
Abstract
Background
Alzheimer’s disease (AD) is characterized by the accumulation of amyloid-β (Aβ) plaques, neurofibrillary tau tangles, and neurodegeneration in the brain parenchyma. Here, we aimed to (i) assess differences in blood and imaging biomarkers used to evaluate neurodegeneration among cognitively unimpaired APOE ε4 homozygotes, heterozygotes, and non-carriers with varying risk for sporadic AD, and (ii) to determine how different cerebral pathologies (i.e., Aβ deposition, medial temporal atrophy, and cerebrovascular pathology) contribute to blood biomarker concentrations in this sample.
Methods
Sixty APOE ε4 homozygotes (n = 19), heterozygotes (n = 21), and non-carriers (n = 20) ranging from 60 to 75 years, were recruited in collaboration with Auria biobank (Turku, Finland). Participants underwent Aβ-PET ([11C]PiB), structural brain MRI including T1-weighted and T2-FLAIR sequences, and blood sampling for measuring serum neurofilament light chain (NfL), plasma total tau (t-tau), plasma N-terminal tau fragments (NTA-tau) and plasma glial fibrillary acidic protein (GFAP). [11C]PiB standardized uptake value ratio was calculated for regions typical for Aβ accumulation in AD. MRI images were analysed for regional volumes, atrophy scores, and volumes of white matter hyperintensities. Differences in biomarker levels and associations between blood and imaging biomarkers were tested using uni- and multivariable linear models (unadjusted and adjusted for age and sex).
Results
Serum NfL concentration was increased in APOE ε4 homozygotes compared with non-carriers (mean 21.4 pg/ml (SD 9.5) vs. 15.5 pg/ml (3.8), p = 0.013), whereas other blood biomarkers did not differ between the groups (p > 0.077 for all). From imaging biomarkers, hippocampal volume was significantly decreased in APOE ε4 homozygotes compared with non-carriers (6.71 ml (0.86) vs. 7.2 ml (0.7), p = 0.029). In the whole sample, blood biomarker levels were differently predicted by the three measured cerebral pathologies; serum NfL concentration was associated with cerebrovascular pathology and medial temporal atrophy, while plasma NTA-tau associated with medial temporal atrophy. Plasma GFAP showed significant association with both medial temporal atrophy and Aβ pathology. Plasma t-tau concentration did not associate with any of the measured pathologies.
Conclusions
Only increased serum NfL concentrations and decreased hippocampal volume was observed in cognitively unimpaired APOEε4 homozygotes compared to non-carriers. In the whole population the concentrations of blood biomarkers were affected in distinct ways by different pathologies.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is the most common cause of dementia, and it is estimated that over 400 million individuals worldwide are in the AD continuum which constituents a decade long asymptomatic phase [1]. AD is characterized by the accumulation of amyloid-β (Aβ) plaques and neurofibrillary tau tangles in the brain parenchyma, accompanied by neurodegeneration [2]. Hereditary factors play a significant role in the prevalence of AD [3], and the epsilon 4 allele of apolipoprotein E gene (APOE ε4) is the strongest genetic risk factor associated with sporadic AD [4]. The APOE ε4 allele has a dose-dependent effect on the prevalence of AD, with individuals carrying one APOE ε4 allele having a three times greater risk for AD, and those with two APOE ε4 alleles having a risk 14.9 times higher when compared with individuals without APOE ε4 alleles [5]. APOE ε4 is well-known for increasing Aβ accumulation in AD [6, 7] and is linked to neurodegeneration in AD as well as other neurodegenerative diseases [8, 9]. Also, it has been found that the APOE ε4 allele affects gray matter volume in the brain, particularly reducing volume in the hippocampus and other regions associated with memory and executive functions [10]. These structural changes, observed even in healthy individuals, have suggested a dose-dependent vulnerability to neurodegeneration associated with the APOE ε4 allele. In addition to the risk of AD, APOE ε4 increases the risk of cardiovascular and cerebrovascular disease [11,12,13].
Current clinically validated biomarkers for AD require cerebrospinal fluid (CSF) sampling or brain imaging. In vivo detection of Aβ and tau pathologies involves CSF measurements (CSF Aβ42/40 and p-tau181, respectively) or the use of positron emission tomography (PET) imaging, while neurodegeneration is typically assessed through magnetic resonance imaging (MRI) or CSF total tau (t-tau) measurements [14]. However, these diagnostic methods are not universally available in all healthcare settings, can be associated with significant patient discomfort, and are often expensive. To address these challenges, the development of easily accessible blood-based biomarkers for AD has shown great promise, and many new biomarkers have recently been introduced [15], although they are not yet established or implemented in clinical practice.
Various blood biomarkers are already available for studying neurodegenerative disorders: CSF neurofilament light chain (NfL) and CSF t-tau are clinically used biomarkers of neuroaxonal injury and neurodegeneration that can also be measured in blood [16, 17]. Plasma NfL is increased in AD and in other neurodegenerative conditions [16], making it unspecific for any particular disease or condition [16, 17]. Effect sizes compared with neurologically healthy controls are lower in AD than in frontotemporal dementia (FTD), some parkinsonian disorders and amyotrophic lateral sclerosis (ALS) [16]. On the other hand, there are confounding effects of age [18, 19] and BMI [20] on plasma NfL levels. Furthermore, NfL levels are also linked to cerebrovascular pathologies [21] including white matter changes [22]. Plasma t-tau has been shown to be associated with neurodegeneration [23, 24], tau-PET [25], and cognitive functions [23, 24, 26], but its use is restricted by wide overlap between diagnostic conditions [27, 28]. To overcome this, other tau biomarkers targeting different epitopes have recently been introduced and studied in relation to neurodegeneration and AD [29,30,31]. One of the latest is a biomarker targeting soluble N-terminal tau fragments (NTA-tau). CSF NTA-tau concentrations are increased in AD and rapidly progressive neurodegenerative diseases, such as Creutzfeldt-Jakob disease (CJD) [23]. Additionally, higher plasma NTA-tau levels have been reported in symptomatic AD patients compared with controls, showing a stronger association with tau PET than Aβ PET or neurodegeneration [29, 32]. Finally, astrocytes are known to be activated in neurodegeneration [33] and in the presence of Aβ [34], and at present, it is possible to measure glial fibrillary acidic protein (GFAP), a marker of astrocytic activation, in blood. Previously, higher plasma GFAP concentration has been associated with the incidence and faster progression of AD [35,36,37,38].
Even though blood biomarkers have been extensively studied during recent years, comparisons evaluating subtle differences in both blood and imaging biomarker measurements between genetic risk groups are not available. In addition, since APOE ε4 is known to increase the risk of Aβ, tau, and vascular pathologies, it is important to further investigate which pathologies contribute to blood biomarker concentrations in these populations. In this study, we aimed to confirm previously reported differences in imaging biomarkers between the different APOE ε4 populations and to evaluate the associations of the aforementioned imaging and blood biomarkers in a cohort comprised of cognitively unimpaired individuals with varying APOE ε4-related genetic risk for sporadic AD. The objectives of our study were twofold: (1) To evaluate the differences in blood and imaging biomarkers among cognitively normal APOE ε4 homozygotes (ε4ε4), heterozygotes (ε4ε3), and non-carriers, and (2) to examine head-to-head within the entire cohort the extent to which different pathologies (Aβ, medial temporal atrophy, cerebrovascular pathology) explain the levels of different blood-based biomarkers.
Materials and methods
Study population
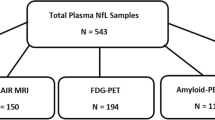
This study was part of a larger research project, and a detailed study protocol has been previously reported [30]. To summarize, the participants of this cross-sectional observational study were recruited in collaboration with the local Auria biobank. Main inclusion criteria were 60–75 years of age and a CERAD total score > 62 points at screening. Exclusion criteria were dementia or cognitive impairment, any neurological or psychiatric disease, diabetes, chronic inflammatory condition, and contraindication for MRI or PET imaging. Our cohort included subjects with one, two, or no APOE ε4 alleles, and all underwent brain PET and MRI imaging, and blood sample collection. The study was approved by the Ethical Committee of the Hospital District of Southwest Finland. All participants signed written informed consent.
Blood biomarkers
Venous EDTA-plasma and serum samples were acquired after a 10–12 h fasting period as reported previously [30] according to in-house standard operating procedures. Samples were gently inverted 5–10 times, centrifuged (2200 × g, 10 min), aliquoted, and stored at − 80◦C prior to biomarker analysis.
All blood biomarkers were measured at the Clinical Neurochemistry Laboratory, University of Gothenburg (Mölndal, Sweden) using the Single molecule array method and HD-X analysers (Quanterix). Commercial kits provided by Quanterix were used to measure single analyte serum NfL (Simoa® NF-light™, #103,186, Quanterix), plasma GFAP (Simoa® GFAP discovery, #102,336, Quanterix), and plasma t-tau (Simoa® Tau Advantage, #101,552, Quanterix). Measurements were performed following the instructions provided by the manufacturer. Briefly, randomized samples were first thawed at room temperature, vortexed (2000 rpm, 10 min, at room temperature), centrifuged (4000 × g, 10 min, at room temperature) and plated. Internal quality control samples were included to the beginning and end of the plate. Calibrators and controls were analysed in duplicates, samples in singlicates. Plasma GFAP concentrations have been reported before [39], and were added here to further investigate the association of GFAP with the biomarkers.
Plasma NTA-tau was measured using an in-house assay, described in detail elsewhere [32, 40]. Briefly, the NTA assay targets soluble N-terminal tau fragments (phosphorylated and non-phosphorylated), and utilizes mouse monoclonal antibodies targeting amino acids (aa) 159–163 (HT7, #MN1000, ThermoScientific, used as a capture antibody) and aa 6–18 (Tau12, BioLegend, used as detector ). Sample handling was done as previously described, and after plating, samples were diluted 1:2 using the assay diluent (Tau 2.0, Quanterix). Calibrators and controls were always analysed as duplicates, samples as singlicates, and non-phosphorylated full-length Tau-441 (SignalChem) was used as an assay calibrator.
Brain imaging
All subjects underwent a structural brain MRI including T1-weighted and T2-FLAIR sequences. Structural brain images were acquired by two different scanners, either with Philips Ingenuity 3.0 T TF PET/MRI (n = 38; Philips Healthcare, Amsterdam, the Netherlands) or Philips Ingenia 3.0 T (n = 22; Philips Healthcare, Amsterdam, the Netherlands).
MRI was used to acquire volumetric variables (hippocampal volume, parahippocampal volume and entorhinal volume), global cortical atrophy score, medial temporal lobe atrophy score, and total volume of white matter hyperintensities. To determine brain Aβ load, [11C]PiB PET scans (n = 60) were acquired from 40 to 90 min post injection (mean injected dose 497 [30] MBq) with an ECAT high-resolution research tomograph (HRRT, Siemens Medical Solutions, Knoxville, TN).
Brain image analysis
PET and MRI image preprocessing and analysis were performed using an automated pipeline at Turku PET Centre [41], which executed the PET data frame by frame realignment, PET-MRI co-registration, FreeSurfer (Freesurfer v6, https://surfer.nmr.mgh.harvard.edu/) region of interest (ROI) parcellation and PET data kinetic modelling. Regional and voxel level [11C]PiB binding was quantified as standardized uptake value ratios (SUVR) calculated for 60 to 90 min post injection using the cerebellar cortex as the reference region. The Aβ PET results have been published earlier in more detail [39]. For this study, a composite neocortical [11C]PiB score was calculated as the volume weighted average of the [11C]PiB region-to-cerebellar cortex SUVRs for the lateral frontal, lateral temporal, and parietal cortices as well as the posterior cingulate, anterior cingulate, and precuneus. This composite [11C]PiB score was used to estimate brain Aβ load and its association with blood biomarkers.
The brain MRI images were also analysed using an automatic cNeuro image analysis tool (Combinostics Oy, Tampere, Finland) to extract detailed data for atrophy scores for different brain regions and volumes of white matter intensities [42,43,44]. Segmentation was done from T1- and T2-weighted MRI, and each voxel was labelled based on which region it belongs to, using multi-atlas segmentation of 133 regions [43]. The structural MRI images were analysed for i) volumes of different brain regions (hippocampus, parahippocampus and entorhinal cortex) and atrophy scores (global cortical atrophy and medial temporal lobe atrophy) as a proxy for neurodegeneration, and iii) volumes of white matter hyperintensities for estimation of existing cerebrovascular pathology. For volumetric analysis, we focused on the hippocampus, parahippocampus, and the entorhinal cortex due to their known association with neurodegeneration related to AD [45]. Global cortical atrophy score (continuous measure, range 0–3) was estimated computationally using voxel-based morphometry for the whole cortex and brain lobes [44]. Medial temporal lobe atrophy score (continuous measure, range 0–4) was estimated computationally from the volumes of the hippocampus and inferior lateral ventricle from the T1-weighted MR images, and an average was calculated from the left and right sides [44]. Volumes of white matter hyperintensities (ml) were segmented from T2-FLAIR images [42]. Voxel -based morphometry analysis was conducted from T1-weighted MRI using SPM12 (http://www.fil.ion.ucl.ac.uk/spm) running on MATLAB R2021b (Math-Works, Natick, MA, USA) to assess group differences in cerebral grey matter at the voxel level.
Statistical analysis
Statistical analyses were performed using JMP Pro 17.0.0 (SAS Institute Inc., Cary, North Carolina, USA). All data following a normal distribution are presented as mean (standard deviation, SD), otherwise as median (interquartile range, IQR). The normality of the data was evaluated visually from the distribution and with the Shapiro-Wilk test. Differences in continuous variables (blood and imaging biomarker levels) between the three APOE groups were tested using linear regression models, adjusting for age and sex. If a significant effect was found, all pairs were compared using the post hoc Tukey-Kramer honest significance test for multiple comparisons. Correlations between the MRI variables and blood biomarker concentrations were evaluated using Spearman’s rank correlation in the whole sample and within the APOE groups.
In addition to group -level differences in biomarker concentrations, we further tested the effect of different pathologies (Aβ, medial temporal atrophy, cerebrovascular pathology) on the blood biomarker concentrations in our cognitively unimpaired sample enriched with APOE ε4 carriers. In all linear regression models, Aβ pathology was estimated by composite neocortical [11C]PiB SUVR, neurodegeneration by the medial temporal lobe atrophy score, and cerebrovascular pathology by total volume of white matter hyperintensities. During this project, two different MRI scanners were used for collecting data; thus, in all models that included MRI -derived variables, the MRI scanner was included as a covariate. First, we performed univariate independent regression models with each of the blood biomarkers (serum NfL, plasma NTA-tau, plasma t-tau, plasma GFAP) as a response variable and each pathology as a predictor (Model 1: Blood biomarker ∼ Pathology marker). Next, we ran the same models adjusting for age, sex and BMI (Model 2: Blood biomarker ∼ Pathology marker + age + sex + BMI). To determine if the different pathologies had an effect simultaneously, we combined all three pathology markers as predictors in the same model (Model 3: Blood biomarker ∼ All pathology markers). In the last model, we again added sex, age, and BMI as covariates to the multivariate model including all pathologies as predictors (Model 4: Blood biomarker ∼ All pathology markers + age + sex + BMI). The normality assumption was evaluated based on the residuals and confirmed visually and calculated using the Shapiro-Wilk test. If the normality assumption was not fulfilled the blood biomarker values were log transformed, after which normality assumption was met. Variance inflation factor values were used to check that the independent variables were not highly correlated with each other. Adjusted R-squared (R2) was used to evaluate how well the models fit the data.
Voxel-wise volumetric differences between the APOE groups were tested using linear regression in statistical parametric mapping (SPM12) to evaluate if structural differences were present also outside the a priori chosen brain regions. Uncorrected p < 0.001 combined with a cluster-level false discovery rate (FDR) correction for multiple comparisons was considered statistically significant in the voxel-based analyses. When significant FDR corrected clusters were found we applied family wise error (FWE) correction with p < 0.05 to see if the results survived the tighter threshold. Voxel wise regression analysis was also done with SPM12 to analyze correlations between the biomarkers and grey matter volumes in the whole study population. All voxel-wise analyses were adjusted for age, sex, MRI scanner, and total intracranial volume.
Results
Participant demographics
Demographic and descriptive data are presented in Table 1. One subject from the APOE ε4ε4 group was excluded from the analysis due to abnormal brain structure. No significant differences in age (p = 0.75), sex (p = 0.91), education (p = 0.37) or BMI (p = 0.86) were observed between the APOE groups. In the whole study population, age correlated positively with serum NfL concentration (Rho = 0.36, p = 0.0051) and global cortical atrophy score (Rho = 0.37, p = 0.0034). On the contrary, age correlated negatively with hippocampal volume (Rho = -0.30, p = 0.018) and total volume of white matter hyperintensities (Rho = -0.27 p = 0.033). Higher BMI correlated with lower serum NfL concentrations (Rho = -0.29, p = 0.024).
Blood biomarkers across APOE groups
Statistically significant differences between the APOE groups were found for serum NfL (p = 0.018), but not for plasma NTA-tau (p = 0.97), plasma t-tau (p = 0.95) or plasma GFAP (p = 0.077) (Fig. 1). Post hoc comparisons between groups showed that serum NfL concentrations were significantly higher in the APOE ε4ε4 (21.4 pg/ml (9.5); mean (SD)) compared with non-carriers (15.5 pg/ml (3.8)) (p = 0.013, Tukey HSD), whereas differences between the APOE ε4ε3 (17.8 pg/ml (7.2)) and APOE ε4ε4 (p = 0.24) or non-carriers (p = 0.37) were not statistically significant.

Serum NfL, plasma NTA-tau and plasma t-tau concentrations in cognitively unimpaired APOE ε4 homozygotes (ε4ε4), heterozygotes (ε4ε3) and noncarriers (ε3ε3). Raw concentrations, median, first and third quartile and range are presented by the box plot, p values are further adjusted for age and sex. P-values below the figures presents overall difference between groups
Structural MRI findings across APOE groups
For imaging markers of neurodegeneration, statistically significant differences among the APOE groups were found only for hippocampal volume (p = 0.041), that was significantly lower in the APOE ε4ε4 (6.71 ml, 0.86; mean, SD) compared with non-carriers (7.2 ml, 0.7, p = 0.029) (Fig. 2). No differences were present between the APOE ε4ε4 and APOE ε4ε3 (7.0 ml, 0.80, p = 0.45) or between the APOE ε4ε3 and non-carriers (p = 0.34). No significant APOE group differences were seen in global cortical atrophy score (p = 0.52), medial temporal lobe atrophy score (p = 0.52), volumes of white matter hyperintensities (p = 0.36), entorhinal volume (p = 0.13) or parahippocampal volume (p = 0.12).

Hippocampal volume, medial temporal and global atrophy scores in cognitively unimpaired APOE ε4 homozygotes (ε4ε4), heterozygotes (ε4ε3) and noncarriers (ε3ε3). Raw concentrations, median, first and third quartile and range are presented by the box plot, p values are further adjusted for age and sex. P-values below the figures presents overall difference between groups
We then further analysed differences in grey matter volume between the APOE groups using voxel-based morphometry. When the significance threshold was set to FWE corrected p < 0.05, lower volume was found in the left hippocampal region of APOE carriers compared with non-carriers (Fig. 3, blue scale). Also, between the ε4ε4 and ε4ε3 carriers, there were similar findings, but also small clusters in the frontal cortex. With a more lenient threshold of FDR corrected p < 0.001, broader reductions in grey matter volumes were found mainly in the hippocampal and parahippocampal regions of APOE ε4ε4 carriers compared with non-carriers (Fig. 3, green scale). Similar findings were also between the ε4ε4 and ε4ε3 carriers, but also in the frontal and right lateral areas.

Significant MRI voxel based morphometry results. (A) ε4ε4 < ε4ε3 (B) ε4ε4 < ε3ε3
Correlations between imaging and blood biomarkers
In the whole study population, higher global cortical atrophy score correlated with higher serum NfL concentrations (Rho = 0.40, p = 0.0017). We then performed additional exploratory analysis stratifying by the APOE group and found that all correlations were significant only in the APOE ε4ε4 group. In the APOE ε4ε4 group, higher global cortical atrophy score correlated with higher serum NfL (Rho = 0.78, p < 0.001) and higher plasma GFAP concentrations (Rho = 0.53, p = 0.024). Higher medial temporal atrophy score correlated with both higher plasma NTA-tau (Rho = 0.61, p = 0.0097) and serum NfL concentrations (Rho = 0.63, p = 0.0049). No significant correlations were seen in the APOE ε4ε3 group or non-carriers. In addition to the ROI based imaging results, we did an exploratory voxel based regression analysis in the whole study population to see if the biomarkers had associations outside the ad hoc chosen brain areas. Here, the significant correlations to smaller grey matter volume were with serum NfL and plasma GFAP (Fig. 4). Serum NfL had negative correlations with grey matter volume in all cortical regions (Fig. 4A). Plasma GFAP had negative correlations with grey matter volume in left insular cortex and right hippocampus (Fig. 4B).

Voxel wise correlations between (A) serum NfL, (B) plasma GFAP and smaller grey matter volume. FDR corrected p < 0.001 in green colour. FWE corrected p < 0.05 in blue colour
Effect of Aβ, medial temporal atrophy, cerebrovascular pathology on blood biomarker concentrations
Next, we tested in the whole cohort, what proportion of the variation in blood biomarker concentrations was explained by Aβ pathology (estimated with [11C]PiB PET), cerebrovascular pathology (estimated with total white matter lesion volume) and medial temporal. Blood biomarkers were set as response variables and pathology markers as predictors in all linear regression models. The blood biomarker values were log transformed, so that the models residuals fulfilled the normal assumption.
In univariate unadjusted (Model 1) and adjusted regression models (Model 2), we first looked at the effect of each pathology separately to a single blood biomarker (serum NfL, plasma NTA-tau, plasma t-tau, plasma GFAP). Cerebrovascular pathology had a positive association with serum NfL concentrations in Model 1 (R2 = 0.18, p = 0. 017, Table 2) and in Model 2, when further adjusting for age, sex, and BMI (R2 = 0.33, p = 0.021, Table 3). Medial temporal atrophy had a similar positive association with serum NfL concentrations in univariate Model 1 (R2 = 0.14, p = 0.025, Table 2) and in adjusted Model 2 (R2 = 0.32, p = 0.049, Table 3).
Medial temporal atrophy was also positively associated with plasma NTA-tau levels both in the unadjusted (R2 = 0.19, p = 0.026, Table 2) and adjusted model (R2 = 0.15, p = 0.022, Table 3).
In addition, medial temporal atrophy had a significant effect on plasma GFAP levels in both the unadjusted (R2 = 0.11, p = 0.022, Table 2) and adjusted model (R2 = 0.15, p = 0.022, Table 3). Aβ pathology was positively associated only with plasma GFAP levels, both alone (R2 = 0.10, p = 0.022, Table 2) and when adjusted for demographic variables (R2 = 0.13, p = 0.022, Table 3).
None of the pathology markers significantly explained variance in plasma t-tau concentrations (Tables 2 and 3).
In the multivariate linear regression models (Model 3 and Model 4, Tables 4 and 5), when we combined all three pathology markers as predictors, the results were aligned with the previously presented models. For serum NfL, both models were statistically significant (R2 = 0.22, p = 0.017 for Model 3, and R2 = 0.32, p < 0.001 for Model 4 including covariates) and in both models, cerebrovascular pathology was the predictor showing a significant effect on the outcome (Model 3, p = 0.0062; Model 4, p = 0.039). In Model 3, medial temporal atrophy also had a significant effect on serum NfL levels (p = 0.042), but this was dampened when further adjusting for age, sex, and BMI (p = 0.076).
For plasma NTA-tau, the Model 3 proved to be significant (R2 = 0.18, p = 0.021, Table 4), but when adjusting for age, sex and BMI the significance diminished (R2 = 0.14, p = 0.083, Table 5) In the Model 3 only medial temporal atrophy contributed significantly to these model (Model 3, p = 0.0063).
For plasma GFAP, both models were significant (R2 = 0.18, p = 0.023 and R2 = 0.21, p = 0.020, Tables 4 and 5). In both models, Aβ pathology and medial temporal atrophy had a similar significant effect on plasma GFAP concentration (Model 3, p = 0.025 and p = 0.026; Model 4, p = 0.013 and p = 0.018 for medial temporal lobe atrophy score, [11C]PiB SUVR, respectively).
Last, in the models explaining plasma t-tau concentrations neither Model 3 or Model 4 were not significant (R2 = 0.081, p = 0.14 and R2 = 0.14, p = 0.082, Tables 4 and 5).
Discussion
In this study, we examined early differences in blood biomarker concentrations, cortical volumes, white matter hyperintensities and grey matter atrophy scores, which serve as biomarkers of neurodegeneration, in a cohort of cognitively unimpaired participants with different numbers of APOE ε4 alleles, and thereby varying risk for sporadic AD. Additionally, we investigated how the different pathologies (cerebrovascular pathology, medial temporal atrophy and Aβ pathology) explained the concentrations of blood biomarkers, using the entire cognitively unimpaired sample enriched with APOE ε4 carriers. Our main findings revealed that serum NfL concentrations were significantly increased, and hippocampal volume was significantly decreased in cognitively unimpaired APOE ε4 homozygotes compared with non-carriers. Second, in the whole sample, we observed different associations of cerebrovascular pathology, medial temporal atrophy and Aβ deposition with the blood biomarkers used to estimate neurodegeneration. Specifically, serum NfL levels were associated with cerebrovascular pathology and medial temporal atrophy, whereas plasma NTA-tau was associated only with medial temporal atrophy. Lastly, plasma GFAP was associated with medial temporal atrophy and Aβ pathology, while plasma t-tau levels could not be explained by any of the assessed pathologies.
The hippocampus is one of the first brain regions where structural changes become visible in the AD continuum [46]. APOE ε4 allele has been shown to affect gray matter volume in the brain in a dose dependent manner, particularly reducing volume in the posterior hippocampus and other regions associated with memory and executive functions [10]. In our study we found similar volumetric differences between APOE4 gene dose groups, in the hippocampus. While ROI analysis revealed a statistically significant difference only between the APOE ε4ε4 group and non-carriers in hippocampal volumes, a visible linear trend related to APOE ε4 allele burden existed across the groups. This finding was further supported by the VBM analysis, which showed a significant difference in hippocampal volume between the APOE ε4ε4 group and non-carriers even when using a strict FWE-corrected threshold, and exhibited a difference between the APOE ε4ε4 and ε4ε3 groups with FDR-corrected threshold. Our findings agree with previous results regarding the loss of hippocampal volume in healthy APOE ε4 carriers [10, 47]. Furthermore, in individuals with mild cognitive impairment (MCI) or AD, hippocampal volume decreases have been observed in relation to APOE ε4 status [48,49,50].
NfL concentrations measured from blood are elevated in AD and other neurodegenerative diseases [16, 17]. Here we found serum NfL concentrations to be elevated already in cognitively unimpaired APOE ε4ε4 carriers compared with non-carriers. This finding is consistent with a recent study that reported differences between cognitively unimpaired non-carriers and both the APOE ε4ε4 and ε4ε3 groups [51]. Previously significant differences in CSF NfL concentration between APOE ε4 carriers and non-carriers from prodromal AD patients has been reported [52]. In contrast to our findings, Mielke (2019) did not observe any effect of APOE ε4 on plasma or CSF NfL levels in cognitively unimpaired subjects [53]. However, the APOE ε4 status was reported only as positive or negative, categorizing subjects as carriers if they had at least one APOE ε4 allele, whereas we evaluated homozygotic and heterozygotic APOE4 carriers separately. Additionally, most subjects were non-carriers, whereas in our study, the different APOE groups were well balanced. If we use the binarized, APOE ε4 positive and negative categorisation, the difference between APOE ε4 carriers and non-carriers in our sample is still statistically significant (p = 0.035).
Higher serum NfL correlated with cortical atrophy and voxel-wise regression analysis were in line with this finding. When we examined this association further with multiple regression models, we found that higher serum NfL concentration was associated with cerebrovascular pathology, estimated from the volume of white matter hyperintensities, and neurodegeneration in the medial temporal lobe. The effect of cerebrovascular pathology seemed to be stronger as it was the only explaining predictor in the model with all pathologies and demographic covariates. This concurs with a study in an elderly population with high prevalence of cerebral small vessel disease burden where plasma NfL was associated with both neurodegenerative and vascular pathologies [54]. White matter hyperintensities are considered markers of cerebral small vessel disease, indicating increased water content and mobility, demyelination, and axonal loss [55], while NfL is a nonspecific marker of axonal damage [16, 56]. Previous studies have demonstrated an increase in CSF NfL levels with an increased number of white matter hyperintensities [57,58,59]. Similar associations have been observed with plasma NfL in neuropathologically confirmed AD [60], AD dementia [61] and serum NfL in neurologically unaffected individuals [18]. These studies have also shown a significant influence of age on blood NfL levels, whereas in our study, the association of white matter hyperintensities with NfL was independent of age. NfL levels are known to rise with age [18, 19], but compared with the previous studies, our population was in a somewhat narrow age range (67 (4.7) years) and was enriched with APOE ε4ε4 carriers. APOE ε4 is associated with increased white matter hyperintensities [13, 62] so in this population, the effect of white matter hyperintensities on serum NfL concentration appeared to be more significant than age. Previous findings suggest that plasma NfL increases in response to amyloid-related neuronal injury in preclinical stages of Alzheimer’s disease, but is related to tau-mediated neurodegeneration in symptomatic patients [22]. However, in our analyses, Aβ-PET did not show any association with serum NfL concentrations, and the biomarker levels were primarily explained by the extent of cerebrovascular pathology. In addition to the association between white matter hyperintensities and NfL, we also found that, neurodegeneration, as estimated here by the medial temporal lobe atrophy score, was associated with higher serum NfL concentrations. In previous studies, plasma NfL has been associated with decreased volumes in the temporal cortex and hippocampal volume in subjects without dementia [63], and CSF NfL to decrease in whole brain and hippocampus volumes [64]. However, this association was weakened when adjusting for simultaneous effects of different pathologies and demographic variables.
NTA-tau is a novel biomarker of soluble, N-terminal tau fragments, recently suggested to be more related to tau tangle pathology (as estimated by tau PET), than Aβ-PET or neurodegeneration [32]. Here, we demonstrated an association between neurodegeneration and NTA-tau in a cognitively normal sample, enriched with “at-risk” individuals [40]. In CSF, NTA-tau has previously been found to be increased in CSF Aβ-positive individuals with MCI due to AD and AD. Similarly, plasma NTA-tau concentrations have been shown to be elevated in AD dementia compared with controls in a clinical pilot cohort [40]. In a study evaluating plasma NTA-tau levels along the AD continuum, it was observed that NTA-tau exhibited a stronger association with tau-PET compared with Aβ -PET. Additionally, plasma NTA-tau predicted tau-PET accumulation in middle to late Braak regions and neurodegeneration in the medial temporal lobe. These findings suggest that NTA-tau could serve as a tool for detecting and monitoring pathological changes associated with middle to late stages of AD [32]. Interestingly, in our study, we found that NTA-tau is associated with medial temporal lobe atrophy already in cognitively unimpaired subjects. This finding may be explained by the fact that our cohort is highly enriched with homozygous APOE ε4 carriers. APOE ε4 is known to be linked to elevated tau accumulation in the medial temporal lobe, and tau itself is an independent driver of neurodegeneration [65,66,67,68,69,70].
In our study, plasma total-tau (t-tau) did not exhibit any correlation with the neuroimaging biomarkers, unlike NTA-tau. Though plasma t-tau was among the earliest blood-based immunoassays used to detect soluble tau in AD, its diagnostic value is limited due to significant overlap in its levels among different diagnostic categories [26, 27]. While CSF t-tau does reflect the intensity of neuronal and axonal degeneration and damage in the brain [71], blood-based t-tau may also arise from peripheral sources, which diminishes its specificity for neurological conditions [72, 73]. Additionally, recent research suggests that only approximately 20% of plasma t-tau originates from the CNS, further challenging its effectiveness as a marker for brain pathology [74].
Plasma GFAP is a protein constituent of astrocyte intermediate filaments and elevated GFAP concentrations are thought to reflect astrocyte activation and reactive gliosis linked with several CNS disorders e.g. neurotrauma, ischemic stroke or neurodegenerative diseases [75]. In our study, we found that GFAP was the only blood biomarker associated with the level of Aβ pathology estimated by PET. This is in line with previous findings where higher plasma GFAP concentrations have been associated with higher Aβ load determined using PET imaging [37, 38, 76, 77] and in genetic forms of AD [78, 79]. Astrocytes are activated and express GFAP in the context of neurodegeneration [33] and in the presence of Aβ [34]. In AD, higher plasma GFAP levels have been associated with the incidence and faster progression of AD [35,36,37,38]. Plasma GFAP is also able to differentiate Aβ-positive and Aβ-negative cognitively unimpaired individuals [35, 37, 38, 76]. Here, also medial temporal atrophy was associated with higher plasma GFAP concentrations. Also, with the voxel-wise analysis, we saw association of higher plasma GFAP to reduced grey matter in the hippocampus. This is likely explained by activation of the astroglia by the subtle neurodegeneration present already in the medial temporal lobe of APOE ε4 carriers. Another explanation could be the type of astrocytes located in the medial temporal lobe as high GFAP expressing astrocytes have been found in the murine hippocampus [80, 81].
The strength of this study is in conducting a comparison of the impact of various pathologies on multiple blood biomarkers used to study neurodegenerative processes. Furthermore, it employs a meticulously selected population with a balanced distribution of subjects across the three APOE-genotypes. This approach enables a precise detection of the effects attributed to the APOE ε4 allele.
There are also limitations in this study. Firstly, it is important to note that the associations between the pathologies and blood biomarkers in the general population may differ from those observed in this study, as the sample used was heavily enriched with APOE ε4 carriers, a third of the sample comprising of individuals who were rare homozygotes. Additionally, the groups were relatively small for between group blood biomarkers comparisons, as the study’s initial power calculations were primarily focused on PET imaging biomarkers, and the small sample size might also limit the generalizability of our findings. Furthermore, the study lacked tau-PET and CSF tau measurements, which prevented the evaluation of the effect of tau-pathology on the blood biomarkers and would have greatly enriched the study.
Conclusions
We investigated the differences associated with the APOE ε4 allele in both imaging and blood biomarkers of neurodegenerative disorders. Our findings revealed that individuals who were APOE ε4 homozygotes exhibited significant grey matter loss in the hippocampus, as well as elevated levels of serum NfL compared with non-carriers. Moreover, within our whole cohort, we demonstrated that Aβ deposition, cerebrovascular pathology, and medial temporal atrophy have distinct influences on the concentrations of the examined blood biomarkers. This study significantly contributes to our understanding of the relationship between the APOE ε4 gene, blood biomarkers, and imaging-based pathology markers in the early stages of the AD pathologic continuum. These findings have the potential to facilitate the development of novel diagnostic and prognostic tools for neurodegenerative diseases, particularly in the preclinical phase.
Data availability
The data are available from the corresponding author on reasonable request.
Abbreviations
- Aβ:
-
Beta-amyloid
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- APOE:
-
Apolipoprotein E gene
- BMI:
-
Body mass index
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- FTD:
-
Frontotemporal dementia
- GFAP:
-
Glial fibrillary acidic protein
- NfL:
-
Neurofilament light chain
- NTA-tau:
-
N-terminal tau fragments
- MRI:
-
Magnetic resonance imaging
- MTA:
-
Medial temporal lobe atrophy
- PET:
-
Positron emission tomography
- ROI:
-
Region of interest
- Simoa:
-
Single molecule array
- SUVR:
-
Standardized uptake value ratio
- t-tau:
-
total tau
- VBM:
-
Volume based morphometry
- WMH:
-
White matter hyperintensities
References
Gustavsson A, Norton N, Fast T, Frölich L, Georges J, Holzapfel D, et al. Global estimates on the number of persons across the Alzheimer’s disease continuum. Alzheimer’s Dement. 2023;19(2):658–70.
CR J. DA B, K B, MC C, B D, SB H, NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement J Alzheimer’s Assoc. 2018;14(4).
Bekris LM, Yu C-E, Bird TD, Tsuang DW. Review article: Genetics of Alzheimer Disease. J Geriatr Psychiatr Neurol. 2010;23(4):213–27.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene Dose of Apolipoprotein E Type 4 Allele and the Risk of Alzheimer’s Disease in Late Onset Families. Volume 261. Science (American Association for the Advancement of Science; 1993. pp. 921–3. 5123.
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. JAMA: J Am Med Association. 1998;279:581–2.
Liu C-C, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Reviews Neurol. 2013;9(2):106–18.
Ossenkoppele R, Jansen WJ, Rabinovici GD, Knol DL, Van Der Flier WM, Van Berckel BNM, et al. Prevalence of amyloid PET positivity in dementia syndromes. JAMA. 2015;313(19):1939.
Yin Y, Wang Z. ApoE and neurodegenerative diseases in Aging. Advances in Experimental Medicine and Biology. Springer Singapore; 2018. pp. 77–92.
Giau VV, Bagyinszky E, An SSA, Kim SY. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr Dis Treat. 2015;11(default):1723–37.
Cacciaglia R, Molinuevo JL, Falcón C, Brugulat-Serrat A, Sánchez-Benavides G, Gramunt N, et al. Effects of APOE-ε4 allele load on brain morphology in a cohort of middle-aged healthy individuals with enriched genetic risk for Alzheimer’s disease. Alzheimer’s Dement. 2018;14(7):902–12.
Mahley RW, Apolipoprotein E. From cardiovascular disease to neurodegenerative disorders. J Mol Med. 2016;94(7):739–46.
Marais AD. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology. 2019;51(2):165–76.
Schilling S, DeStefano AL, Sachdev PS, Choi SH, Mather KA, DeCarli CD, et al. APOE genotype and MRI markers of cerebrovascular disease: systematic review and meta-analysis. Neurology. 2013;81(3):292–300.
Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, et al. Alzheimer’s disease. Lancet. 2021;397(10284):1577–90.
Zetterberg H, Schott JM. Blood biomarkers for Alzheimer’s disease and related disorders. Acta Neurol Scand. 2022;146(1):51–5.
Gaetani L, Blennow K, Calabresi P, Di Filippo M, Parnetti L, Zetterberg H. Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry. 2019;90(8):870–81.
Ashton NJ, Janelidze S, Al Khleifat A, Leuzy A, van der Ende EL, Karikari TK, et al. A multicentre validation study of the diagnostic value of plasma neurofilament light. Nat Commun. 2021;12(1):3400.
Khalil M, Pirpamer L, Hofer E, Voortman MM, Barro C, Leppert D et al. Serum neurofilament light levels in normal aging and their association with morphologic brain changes. Nat Commun. 2020;11(1).
Vermunt L, Otte M, Verberk IMW, Killestein J, Lemstra AW, Van Der Flier WM, et al. Age- and disease‐specific reference values for neurofilament light presented in an online interactive support interface. Ann Clin Transl Neurol. 2022;9(11):1832–7.
Manouchehrinia A, Piehl F, Hillert J, Kuhle J, Alfredsson L, Olsson T, et al. Confounding effect of blood volume and body mass index on blood neurofilament light chain levels. Ann Clin Transl Neurol. 2020;7(1):139–43.
Duering M, Konieczny MJ, Tiedt S, Baykara E, Tuladhar AM, Leijsen EV, et al. Serum neurofilament light chain levels are related to small Vessel Disease Burden. J Stroke. 2018;20(2):228–38.
Benedet AL, Leuzy A, Pascoal TA, Ashton NJ, Mathotaarachchi S, Savard M, et al. Stage-specific links between plasma neurofilament light and imaging biomarkers of Alzheimer’s disease. Brain (London England: 1878). 2020;143(12):3793–804.
Dage JL, Wennberg AMV, Airey DC, Hagen CE, Knopman DS, Machulda MM, et al. Levels of tau protein in plasma are associated with neurodegeneration and cognitive function in a population-based elderly cohort. Alzheimer’s Dement. 2016;12(12):1226–34.
Xiao Z, Wu X, Wu W, Yi J, Liang X, Ding S et al. Plasma biomarker profiles and the correlation with cognitive function across the clinical spectrum of Alzheimer’s disease. Alzheimers Res Ther. 2021;13(1).
Jong-Chan P, Sun-Ho H, Dahyun Y, Min Soo B, Lee JH, Sukjin J, et al. Plasma tau/amyloid-β1–42 ratio predicts brain tau deposition and neurodegeneration in Alzheimer’s disease. Brain (London England: 1878). 2019;142(5):e19–e.
Mattsson N, Zetterberg H, Janelidze S, Insel PS, Andreasson U, Stomrud E, et al. Plasma tau in Alzheimer disease. Neurology. 2016;87(17):1827–35.
Zetterberg H, Wilson D, Andreasson U, Minthon L, Blennow K, Randall J, et al. Plasma tau levels in Alzheimer’s disease. Alzheimers Res Ther. 2013;5(2):9.
Simrén J, Leuzy A, Karikari TK, Hye A, Benedet AL, Lantero-Rodriguez J, et al. The diagnostic and prognostic capabilities of plasma biomarkers in Alzheimer’s disease. Alzheimer’s Dement. 2021;17(7):1145–56.
Snellman A, Lantero-Rodriguez J, Emeršič A, Vrillon A, Karikari TK, Ashton NJ, et al. N-terminal and mid-region tau fragments as fluid biomarkers in neurological diseases. Brain. 2022;145(8):2834–48.
Gonzalez-Ortiz F, Turton M, Kac PR, Smirnov D, Premi E, Ghidoni R et al. Brain-derived tau: a novel blood-based biomarker for Alzheimer’s disease-type neurodegeneration. Brain (London, England: 1878). 2022.
Mengel D, Janelidze S, Glynn RJ, Liu W, Hansson O, Walsh DM. Plasma NT1 tau is a specific and early marker of Alzheimer’s Disease. Ann Neurol. 2020;88(5):878–92.
Lantero-Rodriguez J, Tissot C, Snellman A, Servaes S, Benedet AL, Rahmouni N, et al. Plasma and CSF concentrations of N‐terminal tau fragments associate with in vivo neurofibrillary tangle burden. Alzheimer’s & Dementia; 2023.
Heneka MT, Rodríguez JJ, Verkhratsky A. Neuroglia in neurodegeneration. Brain Res Rev. 2009;63(1):189–211.
Medeiros R, LaFerla FM, Astrocytes. Conductors of the Alzheimer disease neuroinflammatory symphony. Exp Neurol. 2013;239:133–8.
S PC, VL PESKG. V, PR A, Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer’s disease. Translational Psychiatry. 2021;11(1).
C C, K B SJJHHZ. N M-C, Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimers Res Ther. 2021;13(1).
JB P, N M-C SJRS, CE SP. T, Plasma GFAP is an early marker of amyloid-β but not tau pathology in Alzheimer’s disease. Brain. 2021;144(11).
Shir D, Graff-Radford J, Hofrenning EI, Lesnick TG, Przybelski SA, Lowe VJ, et al. Association of plasma glial fibrillary acidic protein (GFAP) with neuroimaging of Alzheimer’s disease and vascular pathology. Volume 14. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring.; 2022. 1.
Snellman A, Ekblad LL, Tuisku J, Koivumäki M, Ashton NJ, Lantero-Rodriguez J et al. APOE ε4 gene dose effect on imaging and blood biomarkers of neuroinflammation and beta-amyloid in cognitively unimpaired elderly. Alzheimers Res Ther. 2023;15(1).
Snellman A, Lantero-Rodriguez J, Emeršič A, Vrillon A, Karikari TK, Ashton NJ, et al. N-terminal and mid-region tau fragments as fluid biomarkers in neurological diseases. Brain (London England: 1878). 2022;145(8):2834–48.
Karjalainen T, Tuisku J, Santavirta S, Kantonen T, Bucci M, Tuominen L, et al. Magia: Robust Automated Image Processing and Kinetic modeling toolbox for PET neuroinformatics. Front Neuroinform. 2020;14:3.
Koikkalainen J, Rhodius-Meester H, Tolonen A, Barkhof F, Tijms B, Lemstra AW, et al. Differential diagnosis of neurodegenerative diseases using structural MRI data. Neuroimage Clin. 2016;11:435–49.
Lötjönen J, Wolz R, Koikkalainen J, Julkunen V, Thurfjell L, Lundqvist R, et al. Fast and robust extraction of hippocampus from MR images for diagnostics of Alzheimer’s disease. NeuroImage. 2011;56(1):185–96.
Koikkalainen JR, Rhodius-Meester HFM, Frederiksen KS, Bruun M, Hasselbalch SG, Baroni M, et al. Automatically computed rating scales from MRI for patients with cognitive disorders. Eur Radiol. 2019;29(9):4937–47.
Chandra A, Dervenoulas G, Politis M. Magnetic resonance imaging in Alzheimer’s disease and mild cognitive impairment. J Neurol. 2019;266(6):1293–302.
Frisoni GB, Fox NC, Jack CR, Scheltens P, Thompson PM. The clinical use of structural MRI in Alzheimer disease. Nat Reviews Neurol. 2010;6(2):67–77.
Veldsman M, Nobis L, Alfaro-Almagro F, Manohar S, Husain M. The human hippocampus and its subfield volumes across age, sex and APOE e4 status. Brain Commun. 2021;3(1):fcaa219–fcaa.
Geroldi C, Pihlajamäki M, Laakso MP, DeCarli C, Beltramello A, Bianchetti A, et al. APOE-epsilon4 is associated with less frontal and more medial temporal lobe atrophy in AD. Neurology. 1999;53(8):1825–32.
Hostage CA, Roy Choudhury K, Doraiswamy PM, Petrella JR. Dissecting the Gene Dose-Effects of the APOE ε4 and ε2 alleles on hippocampal volumes in aging and Alzheimer’s Disease. PLoS ONE. 2013;8(2):e54483.
Novellino F, López ME, Vaccaro MG, Miguel Y, Delgado ML, Maestu F. Association between Hippocampus, Thalamus, and Caudate in mild cognitive impairment APOEε4 carriers: a structural Covariance MRI Study. Front Neurol. 2019;10:1303.
Malek-Ahmadi M, Su Y, Ghisays V, Luo J, Devadas V, Chen Y et al. Plasma NfL is associated with the APOE ε4 allele, brain imaging measurements of neurodegeneration, and lower recall memory scores in cognitively unimpaired late-middle-aged and older adults. Alzheimers Res Ther. 2023;15(1).
Mattsson N, Eriksson O, Lindberg O, Schöll M, Lampinen B, Nilsson M, et al. Effects of APOE ε4 on neuroimaging, cerebrospinal fluid biomarkers, and cognition in prodromal Alzheimer’s disease. Neurobiol Aging. 2018;71:81–90.
Mielke MM, Syrjanen JA, Blennow K, Zetterberg H, Vemuri P, Skoog I, et al. Plasma and CSF neurofilament light. Neurology. 2019;93(3):e252–60.
Chong JR, Hilal S, Ashton NJ, Karikari TK, Reilhac A, Vrooman H et al. Brain atrophy and white matter hyperintensities are independently associated with plasma neurofilament light chain in an Asian cohort of cognitively impaired patients with concomitant cerebral small vessel disease. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring. 2023;15(1).
Wardlaw JM, Valdés Hernández MC, Muñoz-Maniega S. What are white Matter Hyperintensities made of? J Am Heart Association. 2015;4(6):e001140.
Bridel C, Van Wieringen WN, Zetterberg H, Tijms BM, Teunissen CE, Alvarez-Cermeño JC, et al. Diagnostic value of Cerebrospinal Fluid Neurofilament light protein in Neurology. JAMA Neurol. 2019;76(9):1035.
Meeker KL, Butt OH, Gordon BA, Fagan AM, Schindler SE, Morris JC, et al. Cerebrospinal fluid neurofilament light chain is a marker of aging and white matter damage. Neurobiol Dis. 2022;166:105662.
Jonsson M, Zetterberg H, Van Straaten E, Lind K, Syversen S, Edman Å, et al. Cerebrospinal fluid biomarkers of white matter lesions - cross-sectional results from the LADIS study. Eur J Neurol. 2010;17(3):377–82.
Osborn KE, Liu DD, Samuels LR, Moore EE, Cambronero FE, Acosta LMY, et al. Cerebrospinal fluid beta-amyloid(42) and neurofilament light relate to white matter hyperintensities. Neurobiol Aging. 2018;68:18–25.
Ashton NJ, Leuzy A, Lim YM, Troakes C, Hortobágyi T, Höglund K et al. Increased plasma neurofilament light chain concentration correlates with severity of post-mortem neurofibrillary tangle pathology and neurodegeneration. Acta Neuropathol Commun. 2019;7(1).
Walsh P, Sudre CH, Fiford CM, Ryan NS, Lashley T, Frost C, et al. The age-dependent associations of white matter hyperintensities and neurofilament light in early- and late-stage Alzheimer’s disease. Neurobiol Aging. 2021;97:10–7.
Sudre CH, Cardoso MJ, Frost C, Barnes J, Barkhof F, Fox N, et al. APOE ε4 status is associated with white matter hyperintensities volume accumulation rate independent of AD diagnosis. Neurobiol Aging. 2017;53:67–75.
Marks JD, Syrjanen JA, Graff-Radford J, Petersen RC, Machulda MM, Campbell MR et al. Comparison of plasma neurofilament light and total tau as neurodegeneration markers: associations with cognitive and neuroimaging outcomes. Alzheimers Res Ther. 2021;13(1).
Zetterberg H, Skillbäck T, Mattsson N, Trojanowski JQ, Portelius E, Shaw LM, et al. Association of Cerebrospinal Fluid Neurofilament Light Concentration with Alzheimer Disease Progression. JAMA Neurol. 2016;73(1):60.
Baek MS, Cho H, Lee HS, Lee JH, Ryu YH, Lyoo CH. Effect of APOE ε4 genotype on amyloid-β and tau accumulation in Alzheimer’s disease. Alzheimers Res Ther. 2020;12(1).
Therriault J, Benedet AL, Pascoal TA, Mathotaarachchi S, Chamoun M, Savard M, et al. Association of Apolipoprotein E ε4 with medial temporal tau Independent of Amyloid-β. JAMA Neurol. 2020;77(4):470.
La Joie R, Visani AV, Lesman-Segev OH, Baker SL, Edwards L, Iaccarino L, et al. Association of APOE4 and clinical variability in Alzheimer Disease with the pattern of tau- and Amyloid-PET. Neurology. 2021;96(5):E650–61.
Ballatore C, Lee VM-Y, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8(9):663–72.
Iqbal K, Liu F, Gong C-X, Alonso ADC, Grundke-Iqbal I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009;118(1):53–69.
Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegeneration. 2009;4(1):13.
Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Reviews Neurol. 2010;6(3):131–44.
Dugger BN, Whiteside CM, Maarouf CL, Walker DG, Beach TG, Sue LI, et al. The Presence of Select Tau species in Human Peripheral tissues and their relation to Alzheimer’s Disease. J Alzheimers Dis. 2016;51(2):345–56.
Fischer I, Baas PW. Resurrecting the mysteries of Big Tau. Trends Neurosciences (Regular ed). 2020;43(7):493–504.
Barthelemy NR, Horie K, Sato C, Bateman RJ. Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease. J Exp Med. 2020;217(11).
Hol EM, Pekny M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr Opin Cell Biol. 2015;32:121–30.
Benedet AL, Milà-Alomà M, Vrillon A, Ashton NJ, Pascoal TA, Lussier F, et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer Disease Continuum. JAMA Neurol. 2021;78(12):1471.
De Bastiani MA, Bellaver B, Brum WS, Souza DG, Ferreira PCL, Rocha AS, et al. Hippocampal GFAP-positive astrocyte responses to amyloid and tau pathologies. Brain Behav Immun. 2023;110:175–84.
Montoliu-Gaya L, Alcolea D, Ashton NJ, Pegueroles J, Levin J, Bosch B, et al. Plasma and cerebrospinal fluid glial fibrillary acidic protein levels in adults with Down syndrome: a longitudinal cohort study. EBioMedicine. 2023;90:104547.
O’Connor A, Abel E, Benedet AL, Poole T, Ashton N, Weston PSJ, et al. Plasma GFAP in presymptomatic and symptomatic familial Alzheimer’s disease: a longitudinal cohort study. J Neurol Neurosurg Psychiatry. 2023;94(1):90–2.
Batiuk MY, Martirosyan A, Wahis J, de Vin F, Marneffe C, Kusserow C, et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat Commun. 2020;11(1):1220.
Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proceedings of the National Academy of Sciences. 2018;115(8):E1896-E905.
Acknowledgements
The participants of the study are warmly acknowledged for their commitment to the study. The authors would also like to acknowledge the staff of Turku PET Centre data collection for this study.
Funding
This study was funded by the Academy of Finland (#310962 and #341059) and the Finnish Governmental Research Funding (ERVA) for Turku University Hospital. AES was further supported by the Emil Aaltonen Foundation, the Paulo Foundation, and the Orion Research Foundation sr. LLE was supported by the Emil Aaltonen Foundation and the Juho Vainio foundation. JR has received funding from the Academy of Finland (#310962), Sigrid Juselius Foundation. HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2022 − 01018 and #2019–02397), the European Union’s Horizon Europe research and innovation programme under grant agreement No 101053962, Swedish State Support for Clinical Research (#ALFGBG-71320), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809–2016862), the AD Strategic Fund and the Alzheimer’s Association (#ADSF-21-831376-C, #ADSF-21-831381-C, and #ADSF-21-831377-C), the Bluefield Project, the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2022-0270), the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 860197 (MIRIADE), the European Union Joint Programme – Neurodegenerative Disease Research (JPND2021-00694), the National Institute for Health and Care Research University College London Hospitals Biomedical Research Centre, and the UK Dementia Research Institute at UCL (UKDRI-1003). KB is supported by the Swedish Research Council (#2017 − 00915 and #2022 − 00732), the Swedish Alzheimer Foundation (#AF-930351, #AF-939721 and #AF-968270), Hjärnfonden, Sweden (#FO2017-0243 and #ALZ2022-0006), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986 and #ALFGBG-965240), the European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236), the Alzheimer’s Association 2021 Zenith Award (ZEN-21-848495), and the Alzheimer’s Association 2022–2025 Grant (SG-23-1038904 QC).
Author information
Authors and Affiliations
Contributions
M.K., J.O.R and A.S. designed the study concept. M.K. drafted the manuscript. A.S., L.L.E., M.K., JL-R, J.L., S.H., J.L., and R.P. contributed to data collection. M.K. and A.S. analyzed data. M.K., J.O.R, A.S., L.L.E, N.J.A., T.K.K., J.L., H.Z., and K.B., contributed to study design and interpretation of the data. J.O.R., A.S., H.Z. and K.B. supervised the study. All authors read and critically revised the manuscript for its content and approved the final submitted version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethical Committee of the Hospital District of Southwest Finland. All participants signed a written informed consent.
Consent for publication
Not applicable.
Competing interests
HZ has served at scientific advisory boards and/or as a consultant for Abbvie, Acumen, Alector, Alzinova, ALZPath, Annexon, Apellis, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Novo Nordisk, Optoceutics, Passage Bio, Pinteon Therapeutics, Prothena, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). KB has served as a consultant and at advisory boards for Acumen, ALZPath, BioArctic, Biogen, Eisai, Lilly, Moleac Pte. Ltd, Novartis, Ono Pharma, Prothena, Roche Diagnostics, and Siemens Healthineers; has served at data monitoring committees for Julius Clinical and Novartis; has given lectures, produced educational materials and participated in educational programs for AC Immune, Biogen, Celdara Medical, Eisai and Roche Diagnostics; and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program, outside the work presented in this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Koivumäki, M., Ekblad, L., Lantero-Rodriguez, J. et al. Blood biomarkers of neurodegeneration associate differently with amyloid deposition, medial temporal atrophy, and cerebrovascular changes in APOE ε4-enriched cognitively unimpaired elderly. Alz Res Therapy 16, 112 (2024). https://doi.org/10.1186/s13195-024-01477-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-024-01477-w