
Abstract
Background
Transcriptome-wide association study (TWAS) is an influential tool for identifying genes associated with complex diseases whose genetic effects are likely mediated through transcriptome. TWAS utilizes reference genetic and transcriptomic data to estimate effect sizes of genetic variants on gene expression (i.e., effect sizes of a broad sense of expression quantitative trait loci, eQTL). These estimated effect sizes are employed as variant weights in gene-based association tests, facilitating the mapping of risk genes with genome-wide association study (GWAS) data. However, most existing TWAS of Alzheimer's disease (AD) dementia are limited to studying only cis-eQTL proximal to the test gene. To overcome this limitation, we applied the Bayesian Genome-wide TWAS (BGW-TWAS) method to leveraging both cis- and trans- eQTL of brain and blood tissues, in order to enhance mapping risk genes for AD dementia.
Methods
We first applied BGW-TWAS to the Genotype-Tissue Expression (GTEx) V8 dataset to estimate cis- and trans- eQTL effect sizes of the prefrontal cortex, cortex, and whole blood tissues. Estimated eQTL effect sizes were integrated with the summary data of the most recent GWAS of AD dementia to obtain BGW-TWAS (i.e., gene-based association test) p-values of AD dementia per gene per tissue type. Then we used the aggregated Cauchy association test to combine TWAS p-values across three tissues to obtain omnibus TWAS p-values per gene.
Results
We identified 85 significant genes in prefrontal cortex, 82 in cortex, and 76 in whole blood that were significantly associated with AD dementia. By combining BGW-TWAS p-values across these three tissues, we obtained 141 significant risk genes including 34 genes primarily due to trans-eQTL and 35 mapped risk genes in GWAS Catalog. With these 141 significant risk genes, we detected functional clusters comprised of both known mapped GWAS risk genes of AD in GWAS Catalog and our identified TWAS risk genes by protein-protein interaction network analysis, as well as several enriched phenotypes related to AD.
Conclusion
We applied BGW-TWAS and aggregated Cauchy test methods to integrate both cis- and trans- eQTL data of brain and blood tissues with GWAS summary data, identifying 141 TWAS risk genes of AD dementia. These identified risk genes provide novel insights into the underlying biological mechanisms of AD dementia and potential gene targets for therapeutics development.
Similar content being viewed by others


Background
Alzheimer's disease (AD) dementia is a complex neurodegenerative disorder characterized by progressive cognitive decline and memory loss, currently affecting ~7 million Americans aged 65 and older. AD dementia is listed as the seventh-leading cause of death in the United States of America in 2021 [1]. Despite extensive research, the underlying biological mechanisms of AD dementia remain elusive, and effective treatments are still lacking [2]. Recent studies have highlighted the important roles of genomic risk factors of AD dementia [3, 4]. Recent genome-wide association studies (GWAS) identified a total of ~75 genetic risk loci for AD dementia [5]. However, these risk loci still only explain a small portion of the heritability of AD dementia, suggesting additional risk genes or loci might contribute to the genetic etiology of AD dementia. Also, the biological mechanisms underlying the majority of the mapped risk genes remain unknown. Transcriptome-wide association study (TWAS) has emerged as an influential tool for identifying risk genes associated with complex diseases, particularly those with genetic effects mediated through transcriptome [6, 7]. For example, the recent TWAS of AD dementia by Sun et al. identified 53 risk genes by standard two-stage TWAS methods [8].
Standard two-stage TWAS methods first train gene expression prediction models by using reference genetic and transcriptomic data profiled of the same training samples, taking quantitative gene expression traits as response variables and the proximal cis-acting genetic variants as predictors (Stage I). Estimated effect sizes of genetic variants in the gene expression prediction models could be viewed as effect sizes of a broad sense of expression quantitative trait loci (eQTL), which will then be taken as variant weights in gene-based association test to map risk genes with GWAS data (Stage II). TWAS association test in Stage II is equivalent to testing the association between predicted genetically regulated gene expression (GReX) levels and the phenotype of interest in the GWAS data [9].
However, one limitation of most existing TWAS methods is that they only consider cis-eQTL [6, 10, 11], i.e., proximal genetic variants (e.g., located within the 1Mb region around the target gene region), in the gene expression prediction models. The limitation is mainly due to the computational bottleneck of considering genome-wide genetic variants to fit gene expression prediction models for transcriptome-wide ~20K genes per tissue type. The resulting cavity is failing to account for trans-eQTL, i.e., located distal from the target gene region, that have been found playing important roles in biological processes and disease susceptibility [12, 13]. Incorporating trans-eQTL in TWAS is essential as they can reveal additional regulatory mechanisms, help identify additional risk genes, and further our understanding of the underlying biological mechanisms of complex diseases.
To overcome this limitation in studying AD dementia, we employed the Bayesian Genome-wide TWAS (BGW-TWAS) method that incorporated both cis- and trans-eQTL for TWAS [14]. We applied BGW-TWAS to the reference Genotype-Tissue Expression (GTEx) V8 dataset [15] of three tissues –– prefrontal cortex, cortex, and whole blood. The selection of prefrontal cortex and cortex tissues was based on substantial evidence linking their involvement to the progression of AD dementia [16]. The selection of whole blood tissue was due to three reasons: i) a large sample size exists (n=574) for the whole blood tissue in the reference GTEx V8 dataset; ii) gene expression in the whole blood and that of the brain's cortex were found correlated [17]; iii) recent studies have demonstrated that gene expressions in whole blood could serve as biomarkers for AD dementia [18, 19]. For example, multiple studies showed that blood-based transcriptomics biomarkers predict AD risks, such as in Korukonda et. al. Alzheimer’s & Dementia 2021 [20]; Shigemizu et. al. Alzheimer’s Research & Therapy 2020 [21], Abdullah et. al. Informatics in Medicine 2022 [22],and Lee et. al Scientific Reports 2020 [23]. Also, a recent paper by Angioni et. al. J Prev Alzheimers Dis. 2022 [24] discussed the success, challenges, and future directions of deriving blood biomarkers from research use to clinical practice.
In this study, we first estimated cis- and trans- eQTL effect sizes of prefrontal cortex, cortex, and whole blood tissues by BGW-TWAS. Second, we calculated BGW-TWAS p-values using the S-PrediXcan test statistic (gene-based association test) [25], where the estimated eQTL effect sizes were integrated with the summary data of the most recent GWAS of AD dementia (n=~487K) [26]. BGW-TWAS p-values were obtained for transcriptome-wide test genes for each tissue. Third, for each test gene, we used the omnibus aggregated Cauchy association test (ACAT-O) [27, 28] to combine TWAS p-values across three tissues to obtain the omnibus TWAS p-value. The workflow is presented in Fig. 1.

Study workflow
As a result, we identified 85 significant genes with the prefrontal cortex, 82 significant genes with the cortex, and 76 significant genes with the whole blood reference transcriptomic data. Combining BGW-TWAS p-values across these three tissues by ACAT-O, we obtained a total of 141 significant TWAS risk genes for AD dementia, including several well-known AD risk genes and risk genes that not mapped in the GWAS Catalog. Through protein-protein interaction network analysis [29] with these 141 significant TWAS risk genes, we detected gene networks comprised of known AD risk genes such as APOC2, BIN1, CASS4, MS4A4A, MS4A6A, SLC24A4, and MAPT and additional risk genes that are not mapped in the GWAS Catalog. In addition, these 141 significant TWAS risk genes were found enriched with known risk genes of several AD related phenotypes.
Methods
Bayesian genome-wide TWAS (BGW-TWAS)
BGW-TWAS [14] is a recently proposed TWAS method that incorporates both cis- and trans- genetic variants of the target genes as predictors in the gene expression prediction models (Stage I) as follows:
where \({\mathcal{E}}_{g}\) denotes the target gene expression quantitative trait, (\({{\varvec{X}}}_{cis}, {{\varvec{X}}}_{trans}\)) denotes the genotype data matrix of cis- and trans- genetic variants; (\({{\varvec{w}}}_{cis}\), \({{\varvec{w}}}_{trans}\)) denotes the corresponding cis- and trans- effect sizes; and \({\varvec{\epsilon}}\) denotes the errors following a normal distribution with mean 0 and variance \({\sigma }_{\epsilon }^{2}\). Here, we briefly describe the BGW-TWAS method.
BGW-TWAS assumes the Bayesian variable selection regression (BVSR) model [30] to enforce sparse eQTL models, by assuming specific spike-and-slab prior distributions for cis- and trans- effect sizes given by
Here, \(({\pi }_{cis}, {\pi }_{trans})\) denote the respective probability that the corresponding effect size is normally distributed, and \({\delta }_{0}\left({w}_{i}\right)\) is the point mass density function that takes value 0 when \({w}_{i}\ne 0\) and 1 when \({w}_{i}=0\).
BGW-TWAS employs multiple computational strategies to enable efficient computation to account for genome-wide genetic variants as predictors in the gene expression prediction models as in Eq. 1. A previously developed scalable expectation-maximization Markov Chain Monte Carlo (EM-MCMC) algorithm [31] is adapted and used by BGW-TWAS. Genome-wide variants are first segmented into approximately independent genome blocks. Genome blocks containing cis- genetic variants or containing top trans-eQTL with single variant test p-values < \({10}^{-5}\) will be selected for implementing the EM-MCMC algorithm to estimate eQTL effect sizes in the joint multiple regression model as in Eq. 1. The posterior causal probabilities (PCP) for “eQTL” with non-zero posterior effect size estimates will also be estimated. The product of estimated PCPs and effect sizes will represent the expected posterior effect sizes and be used as variant weights in the follow-up gene-based association tests (Stage II). The details of the BGW-TWAS method are referred to the BGW-TWAS paper [14].
The BGW-TWAS tool is memory efficient for first generating single variant eQTL summary data with individual-level reference transcriptomic and genetic data, and then using only these summary eQTL data in the MCMC algorithm. Also, the BGW-TWAS tool implements MCMC algorithm in parallel for multiple genome blocks, which can best utilize high-performance computing clusters with multiple computation cores. In this study, the BGW-TWAS method finished analyzing an average gene with sample size 184 in the reference transcriptomic dataset within 5 minutes in 16 cores. A total of 16 Gb memory was requested and sufficient for all analyses in this study.
Gene-based association test by S-PrediXcan test statistic
With cis- and trans- eQTL weights estimated by BGW-TWAS method and summary-level GWAS data (i.e., Z-scores by single variant tests), we employed the S-PrediXcan [25] approach to calculate the burden type TWAS Z-score test statistic \({Z}_{g}\) per gene as follows:
where \(\widehat{{\beta }_{l}}\) denotes the genetic effect size of variant \(l\) from GWAS, \({Z}_{l}\) denote the corresponding Z-score statistic by single variant GWAS test, and \(\widetilde{{\text{w}}_{\text{lg }}}=\widehat{P{CP}_{l}}{\widehat{w}}_{l}\) is the eQTL weight of variant \(l\) estimated by BGW-TWAS. Here, \({{\varvec{X}}}_{{\varvec{r}}{\varvec{e}}{\varvec{f}}}\) denotes the genotype matrix with genotype covariance matrix \({\varvec{V}}\) from a reference panel. Two-tailed BGW-TWAS p-values can then be obtained from the TWAS Z-score test statistics as in Eq. 3.
Omnibus aggregated cauchy association test (ACAT-O)
ACAT-O is an omnibus test that can combine p-values of multiple tests with respect to the same null hypothesis [27, 28], which employs a linear combination of transformed p-values as the test statistic. Particularly, the ACAT-O method is a general statistical method, which is suitable for combining correlated p-values such as the ones generated with correlated transcriptomic data of multiple tissues. As shown in the following formula,
where \(\{{p}_{i}, i=1,\dots , K\}\) denotes the p-values of K tests and the ACAT-O statistic \({T}_{ACAT-O}\) approximately follows a standard Cauchy distribution under the null hypothesis. The approximate standard Cauchy distribution allows analytical calculations of the ACAT-O p-values, which were shown to be more accurate for combining small/significant p-values [27, 28].
As shown in Fig. 1, in this study, ACAT-O method was used to combine BGW-TWAS p-values (per gene) across three considered tissues to obtain combined TWAS p-values, under the null hypothesis of the combined genetic effects are mediated through the transcriptome of one of these three tissues. Combined TWAS p-values were obtained by using the “ACATO” function from the R package “sumFREGAT” [32].
Since the ACAT-O method only provides an omnibus combined p-value for each test gene, one could refer to the TWAS Z-score test summary data of each tissue to gain information about which tissue contributed most to the significant TWAS risk gene by ACAT-O.
Genotype-tissue expression (GTEx) data
The publicly available GTEx V8 (dbGaP phs000424.v8.p2) data [15] contain whole genome sequencing (WGS) data of 838 human donors and RNA sequencing (RNA-seq) transcriptomic data of 17,382 normal samples from 52 human tissues and two cell lines. We use the transcriptomic data of the prefrontal cortex (n=158), cortex (n=184), and whole blood (n=574) tissues and the corresponding WGS data as reference panel to estimate cis- and trans- eQTL weights by BGW-TWAS. Samples in the GTEx V8 dataset are of European ancestries. The same fully processed, filtered, and normalized transcriptomic data used in the GTEx eQTL analysis were downloaded from the GTEx portal and use in this study. For each tissue, samples with <10 million mapped RNA-seq reads were excluded. For samples with replicates, the replicate with the greatest number of reads was selected. Gene read counts from each sample were normalized using size factors calculated by DESeq2 and log-transformed with an offset of 1. Genes with log-transformed value \(>1\) in \(>10\%\) samples were considered. The resulting gene expression values were centered with mean 0 and standardized with standard deviation 1. The resulting matrix was then hierarchically clustered (based on average and cosine distance), and a chi2 p-value was calculated based on Mahalanobis distance. Clusters with ≥60% samples with Bonferroni-corrected p-values <0.05 were marked as outliers, and their samples were excluded. Genetic variants with missing rate \(<20\%\), minor allele frequency \(>0.01\), and Hardy-Weinberg equilibrium p-value \(>{10}^{-5}\) were considered for fitting the gene expression prediction models.
The fully processed, filtered, and normalized transcriptomic data were adjusted for top five genotype principal components, top probabilistic estimation of expression residuals (PEER) factors [9], sequencing protocol (PCR-based or PCR-free), sequencing platform (Illumina HiSeq 2000 or HiSeq X), and sex, as suggested by the GTEx eQTL data analysis guidelines [15]. The number of top PEER factors [33] used to adjust the gene expression traits depends on the sample sizes in the reference transcriptomic data cohort. We used 30 factors for gene expression traits of the prefrontal cortex and cortex tissues, and 60 factors for the whole blood tissue. Only samples with complete data of these covariates were included in the analyses. Adjusted gene expression quantitative traits were then taken as response variables in the gene expression prediction model.
Summary-level GWAS data of AD dementia
The summary-level GWAS data of AD dementia (i.e., single variant Z-score test statistics obtained by meta-analysis) were generated by the latest GWAS by Bellenguez et al. [5]. The summary-level GWAS data (n=~487K; European ancestry) were generated by meta-analysis with the European Alzheimer & Dementia Biobank consortium (with samples from 15 European countries) and UK Biobank dataset. GWAS of each cohort was conducted with clinically diagnosed cases, ancestry proxy cases, and controls.
Protein-protein interaction network and pathway analysis
STRING (version 12.0) [34] is a bioinformatics web tool that provides information on protein-protein interactions and networks, as well as functional characterization of genes and proteins. The tool integrates different types of evidence from public databases, such as genomic context, high-throughput experiments, and previous knowledge from other databases, to generate reliable predictions of protein interactions and build networks and pathways. Provided with a list of gene names, STRING will construct networks based on the protein-protein interactions of the corresponding proteins, as well as identify phenotypes that have risk genes enriched in the provided list. Proteins corresponding to provided genes are considered as nodes in the protein-protein interaction network. Protein-protein edges represent the predicted functional associations, colored differently to indicate seven categories –– computational interaction predictions from co-expression, “text-mining” of scientific literature, databases of interaction experiments (biochemical/genetic data), known protein complexes or pathways from curated resources, gene co-occurrence, gene fusion and gene neighborhood. Gene co-occurrence, fusion, and neighborhood represent association predictions are based on whole-genome comparisons [35]. Interactions from these resources are critically assessed, scored, and subsequently automatically transferred to less well-studied organisms using hierarchical orthology information [34].
Particularly, the “text-mining” channel is the result of parsing full-text articles from the PMC Open Access Subset (up to April 2022), PubMed abstracts (up to August 2022), as well as summary texts from OMIM [36] and Saccharomyces genome database [37] entry descriptions. These texts are all parsed for co-mentions of protein pairs and assessed against the frequencies of all separate mentions of the respective proteins. An improved deep learning-based relation extraction text mining model was used by STRING v12 [34]. The ‘textmining’ channel significantly increases the number of protein–protein interactions.
Results
BGW-TWAS results of AD dementia
Using BGW-TWAS method, we trained gene expression prediction models for 23,724 genes of prefrontal cortex tissue, 23,900 genes of cortex tissue, and 19,519 genes of whole blood tissue. We identified 85, 82, and 76 TWAS risk genes with significant p-values (with Bonferroni correction) for prefrontal cortex, cortex, and whole blood tissues, respectively. Of these, 20 genes were significant for prefrontal cortex and cortex, 4 were significant for prefrontal cortex and whole blood, 5 were significant for cortex and whole blood, and 13 were significant for all three tissue types (Supplemental Figure 1). Detailed information of the significant TWAS risk genes of these three tissues were provided in Supplemental Files 2-4. Manhattan plots of BGW-TWAS results of these three tissues were presented in Supplemental Figures 2-4.
We also summarized the proportion of trans-eQTL with non-zero weights for all significant genes of three tissues in Supplemental Figure 5. There were 27 (31.8%), 19 (23.2%), and 21 (27.6%) significant genes whose association were driven by ≥50% trans-eQTLs in prefrontal cortex, cortex, and whole blood tissues, respectively. These results demonstrated that trans-eQTL had important contributions to significant TWAS risk genes of all three tissues.
ACAT-O results of AD dementia
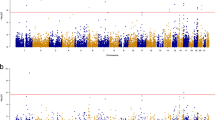
Combining BGW-TWAS p-values across three tissues by ACAT-O, we obtained ACAT-O p-values for a total of 17,449 genes. We identified 141 genes with significant ACAT-O p-values (with Bonferroni correction). As illustrated by the Manhattan plot (Fig. 2), 27 genes were located on chromosome 19 around the well-known APOE locus, which is consistent with prior TWAS findings [8, 38].

Manhattan plot of ACAT-O TWAS p-values for studying AD dementia. The horizontal dashed line represents the Bonferroni corrected significance threshold of the p-values. Orange dots indicate genes that were significant in only one tissue, while red dots highlight genes that were significant in more than one tissue
To compare with TWAS results using only cis-eQTL, we conducted TWAS for all three tissue types using only cis-eQTL with non-zero weights estimated by BGW-TWAS, and then combined these TWAS p-values per gene by ACAT-O. We found that 107 of the 141 significant genes (using both cis- and trans- eQTL) retained their significance using only cis-eQTL, while the remaining 34 genes were not detected (Supplemental Figure 6). Because these 34 (~24%) significant genes were primarily driven by trans-eQTL. We presented example significant TWAS risk genes (63 out of all 141) in Table 1, including these 34 significant genes driven by trans-eQTL and additional 29 risk genes that were significant in both prefrontal cortex and cortex tissues. Summary data of all significant TWAS risk genes were presented in the Supplemental File 1.
Contributing eQTL of significant TWAS risk genes
To investigate how eQTL contributed to significant TWAS risk genes, we plotted eQTL weights estimated by BGW-TWAS for three tissues of example TWAS risk genes in Fig. 3. Specifically, column A in Fig. 3 shows the eQTL weights (in three tissues) of gene ACE whose significance is primarily due to cis-eQTL (circles); and column B shows the eQTL weights of three genes (SNORD22, AP001350.4, and SLC3A2) whose significance is primarily due to trans-eQTL (triangles) in prefrontal cortex, cortex, and whole blood tissues, respectively. As shown in Fig. 3, each dot represents one eQTL with colors ranging from yellow to red to represent the corresponding AD dementia GWAS p-value. We can see that cis- or trans- eQTL colocalizing with potential significant GWAS p-values (colored from yellow to red) are driving the significant TWAS association of the test gene.

Scatter plots of eQTL weights estimated by BGW-TWAS of example TWAS risk genes. Column A: gene ACE (chr17) in three tissues; Column B: gene SNORD22 (chr11), AP001350.4 (chr11), and SLC3A2 (chr11), in prefrontal cortex, cortex, and whole blood tissues, respectively. Y-axis depicts the values of eQTL weights estimated by BGW-TWAS, and the x-axis shows the order of base pair position of the corresponding eQTL. Solid circles denote cis-eQTL, and triangles refer to trans-eQTL. Color legend denotes the -log (GWAS p-value) of the corresponding eQTL
It is noticeable that all the trans-eQTL of the example genes (and for most of the genes we studied) are still on the same chromosome as the test gene. This might be due to both biological mechanisms and methodological limitations. Because the BGM-TWAS method assumes a sparse eQTL model for gene expression and often can only estimate eQTL with relatively large effect sizes, it is possible that trans-eQTL located on the same chromosome as the test gene have relatively larger effect sizes. Thus, the BGW-TWAS method only estimated non-zero weights for these trans-eQTL that were used in the follow-up gene-based association studies.
Known mapped GWAS risk genes in GWAS catalog for AD dementia
Compared to previous GWAS results, 35 out of the 141 TWAS risk genes were reported as mapped risk genes of AD dementia in GWAS Catalog [39], such as known risk genes of ACE, APOC2, BIN1, and CR1 (Supplemental File 1). Particularly, the GReX of CR1 and APOC2 were both found to be negatively associated with risk of AD dementia in prefrontal cortex and cortex. The GReX of BIN1 was positively associated with risk of AD dementia in both cortex and whole blood tissue. The GReX of ACE was positively associated with risk of AD dementia in all three tissues. These findings are consistent with previous studies about genes CR1 [40], APOC2 [41], BIN1 [42], and ACE [43]. Information of upregulation and downregulation of the complete lists of significant genes in the 3 tissues is provided in the Supplemental Files 2-4.
Additional TWAS risk genes of AD dementia
Besides these 35 mapped risk genes reported in GWAS catalog, we identified additional 106 significant TWAS risk genes of AD dementia, including 34 genes whose significance were primarily due to trans-eQTLs. Also, our findings replicated risk genes identified by recent studies of AD that integrated eQTL data with GWAS summary data. For instance, genes OSBP, ZNF296, and ZNF284 were identified by the TWAS tool TIGAR [9, 11] using only cis-eQTL of the prefrontal cortex tissue [44] and GWAS summary data of AD dementia generated by Wightman D.P. et. al in 2021 [26]. A recent study integrating summary-level GWAS and meta-analytic cis-eQTL data found genes NDUFS2 and PRSS36 were related to AD risk [45]. Recent splicing TWAS analyses found that causal splicing introns of gene WDR33 and LRRC37A4P were associated with AD in multiple brain tissues [46]. A recent study that integrated eQTL data of blood tissue and GWAS of late-onset AD (LOAD) by a Bayesian statistical method identified risk gene ZNF226 [47].
We also looked at cis-SNPs within ±1Mb region of our identified TWAS risk genes, using the GWAS results by Bellenguez et al. [5]. We found that 114 genes have at least one cis-SNP with GWAS p-value less than the genome-wide significance threshold (5E-8) and 27 genes have no GWAS significant cis-SNPs (Supplemental File 1). Because our significant TWAS risk genes were identified by testing both cis- and trans-SNPs with non-zero eQTL weights. Those genes without GWAS significant cis-SNPs will not be identified by standard GWAS with the same data. Our findings could be due to the consideration of trans-eQTL and/or burden tests with multiple eQTL. Previous biological studies provided insights of some of our findings with no significant cis-SNPs. For example, it was found that the expression of NRP2, a gene coding the modulating receptors of the vascular endothelial growth factor signaling family, was associated with better cognitive outcomes [48]. Gene OTULIN was found to affect NF-κB-activity in AD patients, subsequently leading to the shrinkage of the entorhinal cortex and the limbic system in early stages of AD [49]. Gene DDR1 is associated with reduced inflammation and vascular fibrosis in AD [50]. In addition, gene TUBB encodes a β-tubulin protein that forms a dimer with alpha tubulin and acts as a structural component of microtubules, where β-tubulin was found aggregating in AD cases [51].
Protein-protein interaction network analysis by STRING
To further understand the underlying biological mechanisms of our identified 141 TWAS risk genes, we constructed protein-protein interaction networks and performed phenotype enrichment analysis using the STRING tool. As shown in Fig. 4, we identified several clusters of genes, including a major cluster composed of known mapped GWAS risk genes of AD such as BIN1, CASS4, MS4A4A, MS4A6A, MS4A3, SLC24A4, INPP5D, FCER1G, APOC2, MAPT, BCL3, RELB, and TRAPPC6A. Importantly, this major gene network is connected through well-known risk genes APOC2, BIN1, and MAPT. Especially, APOC2 is known to be related with both lipids and AD [52]; BIN1 is a key regulator of proinflammatory and neurodegeneration-related activation in microglia [53]; and MAPT encodes the microtubule-associated protein tau (MAPT) which is known as a key AD pathology [54]. Additionally, several of our findings that are not reported in GWAS Catalog and/or have no cis-SNP with significant GWAS p-value were found connected with these known AD risk genes in the main cluster. For example, gene WDR33 was connected to CELF1; genes SHC2 and GAL3ST4 were connected to INPP5D; genes WNT3, PPP5C and NDUFS2 were connected to MAPT.

Protein-protein interaction networks identified with 141 significant TWAS risk genes by the STRING tool. Edge colors show different sources of the identified protein-protein interactions
By pathway enrichment analyses conducted by the STRING tool, five pathways were found to be enriched with our identified TWAS risk genes. Three of these pathways were involved with the CD20-like family such as Cranial nerve maturation, Peptidase A22B, signal peptide peptidase and GTPase GIMA/IAN/Toc. These three pathways included the key known AD risk genes MS4A4A, SLC24A4, MS4A6A, BIN1, and CASS4. The fourth pathway was ADP-ribosylation factor-like protein 17, and LRRC37A/B like protein 1, C-terminal, which included TWAS risk genes LRRC37A2, ARHGAP27, and ARL17A on chromosome 17. The fifth pathway was Cranial nerve maturation, and His Kinase A (phosphoacceptor) domain, including known AD risk genes SLC24A4, CASS4, and BIN1.
Phenotype enrichment analysis
By phenotype enrichment analyses conducted by the STRING tool, we found that our identified 141 TWAS risk genes were enriched with 14 phenotypes (Fig. 5), including AD related phenotypes such as family history of AD (false discovery rate, FDR = 4.20e-18), AD biomarker measurement (FDR = 9.30e-12), mental or behavioral disorders (FDR = 4.93e-09), complete blood cell count (FDR = 6.50e-03), functional laterality (FDR = 8.00e-03), white matter microstructure measurement (FDR = 8.00e-03), non-lobar intracerebral hemorrhage (FDR = 1.29e-02), leukocyte count (FDR = 1.85e-02), myeloid white cell count (FDR = 2.35e-02), and eosinophil count (FDR = 4.83e-02). For example, TWAS risk genes APOC2, BIN1, SLC24A4, BCL3, RELB, and CLPTM1 are known risk genes for family history of AD, AD biomarker measurement, and mental or behavioral disorders; TWAS risk genes MAPT, WNT3, and LINC02210 were related to white matter microstructure measurement; TWAS risk genes MAPT, TUBB, PPP5C, FCER1G, INPP5D, NDUFS2, NDUFAF6, and BCL3 are common risk genes for complete blood cell count, leukocyte count, myeloid white cell count, and eosinophil count.

Phenotype enrichment analysis with 141 significant TWAS risk genes by the STRING tool. The -log10 of the false discovery rates (FDR, x-axis) for testing the enrichment of known risk genes of the corresponding phenotype (y-axis) were plotted
Interestingly, the enriched phenotypes of functional laterality, intracerebral hemorrhage, white matter microstructure measurement, complete blood cell count, leukocyte count, myeloid white cell count, and eosinophil count have been reported to be associated with AD by previous studies. According to a high-resolution MRI study, lack of the laterality shift in limbic system and early loss of asymmetry in entorhinal cortex could be biomarkers to identify preclinical AD [55]. Several studies found that the risk of dementia increased significantly after intracerebral hemorrhage [56, 57]. A study observed microstructural damage of white matter in AD brains [58]. Chronic inflammation has been proposed as a significant risk factor in AD pathogenesis [59]. Increased levels of complete blood cell count and immune cell count, including leukocyte, myeloid white cell, and eosinophil, have been observed in the brains of AD patients [60,61,62,63].
Further, we conducted separate phenotype enrichment analyses by using tissue-specific TWAS risk genes that are of upregulated (with positive TWAS Z-score) and downregulated (with negative TWAS Z-score) groups (Supplemental Figure 7). We found that mental or behavioral disorders and functional laterality were enriched in the upregulated genes in prefrontal cortex, cortex, and whole blood. Additionally, family history of AD was enriched in the upregulated genes in cortex and whole blood, and the downregulated genes in whole blood; AD biomarker measurement was enriched in the downregulated genes in whole blood. No phenotype was found enriched in the downregulated genes in prefrontal cortex and cortex.
Discussion
In this study, we applied the BGW-TWAS method to the reference GTEx V8 data of three tissues (prefrontal cortex, cortex, and whole blood) to leverage both cis- and trans-acting eQTL for studying AD dementia. With the latest GWAS summary data of AD dementia [5], we identified 85 significant genes with the prefrontal cortex, 82 significant genes with the cortex, and 76 significant genes with the whole blood reference transcriptomic data. Combining BGW-TWAS p-values by ACAT-O, we obtained 141 significant TWAS risk genes of AD dementia, including 35 mapped GWAS risk genes in GWAS Catalog and 34 TWAS risk genes primarily driven by trans-eQTL. As expected, most of the genes exhibiting significance due to trans-eQTL were not detected by previous GWAS and TWAS which failed to account for trans-eQTL. Interestingly, previous studies reported AD-related biological functions for our identified TWAS risk genes that have no cis-SNP with significant GWAS p-value, such as NRP2, OTULIN, DDR1, and TUBB [48,49,50,51].
Through protein-protein interaction network analysis using the STRING tool, we identified network clusters containing mapped GWAS risk genes of AD in GWAS Catalog and our identified TWAS risk genes that are either driven by trans-eQTL or/and have no cis-SNP with significant GWAS p-value. A total of 46 identified TWAS risk genes that are not reported as mapped risk genes in GWAS catalog, are connected with mapped AD GWAS risk genes in the identified protein-protein interaction clusters. Our findings are consistent with previous studies highlighting the critical involvement of APOC2, BIN1, and MAPT in AD [52,53,54], as evidenced by their extensive connectivity with other mapped GWAS risk genes of AD and TWAS risk genes within the major network cluster. The identified protein-protein interaction networks also underline the important function of genes in the CD20-like family. Since the “text-mining” channel (see Methods) used by the STRING tool identified the majority of the protein-protein interactions among our identified TWAS risk genes, further functional studies by biological experiments are still needed to validate these interactions.
We also identified 14 phenotypes whose known risk genes were enriched in our identified 141 TWAS risk genes. Especially, brain lesions such as functional laterality, intracerebral hemorrhage, and white matter microstructure have been reported to be associated with AD by previous studies [55,56,57,58]. Biomarkers such as complete blood cell count, leukocyte count, myeloid white cell count, and eosinophil count were shown to be related to AD [60,61,62,63], suggesting a complex interplay among genetic, transcriptomic, metabolic, and inflammatory risk factors in the pathogenesis of AD. Shared risk genes between these biomarkers and AD have also been reported by previous studies. For example, gene BCL3 and INPP5D were reported with pleiotropy effects on AD and inflammation biomarkers [64, 65]. Further biological experiments are still needed to understand the roles of these TWAS risk genes in the biological mechanism of AD.
Nevertheless, our study has several limitations. First, due to the computation burden of running BGW-TWAS tool, we only applied BGW-TWAS to reference GTEx V8 data of three tissues (prefrontal cortex, cortex, and whole blood). Other tissues within and outside the brain such as hippocampus, muscle, and spinal cord, are also known to play crucial roles in the biological mechanisms of AD dementia [66,67,68,69,70]. Failing to consider all available tissues in GTEx V8 data may fail to capture a full spectrum of genetically regulated gene expression associated with AD dementia. Our ongoing work includes further improving the computation efficiency of the BGW-TWAS tool and applying it to all available tissues in GTEx V8.
Second, since samples in the summary-level GWAS data of AD dementia (generated by Bellenguez et. al.) and the GTEx reference dataset are all European ancestries, our TWAS significant gene findings are also limited to the European population. Conducting similar TWAS analyses with reference transcriptomic data and GWAS summary data of other populations would be needed to study the disparity of AD risks in different populations.
Third, the BVSR model employed in the BGW-TWAS method inherently assumes a sparse model implying that only a small number of eQTL have true causal effects on gene expression. Although this assumption can be computationally advantageous, it may not always accurately represent the underlying genetic architecture of complex gene expression quantitative traits. In our future work, we will also investigate using other statistical models for genome-wide TWAS analysis.
Conclusions
In conclusion, our study highlights the importance of considering both cis- and trans-eQTL in TWAS analysis as it can help identify significant risk genes that would have been missed by using only cis-eQTL. We identified several well-known AD risk genes as well as genes that are not reported in GWAS Catalog but interconnected with known AD risk genes in the same protein-protein interaction networks. As a genome-wide TWAS, our study is the first to utilize both cis- and trans- eQTLs of multiple tissues for AD risk gene identification. Our results provide further insights into the underlying biological mechanisms of AD dementia and a list of potential gene targets for the development of therapeutics for treating AD dementia.
Availability of data and materials
The RNAseq transcriptomic and WGS genetic data of GTEx V8 are publicly available from dbGAP (phs000424.v8.p2) and the GTEx portal website (https://gtexportal.org/home/). The summary-level GWAS data of AD dementia was generated by Bellenguez et. al., which are publicly available at https://www.ebi.ac.uk/gwas/studies/GCST90027158. The BGW-TWAS tool is available freely on GitHub (https://github.com/yanglab-emory/BGW-TWAS). The STRING tool is available online (https://string-db.org/). Summary data of our trained gene expression prediction models by BGW-TWAS method and our TWAS results are publicly available on SYNAPSE (https://doi.org/10.7303/syn54950908).
Abbreviations
- AD:
-
Alzheimer’s disease
- GWAS:
-
Genome-wide association studies
- TWAS:
-
Transcriptome-wide association study
- BGW-TWAS:
-
Bayesian Genome-wide TWAS
- eQTL:
-
Expression quantitative trait loci
- GTEx:
-
Genotype-Tissue Expression
- ACAT-O:
-
Omnibus aggregated Cauchy association test
- GReX:
-
Genetically regulated gene expression
- BVSR:
-
Bayesian variable selection regression
- EM-MCMC:
-
Expectation-maximization Markov Chain Monte Carlo
- PCP:
-
Posterior causal probabilities
- WGS:
-
Whole genome sequencing
- TPM:
-
Transcripts Per Million
References
2022 Alzheimer's disease facts and figures. Alzheimers Dement. 2022;18(4):700-89.
Long JM, Holtzman DM. Alzheimer Disease: an update on pathobiology and treatment strategies. Cell. 2019;179(2):312–39.
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63(2):168–74.
Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51(3):404–13.
Bellenguez C, Kucukali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet. 2022;54(4):412–36.
Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47(9):1091–8.
Zhao B, Shan Y, Yang Y, Yu Z, Li T, Wang X, Luo T, Zhu Z, Sullivan P, Zhao H, Li Y, Zhu H. Transcriptomewide association analysis of brain structures yields insights into pleiotropy with complex neuropsychiatric traits. Nat Commun. 2021;12(1):2878. https://doi.org/10.1038/s41467-021-23130-y.
Sun Y, Zhu J, Zhou D, Canchi S, Wu C, Cox NJ, et al. A transcriptome-wide association study of Alzheimer’s disease using prediction models of relevant tissues identifies novel candidate susceptibility genes. Genome Med. 2021;13(1):141.
Parrish RL, Gibson GC, Epstein MP, Yang J. TIGAR-V2: Efficient TWAS tool with nonparametric Bayesian eQTL weights of 49 tissue types from GTEx V8. HGG Adv. 2022;3(1):100068.
Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48(3):245–52.
Nagpal S, Meng X, Epstein MP, Tsoi LC, Patrick M, Gibson G, et al. TIGAR: an improved bayesian tool for transcriptomic data imputation enhances gene mapping of complex traits. Am J Hum Genet. 2019;105(2):258–66.
Vosa U, Claringbould A, Westra HJ, Bonder MJ, Deelen P, Zeng B, et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet. 2021;53(9):1300–10.
Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45(10):1238–43.
Luningham JM, Chen J, Tang S, De Jager PL, Bennett DA, Buchman AS, et al. Bayesian genome-wide TWAS method to leverage both cis- and trans-eQTL information through summary statistics. Am J Hum Genet. 2020;107(4):714–26.
Consortium GT. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369(6509):1318–30.
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189.
Sullivan PF, Fan C, Perou CM. Evaluating the comparability of gene expression in blood and brain. Am J Med Genet B Neuropsychiatr Genet. 2006;141B(3):261–8.
Lunnon K, Keohane A, Pidsley R, Newhouse S, Riddoch-Contreras J, Thubron EB, et al. Mitochondrial genes are altered in blood early in Alzheimer’s disease. Neurobiol Aging. 2017;53:36–47.
Souza VC, Morais GS Jr, Henriques AD, Machado-Silva W, Perez DIV, Brito CJ, et al. Whole-blood levels of MicroRNA-9 are decreased in patients with late-onset Alzheimer Disease. Am J Alzheimers Dis Other Demen. 2020;35:1533317520911573.
Korukonda A, Dutkowski J, Ronen R, Sachdev P. Developing a blood-derived gene expression biomarker specific for Alzheimer’s disease. Alzheimer's Dement. 2021;17:e053842.
Shigemizu D, Mori T, Akiyama S, Higaki S, Watanabe H, Sakurai T, et al. Identification of potential blood biomarkers for early diagnosis of Alzheimer’s disease through RNA sequencing analysis. Alzheimers Res Ther. 2020;12(1):87.
Mohammad Nasir Abdullah YBW, Abu Bakar Abdul Majeed, Yuslina Zakaria, Norshahida Shaadan. Identification of blood-based transcriptomics biomarkers for Alzheimer's disease using statistical and machine learning classifier. Inform Medi Unlock. 2022;33:101083.
Lee T, Lee H. Prediction of Alzheimer’s disease using blood gene expression data. Sci Rep. 2020;10(1):3485.
Angioni D, Delrieu J, Hansson O, Fillit H, Aisen P, Cummings J, et al. Blood biomarkers from research use to clinical practice: what must Be done? A report from the EU/US CTAD task force. J Prev Alzheimers Dis. 2022;9(4):569–79.
Barbeira AN, Dickinson SP, Bonazzola R, Zheng J, Wheeler HE, Torres JM, et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun. 2018;9(1):1825.
Wightman DP, Jansen IE, Savage JE, Shadrin AA, Bahrami S, Holland D, et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet. 2021;53(9):1276–82.
Liu Y, Chen S, Li Z, Morrison AC, Boerwinkle E, Lin X. ACAT: a fast and powerful p value combination method for rare-variant analysis in sequencing studies. Am J Hum Genet. 2019;104(3):410–21.
Liu Y, Xie J. Cauchy combination test: a powerful test with analytic p-value calculation under arbitrary dependency structures. J Am Stat Assoc. 2020;115(529):393–402.
Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605–12.
Yongtao Guan MS. Bayesian variable selection regression for genome-wide association studies and other large-scale problems. Ann Appl Stat. 2011;5(3):1780–815.
Yang J, Fritsche LG, Zhou X, Abecasis G, International Age-Related Macular Degeneration Genomics C. A Scalable Bayesian Method for Integrating Functional Information in Genome-wide Association Studies. Am J Hum Genet. 2017;101(3):404-16.
Svishcheva GR, Belonogova NM, Zorkoltseva IV, Kirichenko AV, Axenovich TI. Gene-based association tests using GWAS summary statistics. Bioinformatics. 2019;35(19):3701–8.
Stegle O, Parts L, Durbin R, Winn J. A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Comput Biol. 2010;6(5):e1000770.
Szklarczyk D, Kirsch R, Koutrouli M, Nastou K, Mehryary F, Hachilif R, et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51(D1):D638–46.
Hu T, Parrish RL, Buchman AS, Tasaki S, Bennett DA, Seyfried NT, et al. Omnibus proteome-wide association study (PWAS-O) identified 43 risk genes for Alzheimer’s disease dementia. medRxiv. 2022:2022.12.25.22283936.
Amberger JS, Bocchini CA, Scott AF, Hamosh A. OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019;47(D1):D1038-D43.
Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, et al. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 2012;40(Database issue):D700-5.
Tang S, Buchman AS, De Jager PL, Bennett DA, Epstein MP, Yang J. Novel Variance-Component TWAS method for studying complex human diseases with applications to Alzheimer’s dementia. PLoS Genet. 2021;17(4):e1009482.
MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45(D1):D896–901.
Mahmoudi R, Kisserli A, Novella JL, Donvito B, Drame M, Reveil B, et al. Alzheimer's disease is associated with low density of the long CR1 isoform. Neurobiol Aging. 2015;36(4):1766 e5- e12.
Shao Y, Shaw M, Todd K, Khrestian M, D’Aleo G, Barnard PJ, et al. DNA methylation of TOMM40-APOE-APOC2 in Alzheimer’s disease. J Hum Genet. 2018;63(4):459–71.
Chapuis J, Hansmannel F, Gistelinck M, Mounier A, Van Cauwenberghe C, Kolen KV, et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry. 2013;18(11):1225–34.
Reveret L, Leclerc M, Emond V, Tremblay C, Loiselle A, Bourassa P, et al. Higher angiotensin-converting enzyme 2 (ACE2) levels in the brain of individuals with Alzheimer’s disease. Acta Neuropathol Commun. 2023;11(1):159.
De Jager PL, Ma Y, McCabe C, Xu J, Vardarajan BN, Felsky D, et al. A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Sci Data. 2018;5:180142.
Lee B, Yao X, Shen L, Alzheimer's Disease Neuroimaging I. Integrative analysis of summary data from GWAS and eQTL studies implicates genes differentially expressed in Alzheimer's disease. BMC Genomics. 2022;23(Suppl 4):414.
Sun Y, Bae YE, Zhu J, Zhang Z, Zhong H, Yu J, et al. A splicing transcriptome-wide association study identifies novel altered splicing for Alzheimer’s disease susceptibility. Neurobiol Dis. 2023;184:106209.
Rao S, Ghani M, Guo Z, Deming Y, Wang K, Sims R, et al. An APOE-independent cis-eSNP on chromosome 19q13.32 influences tau levels and late-onset Alzheimer's disease risk. Neurobiol Aging. 2018;66:178 e1- e8.
Seto M, Dumitrescu L, Mahoney ER, Sclafani AM, De Jager PL, Menon V, et al. Multi-omic characterization of brain changes in the vascular endothelial growth factor family during aging and Alzheimer’s disease. Neurobiol Aging. 2023;126:25–33.
Kaltschmidt B, Helweg LP, Greiner JFW, Kaltschmidt C. NF-kappaB in neurodegenerative diseases: recent evidence from human genetics. Front Mol Neurosci. 2022;15:954541.
Stevenson M, Varghese R, Hebron ML, Liu X, Ratliff N, Smith A, et al. Inhibition of discoidin domain receptor (DDR)-1 with nilotinib alters CSF miRNAs and is associated with reduced inflammation and vascular fibrosis in Alzheimer’s disease. J Neuroinflammation. 2023;20(1):116.
Puig B, Ferrer I, Luduena RF, Avila J. BetaII-tubulin and phospho-tau aggregates in Alzheimer's disease and Pick's disease. J Alzheimers Dis. 2005;7(3):213-20; discussion 55-62.
Carter CJ. Convergence of genes implicated in Alzheimer’s disease on the cerebral cholesterol shuttle: APP, cholesterol, lipoproteins, and atherosclerosis. Neurochem Int. 2007;50(1):12–38.
Sudwarts A, Ramesha S, Gao T, Ponnusamy M, Wang S, Hansen M, et al. BIN1 is a key regulator of proinflammatory and neurodegeneration-related activation in microglia. Mol Neurodegener. 2022;17(1):33.
Ma RH, Zhang Y, Hong XY, Zhang JF, Wang JZ, Liu GP. Role of microtubule-associated protein tau phosphorylation in Alzheimer’s disease. J Huazhong Univ Sci Technolog Med Sci. 2017;37(3):307–12.
Long X, Zhang L, Liao W, Jiang C, Qiu B, Alzheimer's Disease Neuroimaging I. Distinct laterality alterations distinguish mild cognitive impairment and Alzheimer's disease from healthy aging: statistical parametric mapping with high resolution MRI. Hum Brain Mapp. 2013;34(12):3400-10.
Moulin S, Labreuche J, Bombois S, Rossi C, Boulouis G, Henon H, et al. Dementia risk after spontaneous intracerebral haemorrhage: a prospective cohort study. Lancet Neurol. 2016;15(8):820–9.
Puppala GK, Gorthi SP, Chandran V, Gandeti R, Rao SS. Prevalence of Cognitive Impairment and Dementia After Intracerebral Hemorrhage. Ann Indian Acad Neurol. 2023;26(6):952–7.
Chen Y, Wang Y, Song Z, Fan Y, Gao T, Tang X. Abnormal white matter changes in Alzheimer’s disease based on diffusion tensor imaging: A systematic review. Ageing Res Rev. 2023;87:101911.
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405.
Luo J, Thomassen JQ, Nordestgaard BG, Tybjaerg-Hansen A, Frikke-Schmidt R. Blood leukocyte counts in Alzheimer Disease. JAMA Netw Open. 2022;5(10):e2235648.
Garcia-Culebras A, Cuartero MI, Pena-Martinez C, Moraga A, Vazquez-Reyes S, de Castro-Millan FJ, et al. Myeloid cells in vascular dementia and Alzheimer’s disease: Possible therapeutic targets? Br J Pharmacol. 2024;181(6):777–98.
van der Willik KD, Fani L, Rizopoulos D, Licher S, Fest J, Schagen SB, et al. Balance between innate versus adaptive immune system and the risk of dementia: a population-based cohort study. J Neuroinflamm. 2019;16(1):68.
Fang Y, Doyle MF, Alosco ML, Mez J, Satizabal CL, Qiu WQ, et al. Cross-sectional association between blood cell phenotypes, cognitive function, and brain imaging measures in the community-based Framingham heart study. J Alzheimers Dis. 2022;87(3):1291–305.
Liu H, Zeng L, Yang Y, Guo C, Wang H. Bcl-3: a double-edged sword in immune cells and inflammation. Front Immunol. 2022;13:847699.
Terzioglu G, Young-Pearse TL. Microglial function, INPP5D/SHIP1 signaling, and NLRP3 inflammasome activation: implications for Alzheimer’s disease. Mol Neurodegener. 2023;18(1):89.
Rao YL, Ganaraja B, Murlimanju BV, Joy T, Krishnamurthy A, Agrawal A. Hippocampus and its involvement in Alzheimer's disease: a review. 3 Biotech. 2022;12(2):55.
Boyle PA, Buchman AS, Wilson RS, Leurgans SE, Bennett DA. Association of muscle strength with the risk of Alzheimer disease and the rate of cognitive decline in community-dwelling older persons. Arch Neurol. 2009;66(11):1339–44.
Daghlas I, Nassan M, Gill D. Genetically proxied lean mass and risk of Alzheimer’s disease: mendelian randomisation study. BMJ Med. 2023;2(1):e000354.
Xie Q, Zhao WJ, Ou GY, Xue WK. An Overview of Experimental and Clinical Spinal Cord Findings in Alzheimer's Disease. Brain Sci. 2019;9(7):168. https://doi.org/10.3390/brainsci9070168.
Horie K, Barthelemy NR, Sato C, Bateman RJ. CSF tau microtubule binding region identifies tau tangle and clinical stages of Alzheimer’s disease. Brain. 2021;144(2):515–27.
Acknowledgements
We would like to thank Dr. Michael P. Epstein from the Center for Computational and Quantitative Genetics, Department of Human Genetics, Emory University School of Medicine, for his helpful comments on this work.
Funding
S.G. and J.Y. are supported by grant R35GM138313 provided by the National Institute of General Medical Sciences, National Institute of Health.
Author information
Authors and Affiliations
Contributions
S.G. analyzed the data, drafted and revised the manuscript. J.Y. designed the study and revised the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All genomic data analyzed in this study are either de-identified or summary statistics, which are not considered as human data according to the guidelines of National Institute of Health.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Guo, S., Yang, J. Bayesian genome-wide TWAS with reference transcriptomic data of brain and blood tissues identified 141 risk genes for Alzheimer’s disease dementia. Alz Res Therapy 16, 120 (2024). https://doi.org/10.1186/s13195-024-01488-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-024-01488-7