu Liu & Xinzhong Hu: Conceptualization, Methodology, Writing - Review & Editing, Funding acquisition; Zhen Ma & Tian Ren: Validation, Investigation. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
N/A
Consent for publication
N/A
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Liu, T., Kang, J., Liu, L. et al. Microbial community diversity of traditional dough starter (Jiaozi) from two provinces in northwest China. Ann Microbiol 70, 18 (2020). https://doi.org/10.1186/s13213-020-01544-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13213-020-01544-1