
Abstract
Marked improvements in survival and health outcome for people infected with HIV have occurred since the advent of combination antiretroviral therapy over a decade ago. Yet HIV-associated neurocognitive disorders continue to occur with an alarming prevalence. This may reflect the fact that infected people are now living longer with chronic infection. There is mounting evidence that HIV exacerbates age-associated cognitive decline. Many middle-aged HIV-infected people are experiencing cognitive decline similar that to that found among much older adults. An increased prevalence of vascular and metabolic comorbidities has also been observed and is greatest among older adults with HIV. Premature age-associated neurocognitive decline appears to be related to structural and functional brain changes on neuroimaging, and of particular concern is the fact that pathology indicative of neurodegenerative disease has been shown to occur in the brains of HIV-infected people. Yet notable differences also exist between the clinical presentation and brain disturbances occurring with HIV and those occurring in neurodegenerative conditions such as Alzheimer’s disease. HIV interacts with the aging brain to affect neurological structure and function. However, whether this interaction directly affects neurodegenerative processes, accelerates normal cognitive aging, or contributes to a worsening of other comorbidities that affect the brain in older adults remains an open question. Evidence for and against each of these possibilities is reviewed.
Similar content being viewed by others


Introduction
HIV continues to be a major public health problem [1]. During the early years of the HIV epidemic, the cognitive and functional consequences of HIV were devastating for patients and their families [2]. HIV-associated encephalopathy and dementia were among the most common diagnoses in people with AIDS at the time of death [3,4].
The proportion of HIV-infected people who are older than 45 years of age is approaching 50% in the US and other developed nations and this is due in large part to the effectiveness of antiretroviral therapies [5]. HIV-infected adults over age 55 comprise the fastest-growing age group in the HIV-positive population [6], and advanced age at the time of seroconversion increases the risk for neurocognitive impairment [7]. These epidemiological trends point to the potential significance of the effects of HIV on the aging brain.
Neurocognitive manifestations
Prior to the availability of antiretroviral drugs, dementia occurred in over 20% of HIV-infected people [4]. The term AIDS dementia complex (ADC) was coined as a diagnosis of severe decline secondary to HIV, typically involving areas of cognitive, motor, and behavioral function [3]. Patients with severe ADC usually experienced the greatest impairments in attention, working memory, and executive functions, along with fine motor and information processing speed [8,9]. Primary amnestic disturbances did not typically occur, and language, semantics, comprehension, visual-spatial processing, and other sensory and perceptual functions were usually preserved. Although brain disturbances due to opportunistic infections acquired during periods of severe immunosuppression were common [10], ADC was shown to be directly related to HIV infection, predominantly involving macrophages, in the absence of opportunistic infection [11].
In recent years, diagnostic classification of HIV-associated neurocognitive disorder (HAND) was developed as an alternative to ADC staging. HAND encompasses a range of cognitive impairment from mild cognitive difficulties with no functional impairment (asymptomatic neurocognitive impairment, or ANI) to cognitive difficulties with mild functional impairment (mild neurocognitive disorder, or MND) to dementia with significant functional impairment (HIV-associated dementia, or HAD). The advent of combination antiretroviral therapy (cART) in the late 1990s led to reductions in HIV-associated mortality and morbidity [5] and a precipitous decline in incidence of dementia [12]. Overall, cART use led to improved cognitive functioning [13] and reduced neurological damage [14]. Yet HAND continues to occur in 30% to 50% of infected people [8,15-17]. In the cART era, as before, cognitive and motor slowing are major elements of HAND, along with impairments of attention, working memory, and executive functioning [17]. Learning efficiency is reduced, along with memory retrieval, although primary amnestic disturbances are still rare. Though less severe than dementia, ANI and MND affect occupational and psychosocial functioning, quality of life, and health outcomes [9,18,19].
Age-associated cognitive decline in HIV
For several reasons, the effect of HIV on the aging brain has become the subject of much greater concern over the past decade. First, HIV has become a chronic illness, with infected people now having nearly normal life expectancy [20]. Second, there has been a significant increase in the number of older adults living with HIV. Third, although cART has been very effective in reducing viral replication and AIDS and restoring immunological function, HAND remains prevalent. Finally, there is mounting evidence that HIV and aging may interact to adversely affect the brain and neurocognitive functions.
Advanced age is among demographic factors associated with reduced neurocognitive performance and susceptibility to HAND in HIV-infected people [21-24], as greater neurocognitive impairment exists among older HIV-infected adults relative to normative data and compared with younger infected individuals. Although greater cognitive and neurological deficits in older people with HIV may result from independent additive effects of the pathophysiological mechanisms of aging and HIV [25,26], longitudinal studies show significant interaction effects of HIV and age [27,28], suggesting that the mechanisms are synergistic. For example, Seider and colleagues [27] showed that older people with HIV showed significant memory decline in 1 year, but no decline was seen in younger people with HIV or in seronegative controls regardless of age (Figure 1). These data indicate that HIV is associated with accelerated cognitive aging such that people with HIV in their 50s and 60s are functioning cognitively more like people typically do in their 70s and 80s. It is interesting to note that problems with learning and memory are reported to a greater extent in the cART era [15,16,29-31], indicating a change in the typical presentation of HAND in older adults with HIV. These changes as well as neuroimaging and neuropathological findings described below raise the question of whether typical age-related neurodegenerative diseases, particularly Alzheimer’s disease (AD), are affecting the development of HAND. In the sections that follow, evidence for and against the idea that AD is contributing to HAND will be discussed, as will research findings that address some of the mechanisms underlying HAND and how they may escalate as infected people reach advanced age.

Change in delayed recall performance on the Hopkins Verbal Learning Test Revised (HVLT-R) as a function of age for HIV + and HIV − groups. The HIV+ group showed performance declines with increasing age, whereas the HIV− group had either stable or slight improvements in recall over 1 year, showing a clear interaction between age and HIV effects on verbal recall. Results are displayed as means with 95% confidence bands. Change is defined as the difference between baseline and follow-up scores (12 months - 0 months).
Neuroimaging
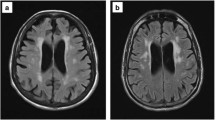
HIV-associated cortical and subcortical volume reductions, white matter changes, metabolite abnormalities, and regional glucose metabolism that vary relative to HIV clinical factors (for example, viral load, nadir CD4), and HAND severity are evident on magnetic resonance imaging (MRI), magnetic resonance spectroscopy (MRS), and positron emission tomography (PET) [32-34]. Although neuroimaging abnormalities are usually most significant in cases of opportunistic brain infection, HIV also has direct and indirect effects on brain structure and function. Historically, research focused on the basal ganglia and cerebral white matter, regions considered to be particularly vulnerable to HIV [35]. Yet when compared with seronegative controls, people with HIV also show reduced grey matter and cortical thinning [32-34,36], especially in frontal and temporal regions [37]. A study of asymptomatic individuals with HIV revealed decreased frontal grey matter volumes in the absence of other brain changes [38], suggesting that cortical atrophy begins in frontal areas. White matter hyperintensities (WMHs) on MRI reflect white matter damage, and studies show WMHs occurring at younger ages in people with HIV than they do in adults aging without HIV [39,40]. Case-controlled diffusion tensor imaging (DTI) studies show that HIV is associated with lower white matter integrity globally [41] and in specific areas such as the corpus callosum, internal capsules, and the frontal and parietal lobes [42,43]. There is evidence that the earliest HIV-associated white matter effects occur in the frontal lobes [44], with more widespread damage occurring as the disease increases in severity [43,45]. MRS studies show increased myoinositol (MI), choline (Cho), and total creatine (Cr) in brain disorders with chronic inflammation and glial activation, including HIV [32,34,46,47]. N-acetylaspartate (NAA), a marker of neuronal integrity that decreases in response to neuronal damage, has been shown to be lower in people with HIV compared with age-matched controls, especially in the basal ganglia and frontal white matter [46,48]. Although NAA associations with plasma HIV-RNA show that neuronal injury is related to current viral replication, neuronal injury is also found in virologically suppressed patients and may be attributed to the effects of chronic immune activation and inflammation. PET studies have shown that glucose hypometabolism in the frontal cortex, suggesting deficient functioning, and basal ganglia hypermetabolism occur in HIV [49]. Increased basal ganglia metabolism may seem counterintuitive, although HIV-infected astrocytes require increased glucose to proliferate [50], and the basal ganglia are known to be particularly vulnerable to HIV [35].
The neuroimaging abnormalities that have been observed historically among HIV-infected people are similar to those observed among older adults without HIV. As people reach advanced age, cortical and subcortical volumes gradually decrease [51]. Additionally, older age in healthy cohorts has consistently been found to be one of the most important independent predictors of greater WMH volume [52]. WMHs in frontal and parietal regions have been especially associated with older age and greater cognitive dysfunction [53], and longitudinal studies show greater age-related volumetric decline in anterior versus posterior white matter regions [54]. Declines in DTI measures of white matter integrity also occur with increased age [55], with anterior regions showing the greatest changes [56]. Furthermore, MRS research indicates that there is an age-related decline in NAA and increases in Cho and Cr [57]. Finally, PET research shows age-related declines in glucose metabolism, beginning with frontal lobe changes [58].
cART-era research shows that the neuropathology of HIV appears to be changing in that it now involves cortical as well as subcortical structures [33]. This signifies that HIV may progress to involve processes that bear a greater resemblance to age-related neurodegenerative diseases, such as AD, of which cortical atrophy and ventricular enlargement are hallmarks [59]. As in HIV, WMHs have been shown to occur in frontal and parietal lobes in AD, and the degree of WMH in parietal lobes and posterior periventricular areas corresponds with level of cognitive impairment [60]. AD is also associated with widespread DTI abnormalities [61]. However, cortical changes in AD cases are typically more pronounced than in cases of HIV, and hippocampal atrophy occurs early and ubiquitously in AD whereas the hippocampus is not as vulnerable in HIV [62,63]. Also, unlike in HIV, in AD the largest DTI effects are in hippocampal areas. A recent review of MRS abnormalities in AD showed NAA decreases and MI increases similar to HIV. In AD, decreased NAA is generally found in all major lobes of the brain as well as the medial temporal lobe and the posterior cingulate gyrus [64]. MI increases are also common, and changes in NAA and MI are associated with level of AD neuropathology. PET research in AD shows parietal, temporal, and posterior cingulate glucose metabolism decreases that predict cortical volume loss [65], with decreases in the frontal cortex as the disease progresses [66], whereas in HIV frontal hypometabolism is seen early on in the disease.
Neuropathology and pathophysiology
HIV enters the brain soon after infection, and the brain continues to be a reservoir for HIV even among patients who receive cART [67]. The absence of circulating HIV-RNA in the blood and cerebrospinal fluid (CSF) does not guarantee that infected people are free of the virus, its adverse immunological effects, or risk for HAND [68]. Persistent and progressive neuronal loss occurs in people with chronic HIV despite successful viral suppression by cART [69], suggesting that they are developing a concurrent neurodegenerative disorder in the setting of stable HIV infection, that HIV is causing neurodegenerative changes or that both are occurring.
Beta-amyloid
Abnormal beta-amyloid (Aβ) accumulation is a hallmark of AD that has been found to occur in HIV [70,71]. Aβ abnormalities are more consistent in AD than in HIV, particularly among younger HIV-infected people. Increasing age is a risk factor for Aβ deposition in HIV, but recent evidence suggests that HIV and aging independently affect Aβ deposition [72], whereas AD and aging clearly interact. In HIV, plaques tend to be diffuse, and amyloid depositions commonly occur in neuronal somas as well as in extracellular plaques and along axonal tracks [70,71,73]. In AD, however, plaques are neuritic and occur particularly in extracellular space [74]. Neuropathology studies suggest that, in HIV, Aβ aggregates preferentially in the hippocampus, frontal lobe, and basal ganglia [70,75]. The location of Aβ accumulation varies in AD but tends to occur in neocortical areas first [74]. There is some evidence that long-term cART use may contribute to Aβ accumulation [70].
Aggregated Aβ can also occur in older people without cognitive disturbances, but whereas it is ubiquitous and extensive in AD [76], it is not a fundamental aspect of normal cognitive aging. Aβ aggregates in similar brain areas in healthy aging as in AD but usually more slowly and with less neurotoxicity [77]. Overall, although Aβ is highly associated with AD, evidence is limited to suggest that it is a driving force in HAND.
Phosphorylated tau
Neurofibrillary tangles composed of hyperphosphorylated tau (pTau) are another hallmark of AD that occurs in people with HIV. Unlike amyloid plaques, pTau occurs in the majority of older adults. However, elevated pTau occurs at earlier ages in people with HIV than in healthy controls [78]. Although pTau levels appear to be unrelated to HIV viral levels in the brain [79], pTau is associated with microglial activation. Tau phosphorylation in HIV may result from pro-inflammatory cytokines and viral proteins that alter amyloidosis, which precede the formation of tau tangles [80]. Higher levels are also associated with antiretroviral treatment [78]. In the context of HIV, pTau is generally found in the hippocampus and entorhinal cortex and later spreads to surrounding areas [78], which mirrors the pattern seen in normal aging and AD [81].
Cerebrospinal fluid markers
CSF concentrations of pTau and Aβ correspond with concentrations in the brain, although for Aβ an inverse relationship exists, reflecting problems with clearance. Elevated pTau and decreased Aβ have been reported in the CSF of people with symptomatic HIV [82], mirroring the pattern found in people with AD, although this finding has been inconsistent, particularly for total tau and pTau [83]. In one study, decreased CSF Aβ, but not elevated total tau or pTau, was found in people with HAND [84]. In contrast, elevated CSF pTau was shown in patients with asymptomatic HIV compared with controls in a recent study [85], and recent findings yet to be published by our group indicate elevated CSF pTau among older HIV-infected people with HAND. Accordingly, similarities exist between HIV and AD with respect to CSF Aβ and tau [82], although greater disturbances are found in AD, particularly compared with young adults with neuroasymptomatic HIV.
Blood–brain barrier disturbances
The permeability of the blood–brain barrier (BBB) is altered in HIV, allowing leakage of toxic substances, including infected macrophages from the blood into the brain parenchyma. HIV affects neuronal endocytosis, which alters the integrity of the microvascular endothelial cells that compose the BBB [86]. HIV-induced disruption of the tight cell junctions and upregulation of adhesion molecules facilitate BBB passage [87]. BBB dysfunction has been linked to Aβ accumulation in HIV and other diseases resulting from a failure to filter amyloid peptides [88]. HIV has been shown to increase in vitro intracellular Aβ accumulation in microvascular endothelial cells [89]. BBB dysfunction is associated with neurodegeneration in AD, acting as both a cause and a consequence of cerebral Aβ accumulation, and AD and HIV share several common pathophysiological mechanisms that affect BBB permeability and Aβ accumulation [88,90].
Risk factors and pathophysiological mechanisms
Genetic predisposition
The apolipoprotein-E ε4 allele (ApoEε4) is a well-established AD risk factor [91] that has been associated with increased amyloid accumulation, reduced brain volumes, impaired neurocognitive functioning, and accelerated systemic progression of HIV [92,93]. ApoEε4 has been shown to increase cell susceptibility to HIV infection in vitro [93]. ApoEε4 has also been linked to reduced cognitive performance in HIV compared with age-matched seronegative ApoEε4+ participants [92], although some research does not support a significant association between ApoEε4 and HAND [94]. The relationship between ApoEε4 and cognitive functioning is more robust in AD than in HIV, as carriers with two alleles have up to a 90% chance of having AD by age 80, and ApoEε4 has been said to account for the majority of the risk associated with developing AD [91]. Although pre-existing genetic factors may influence the impact of HIV on neurological structure and function, HIV also causes epigenetic changes that may contribute to neurodegeneration and cognitive impairment as well [95].
Cerebral metabolism
Converging lines of evidence indicate that cerebral metabolite disturbances are common among HIV-infected individuals and contribute to neurocognitive and brain abnormalities [47,87,96]. Mitochondrial disturbances in these individuals cause oxidative stress through the overproduction of reactive oxygen species (ROS), which affects viral replication, inflammation, immune function, sensitivity to drug toxicities, and HAND development [87,96,97]. The oxidative stress and cell damage caused by ROS have been proposed as a major driver underlying brain aging [98] and may also contribute to HIV effects on the aging brain, along with abnormal insulin signaling [99]. Mitochondrial dysfunction has also been associated with increased neuroinflammation, glutamate overproduction, and calcium accumulation, all of which can be neurotoxic [100]. Similarly, alterations in brain mitochondrial function, glucose metabolism, and oxygen utilization have been implicated in AD [101,102]. Oxidative stress occurs at early stages of AD and may promote the formation of Aβ plaques and tau tangles [101].
Inflammation and neuroimmunological disturbances
HIV spreads from infected monocytes to uninfected cerebral microglia and astrocytes, activating inflammatory immune responses involving the release of cytokines, chemokines, and ROS. Chronic neuroinflammation resulting from prolonged glial and astrocyte activation has been shown to lead to neuronal dysfunction and death [87,96] and has been linked to HIV-associated brain abnormalities [97].
Regional microglial activation measured by PET has been shown to correspond to executive dysfunction in HIV [103], consistent with autopsy findings showing frontal cortical accumulation of DNA oxidative damage induced by ROS in people with AIDS [104]. Increased glial activation was found in cases of neuroasymptomatic HIV, with significant increases in frontal and parietal activation among people with HAD, suggesting that excessive glial activation and neuroinflammation precede cognitive decline [105]. PET studies show that widespread microglial activation also occurs in AD and is linked to cognitive dysfunction [106]. Similar immunological responses also occur, with Aβ accumulation leading to astrocyte upregulation and inflammatory response [90]. Neurofibrillary tangles and neuronal degeneration also promote neuroinflammation.
Neurotoxicity
HIV-associated brain dysfunction is potentiated by a cascade of excitotoxic and apoptotic processes that amplify immunologic and inflammatory responses to the virus [87,96,99]. T-cell depletion and apoptosis are affected directly by HIV gene expression and indirectly by apoptosis in uninfected cells. Among the substances that have been implicated in HIV-associated neurotoxicity are trans-activator of transcription (Tat), glycoproteins (such as gp120), and complementary proteins (such as Fas). Both Tat and gp120 impair glutamate uptake by astrocytes, causing glutamate excitotoxicity, which leads to inflammation and apoptosis. They also cause calcium accumulation, which has similar neurotoxic effects. Furthermore, Tat can induce astrocytosis and neuronal death and interacts with amyloid precursor protein to increase Aβ [107]. These viral structure and regulatory proteins also cause cerebral mitochondrial dysfunction and ROS overproduction, causing oxidative toxicity that, as previously described, contributes to BBB dysfunction and tissue damage [97,100]. Neurotoxicity has also been implicated in AD, other neurodegenerative diseases, and normal brain aging [90,108].
Neurotoxicity may also result from the antiretroviral drugs used to treat HIV [109], particularly certain nucleoside analog reverse transcriptase inhibitors. Certain antiretroviral drugs penetrate the BBB and enter the brain more easily than others, making them good candidates to treat HIV-associated brain dysfunction [110]. Yet cART-treated HIV patients show higher levels of cerebral Aβ and pTau than cART-naïve patients in recent studies [70,78]. Findings have been mixed [30,84], but overall it seems unlikely that cART is the major cause of brain dysfunction in most patients. Nonetheless, more research on cART-associated neurotoxicity is needed, especially given the chronic cART use among people aging with HIV and the large number of new drugs under development.
Neurotoxicity also occurs indirectly as a result of infection of other organ systems outside of the brain, such as gut, liver, and vascular systems. For example, HIV causes leaky gut syndrome by infecting the gut and altering the permeability of the intestinal lining, enabling bacteria and toxins to enter the blood, which causes systemic and ultimately cerebral inflammation [111]. Hepatic ceramides produced in response to HIV have also been linked to metabolic syndrome, apoptosis, and neurodegeneration [112].
Vascular and metabolic comorbidities
Some comorbid conditions, like chronic substance abuse, contribute to HIV transmission, functional outcomes, and cognitive problems in their own right, largely independent of the direct effects of HIV [113]. Others, like hepatitis C, exacerbate the neurocognitive effects of HIV through similar mechanisms [29,42]. Vascular and metabolic comorbidities, including diabetes, metabolic syndrome, obesity, and vascular disease, are now occurring with increased prevalence as chronically HIV-infected people age [114], and there is mounting evidence that HIV contributes to their development or expression [115]. Each of these conditions can adversely affect neurocognitive functioning [116,117]. For example, abnormal glucose metabolism leads to hyperglycemia and hyperinsulinemia, which induce ROS production, tau hyperphosphorylation, amyloid oligomerization, and widespread brain microangiopathy, and can lead to reduced Aβ clearance [116]. Thus, vascular cognitive impairment may be an important component of HAND caused by contribution of HIV to the development of vascular comorbidities. Still, the unique contribution of vascular cognitive impairment to HAND may be difficult to ascertain. It should also be emphasized that vascular risk factors are highly prevalent in the elderly, and there is strong evidence that these can be associated with vascular cognitive impairment, even in the absence of discrete cerebrovascular events [118]. Epidemiological studies have long suggested that these conditions increase risk for developing AD [116,117,119], and increased vascular risk is associated with greater amyloid burden in both HIV [120] and AD [111,121]. Given the modifiable nature of vascular and metabolic risk factors, these may ultimately be important targets for treatment as a way of preventing or diminishing cognitive dysfunction in HIV.
Premature cognitive aging, neurodegenerative disease, or both?
The findings described above illustrate multiple commonalities as well as notable differences between the cognitive disturbances and brain dysfunction that occur secondary to HIV versus AD and other neurodegenerative diseases (Table 1). The fact that many people living with chronic HIV are experiencing cognitive and neurological decline during mid-life and resembling the neurological functioning of older adults provides compelling evidence that premature cognitive aging is occurring despite cART effectively reducing HIV-associated morbidity. Yet obvious differences exist with respect to the clinical course and cognitive domains affected in HIV and AD. Progressive cognitive decline resulting in severe dementia is ubiquitous in AD. Eventually, patients with AD experience profound cognitive dysfunction that affects memory encoding and storage, language, and higher-order intellectual abilities. This is not the case with HAND, and severe dementia is currently rare among people whose HIV is well controlled. Different trajectories of cognitive decline also exist for HIV and AD, and the usual age of onset is much younger in HAND than in AD. Furthermore, the correspondence between viral pathogen, immunological disturbance, and cognitive decline that is an essential feature of HAND does not exist for AD or most other neurodegenerative disorders. These clinical considerations might lead one to conclude that HIV and AD affect the brain and cognition in very different ways.
Although AD and HIV share some structural, functional, and metabolic brain abnormalities and neuropathology, there are important differences. Aβ accumulation occurs at much higher rates in AD than in HIV or normal aging, while pTau findings are inconclusive. Furthermore, neurodegeneration generally occurs in hippocampal regions first in AD, whereas HIV shows a fronto-subcortical pattern. AD and HIV share some common pathophysiological mechanisms, including altered BBB activity, oxidative stress, and neuroinflammation, but HIV has a number of more specific brain effects caused by toxic glycoproteins, such as gp120, ongoing viral replication in certain brain areas despite overall systemic and CSF viral suppression, HIV co-receptors on lymphocytes, macrophages, neurons and microglial cells, and genetic or epigenetic alterations in response to the virus that may cause neuronal damage. These specific pathophysiological mechanisms obviously differentiate HIV from AD. The extent to which AD is caused by related mechanisms remains to be determined.
The verdict
Based on existing evidence, several conclusions can be reached. HIV causes premature cognitive and brain aging. These effects are caused by direct damage from the virus as well as indirectly through increased risk of cardiovascular disease, chronic drug use, and potentially toxic long-term antiretroviral use. There has been some controversy over whether HIV causes neurodegeneration as such, and over whether HAND should be considered a neurodegenerative disease. Evidence for neurodegeneration is provided by recent longitudinal studies showing declines in memory and other cognitive functions over time as well as relatively high rates of HIV-infected people transitioning from being asymptomatic to having HAND. Brain pathophysiology also suggests that HIV causes neurodegenerative changes, at least in some people. Despite the many commonalities between HIV and AD, it seems unlikely that HIV causes AD per se. Besides differences in their usual clinical, cognitive, and neuropathological presentation, HIV is clearly caused by virus, whereas AD is not. Nonetheless, common pathophysiological pathways exist in HIV and typical neurodegenerative diseases that contribute to accelerated age-associated cognitive decline. In the current era of cART, when HIV can be well controlled from the time of diagnosis, it may be that people will not show the same effects of chronic infection as they age, or at least not to the same degree. Future research is necessary to address this question, examining newer cohorts of HIV-infected people who have not experienced severe immunocompromise. Beyond targeting viral replication with cART, targeting vascular and metabolic factors will likely be important for delaying or mitigating HAND. Future studies should examine whether modifying these factors through clinical interventions results in improved cognitive function in people with HIV.
Abbreviations
- AD:
-
Alzheimer’s disease
- ADC:
-
AIDS dementia complex
- ANI:
-
Asymptomatic neurocognitive impairment
- ApoEε4:
-
Apolipoprotein-E ε4
- Aβ:
-
Beta-amyloid
- BBB:
-
Blood–brain barrier
- cART:
-
Combination antiretroviral therapy
- Cho:
-
Choline
- Cr:
-
Creatine
- CSF:
-
Cerebrospinal fluid
- DTI:
-
Diffusion tensor imaging
- gp120:
-
Glycoprotein 120
- HAD:
-
HIV-associated dementia
- HAND:
-
HIV-associated neurocognitive disorder
- MI:
-
Myoinositol
- MND:
-
Mild neurocognitive disorder
- MRI:
-
Magnetic resonance imaging
- MRS:
-
Magnetic resonance spectroscopy
- NAA:
-
N-acetylaspartate
- PET:
-
Positron emission tomography
- pTau:
-
Hyperphosphorylated tau
- ROS:
-
Reactive oxygen species
- Tat:
-
Trans-activator of transcription
- WMH:
-
White matter hyperintensity
References
Centers for Disease Control and Prevention. HIV surveillance report, 2011. Vol. 23. 2013:1–11. http://www.cdc.gov/hiv/topics/surveillance/resources/reports/. Accessed 14 May 2014.
Holland JC, Tross S. The psychosocial and neuropsychiatric sequelae of the acquired immunodeficiency syndrome and related disorders. Ann Intern Med. 1985;103:760–4.
Navia BA, Jordan BD, Price RW. The AIDS dementia complex: I. Clinical features. Ann Neurol. 1986;19:517–24.
Dorrell L, Snow MH, Ong EL. Mortality and survival trends in patients with AIDS in north east England from 1984–1992. J Infect. 1995;30:23–7.
Antiretroviral Therapy Cohort Collaboration. Life expectancy of individuals on combination antiretroviral therapy in high-income countries: a collaborative analysis of 14 cohort studies. Lancet. 2008;372:293–9.
Hall HI, Song R, Rhodes P, Prejean J, An Q, Lee LM, et al. Estimation of HIV incidence in the United States. JAMA. 2008;300:520–9.
Bhaskaran K, Mussini C, Antinori A, Walker AS, Dorrucci M, Sabin C, et al. Changes in the incidence and predictors of human immunodeficiency virus-associated dementia in the era of highly active antiretroviral therapy. Ann Neurol. 2008;63:213–21.
Heaton RK, Grant I, Butters N, White DA, Kirson D, Atkinson JH, et al. The HNRC 500–neuropsychology of HIV infection at different disease stages. HIV Neurobehavioral Research Center. J Int Neuropsychol Soc. 1995;1:231–51.
Sacktor N, Bacellar H, Hoover D, Nance-Sproson T, Selness O, Miller E, et al. Psychomotor slowing in HIV infection: a predictor of dementia, AIDS & death. J Neurovirol. 1996;2:404–10.
Neuenburg JK, Brodt HR, Herndier BG, Bickel M, Bacchetti P, Price RW, et al. HIV-related neuropathology, 1985 to 1999: rising prevalence of HIV encephalopathy in the era of highly active antiretroviral therapy. J Acquir Immune Defic Syndr. 2002;31:171–7.
Brew BJ, Rosenblum M, Cronin K, Price RW. AIDS dementia complex and HIV-1 brain infection: clinical-virological correlations. Ann Neurol. 1995;38:563–70.
Portegies P, de Gans J, Lange JM, Derix MM, Speelman H, Bakker M, et al. Declining incidence of AIDS dementia complex after introduction of zidovudine treatment. BMJ. 1989;299:819–21.
Cohen RA, Boland R, Paul R, Tashima KT, Schoenbaum EE, Celentano DD, et al. Neurocognitive performance enhanced by highly active antiretroviral therapy in HIV-infected women. AIDS. 2001;15:341–5.
McArthur JC, McDermott MP, McClernon D, St Hillaire C, Conant K, Marder K, et al. Attenuated central nervous system infection in advanced HIV/AIDS with combination antiretroviral therapy. Arch Neurol. 2004;61:1687–96.
Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, et al. HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirol. 2002;8:136–42.
Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;17:3–16.
Carey CL, Woods SP, Gonzalez R, Conover E, Marcotte TD, Grant I, et al. Predictive validity of global deficit scores in detecting neuropsychological impairment in HIV infection. J Clin Exp Neuropsychol. 2004;26:307–19.
Osowiecki DM, Cohen RA, Morrow KM, Paul RH, Carpenter CC, Flanigan T, et al. Neurocognitive and psychological contributions to quality of life in HIV-1-infected women. AIDS. 2000;14:1327–32.
Heaton RK, Marcotte TD, Mindt MR, Sadek J, Moore DJ, Bentley H, et al. The impact of HIV-associated neuropsychological impairment on everyday functioning. J Int Neuropsychol Soc. 2004;10:317–31.
Lai DJ, Tarwater PM, Hardy RJ. Measuring the impact of HIV/AIDS, heart disease and malignant neoplasms on life expectancy in the USA from 1987 to 2000. Public Health. 2006;120:486–92.
Wendelken LA, Valcour V. Impact of HIV and aging on neuropsychological function. J Neurovirol. 2012;18:256–63.
Valcour VG, Shikuma CM, Watters MR, Sacktor NC. Cognitive impairment in older HIV-1-seropositive individuals: prevalence and potential mechanisms. AIDS. 2004;18:S79–86.
Becker JT, Lopez OL, Dew MA, Aizenstein HJ. Prevalence of cognitive disorders differs as a function of age in HIV virus infection. AIDS. 2004;18:S11–8.
Wilkie FL, Goodkin K, Khamis I, van Zuilen MH, Lee D, Lecusay R, et al. Cognitive functioning in younger and older HIV-1-infected adults. J Acquir Immune Defic Syndr. 2003;33:S93–S105.
Becker JT, Maruca V, Kingsley LA, Sanders JM, Alger JR, Barker PB, et al. Factors affecting brain structure in men with HIV disease in the post-HAART era. Neuroradiology. 2012;54:113–21.
Ances BM, Ortega M, Vaida F, Heaps J, Paul R. Independent effects of HIV, aging, and HAART on brain volumetric measures. J Acquir Immune Defic Syndr. 2012;59:469–77.
Seider TR, Luo X, Gongvatana A, Devlin KN, de la Monte SM, Chasman JD, et al. Verbal memory declines more rapidly with age in HIV infected versus uninfected adults. J Clin Exp Neuropsychol. 2014;36:356–67.
Morgan EE, Woods SP, Smith C, Weber E, Scott JC, Grant I. Lower cognitive reserve among individuals with syndromic HIV-associated neurocognitive disorders (HAND). AIDS Behav. 2012;16:2279–85.
Devlin KN, Gongvatana A, Clark US, Chasman JD, Westbrook ML, Tashima KT, et al. Neurocognitive effects of HIV, hepatitis C, and substance use history. J Int Neuropsychol Soc. 2012;18:68–78.
Harezlak J, Buchthal S, Taylor M, Schifitto G, Zhong J, Daar E, et al. Persistence of HIV-associated cognitive impairment, inflammation, and neuronal injury in era of highly active antiretroviral treatment. AIDS. 2011;25:625–33.
Heaton RK, Clifford DB, Franklin Jr DR, Woods SP, Ake C, Vaida F, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. 2010;75:2087–96.
Cohen RA, Harezlak J, Gongvatana A, Buchthal S, Schifitto G, Clark U, et al. Cerebral metabolite abnormalities in human immunodeficiency virus are associated with cortical and subcortical volumes. J Neurovirol. 2010;16:435–44.
Cohen RA, Harezlak J, Schifitto G, Hana G, Clark U, Gongvatana A, et al. Effects of nadir CD4 count and duration of human immunodeficiency virus infection on brain volumes in the highly active antiretroviral therapy era. J Neurovirol. 2010;16:25–32.
Hua X, Boyle CP, Harezlak J, Tate DF, Yiannoutsos CT, Cohen R, et al. Disrupted cerebral metabolite levels and lower nadir CD4 + counts are linked to brain volume deficits in 210 HIV-infected patients on stable treatment. Neuroimage Clin. 2013;3:132–42.
Aylward EH, Henderer JD, McArthur JC, Brettschneider PD, Harris GJ, Barta PE, et al. Reduced basal ganglia volume in HIV-1 associated dementia: results from quantitative neuroimaging. Neurology. 1993;43:2099–104.
Thompson PM, Dutton RA, Hayashi KM, Toga AW, Lopez OL, Aizenstein HJ, et al. Thinning of the cerebral cortex visualized in HIV/AIDS reflects CD4+ T lymphocyte decline. Proc Natl Acad Sci U S A. 2005;102:15647–52.
Becker JT, Bajo R, Fabrizio M, Sudre G, Cuesta P, Aizenstein HJ, et al. Functional connectivity measured with magnetoencephalography identifies persons with HIV disease. Brain Imaging Behav. 2012;6:366–73.
Towgood KJ, Pitkanen M, Kulasegaram R, Fradera A, Kumar A, Soni S, et al. Mapping the brain in younger and older asymptomatic HIV-1 men: frontal volume changes in the absence of other cortical or diffusion tensor abnormalities. Cortex. 2012;48:230–41.
Seider T, Cohen RA, Correia S, Gongvatana A, Cummings T, Tashima KT, et al. Synergistic effects of age and HIV on white matter hyperintensities [abstract]. Annual American Academy of Clinical Neuropsychology Conference. 25–28 June 2014; New York, NY.
McMurtray A, Nakamoto B, Shikuma C, Valcour V. Cortical atrophy and white matter hyperintensities in HIV: the Hawaii Aging with HIV Cohort Study. J Stroke Cerebrovasc Dis. 2008;17:212–7.
Tate DF, Conley J, Paul RH, Coop K, Zhang S, Zhou W, et al. Quantitative diffusion tensor imaging tractography metrics are associated with cognitive performance among HIV-infected patients. Brain Imaging Behav. 2010;4:68–79.
Gongvatana A, Cohen RA, Correia S, Devlin KN, Miles J, Kang H, et al. Clinical contributors to cerebral white matter integrity in HIV-infected individuals. J Neurovirol. 2011;17:477–86.
Gongvatana A, Schweinsburg BC, Taylor MJ, Theilmann RJ, Letendre SL, Alhassoon OM, et al. White matter tract injury and cognitive impairment in human immunodeficiency virus-infected individuals. J Neurovirol. 2009;15:187–95.
Pomara N, Crandall DT, Choi SJ, Johnson G, Lim KO. White matter abnormalities in HIV-1 infection: a diffusion tensor imaging study. Psychiatry Res. 2001;106:15–24.
Ragin AB, Wu Y, Storey P, Cohen BA, Edelman RR, Epstein LG. Diffusion tensor imaging of subcortical brain injury in patients infected with human immunodeficiency virus. J Neurovirol. 2005;11:292–8.
Harezlak J, Cohen R, Gongvatana A, Taylor M, Buchthal S, Schifitto G, et al. Predictors of CNS injury as measured by proton magnetic resonance spectroscopy in the setting of chronic HIV infection and CART. J Neurovirol. 2014;20:294–303.
Paul RH, Ernst T, Brickman AM, Yiannoutsos CT, Tate DF, Cohen RA, et al. Relative sensitivity of magnetic resonance spectroscopy and quantitative magnetic resonance imaging to cognitive function among nondemented individuals infected with HIV. J Int Neuropsychol Soc. 2008;14:725–33.
Cysique LA, Moffat K, Moore DM, Lane TA, Davies NW, Carr A, et al. HIV, vascular and aging injuries in the brain of clinically stable HIV-infected adults: a (1)H MRS study. PLoS One. 2013;8:e61738.
von Giesen HJ, Antke C, Hefter H, Wenserski F, Seitz RJ, Arendt G. Potential time course of human immunodeficiency virus type 1-associated minor motor deficits: electrophysiologic and positron emission tomography findings. Arch Neurol. 2000;57:1601–7.
Palmer CS, Ostrowski M, Gouillou M, Tsai L, Yu D, Zhou J, et al. Increased glucose metabolic activity is associated with CD4+ T-cell activation and depletion during chronic HIV infection. AIDS. 2014;28:297–309.
Raz N, Ghisletta P, Rodrigue KM, Kennedy KM, Lindenberger U. Trajectories of brain aging in middle-aged and older adults: regional and individual differences. Neuroimage. 2010;51:501–11.
Longstreth Jr WT, Manolio TA, Arnold A, Burke GL, Bryan N, Jungreis CA, et al. Clinical correlates of white matter findings on cranial magnetic resonance imaging of 3301 elderly people. The Cardiovascular Health Study. Stroke. 1996;27:1274–82.
Meier IB, Manly JJ, Provenzano FA, Louie KS, Wasserman BT, Griffith EY, et al. White matter predictors of cognitive functioning in older adults. J Int Neuropsychol Soc. 2012;18:414–27.
Resnick SM, Pham DL, Kraut MA, Zonderman AB, Davatzikos C. Longitudinal magnetic resonance imaging studies of older adults: a shrinking brain. J Neurosci. 2003;23:3295–301.
Pfefferbaum A, Sullivan EV, Hedehus M, Lim KO, Adalsteinsson E, Moseley M. Age-related decline in brain white matter anisotropy measured with spatially corrected echo-planar diffusion tensor imaging. Magn Reson Med. 2000;44:259–68.
Grieve SM, Williams LM, Paul RH, Clark CR, Gordon E. Cognitive aging, executive function, and fractional anisotropy: a diffusion tensor MR imaging study. AJNR Am J Neuroradiol. 2007;28:226–35.
Haga KK, Khor YP, Farrall A, Wardlaw JM. A systematic review of brain metabolite changes, measured with 1H magnetic resonance spectroscopy, in healthy aging. Neurobiol Aging. 2009;30:353–63.
Loessner A, Alavi A, Lewandrowski KU, Mozley D, Souder E, Gur RE. Regional cerebral function determined by FDG-PET in healthy volunteers: normal patterns and changes with age. J Nucl Med. 1995;36:1141–9.
Ott BR, Cohen RA, Gongvatana A, Okonkwo OC, Johanson CE, Stopa EG, et al. Brain ventricular volume and cerebrospinal fluid biomarkers of Alzheimer’s disease. J Alzheimers Dis. 2010;20:647–57.
Brickman AM. Contemplating Alzheimer’s disease and the contribution of white matter hyperintensities. Curr Neurol Neurosci Rep. 2013;13:415.
Sexton CE, Kalu UG, Filippini N, Mackay CE, Ebmeier KP. A meta-analysis of diffusion tensor imaging in mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging. 2011;32:2322. e2325-2318.
Frings L, Yew B, Flanagan E, Lam BY, Hull M, Huppertz HJ, et al. Longitudinal grey and white matter changes in frontotemporal dementia and Alzheimer’s disease. PLoS One. 2014;9:e90814.
Stout JC, Ellis RJ, Jernigan TL, Archibald SL, Abramson I, Wolfson T, et al. Progressive cerebral volume loss in human immunodeficiency virus infection: a longitudinal volumetric magnetic resonance imaging study. HIV Neurobehavioral Research Center Group. Arch Neurol. 1998;55:161–8.
Zhang N, Song X, Bartha R, Beyea S, D’Arcy R, Zhang Y, et al. Advances in High-Field Magnetic Resonance Spectroscopy in Alzheimer’s Disease. Curr Alzheimer Res. 2014;11:367–88.
Adriaanse SM, van Dijk KR, Ossenkoppele R, Reuter M, Tolboom N, Zwan MD, et al. The effect of amyloid pathology and glucose metabolism on cortical volume loss over time in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2014;41:1190–8.
Mosconi L, Berti V, Glodzik L, Pupi A, De Santi S, de Leon MJ. Pre-clinical detection of Alzheimer’s disease using FDG-PET, with or without amyloid imaging. J Alzheimers Dis. 2010;20:843–54.
Masliah E, DeTeresa RM, Mallory ME, Hansen LA. Changes in pathological findings at autopsy in AIDS cases for the last 15 years. AIDS. 2000;14:69–74.
Cysique LA, Brew BJ, Halman M, Catalan J, Sacktor N, Price RW, et al. Undetectable cerebrospinal fluid HIV RNA and beta-2 microglobulin do not indicate inactive AIDS dementia complex in highly active antiretroviral therapy-treated patients. J Acquir Immune Defic Syndr. 2005;39:426–9.
Gongvatana A, Harezlak J, Buchthal S, Daar E, Schifitto G, Campbell T, et al. Progressive cerebral injury in the setting of chronic HIV infection and antiretroviral therapy. J Neurovirol. 2013;19:209–18.
Green DA, Masliah E, Vinters HV, Beizai P, Moore DJ, Achim CL. Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. AIDS. 2005;19:407–11.
Brew BJ, Crowe SM, Landay A, Cysique LA, Guillemin G. Neurodegeneration and ageing in the HAART era. J Neuroimmune Pharmacol. 2009;4:163–74.
Ortega M, Ances BM. Role of HIV in amyloid metabolism. J Neuroimmune Pharmacol. 2014;9:483–91.
Everall I, Vaida F, Khanlou N, Lazzaretto D, Achim C, Letendre S, et al. Cliniconeuropathologic correlates of human immunodeficiency virus in the era of antiretroviral therapy. J Neurovirol. 2009;15:360–70.
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404.
Nebuloni M, Pellegrinelli A, Ferri A, Bonetto S, Boldorini R, Vago L, et al. Beta amyloid precursor protein and patterns of HIV p24 immunohistochemistry in different brain areas of AIDS patients. AIDS. 2001;15:571–5.
Fjell AM, McEvoy L, Holland D, Dale AM, Walhovd KB. What is normal in normal aging? Effects of aging, amyloid and Alzheimer’s disease on the cerebral cortex and the hippocampus. Prog Neurobiol. 2014;117:20–40.
Piccini A, Russo C, Gliozzi A, Relini A, Vitali A, Borghi R, et al. beta-amyloid is different in normal aging and in Alzheimer disease. J Biol Chem. 2005;280:34186–92.
Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Accelerated Tau deposition in the brains of individuals infected with human immunodeficiency virus-1 before and after the advent of highly active anti-retroviral therapy. Acta Neuropathol. 2006;111:529–38.
Smith DB, Simmonds P, Bell JE. Brain viral burden, neuroinflammation and neurodegeneration in HAART-treated HIV positive injecting drug users. J Neurovirol. 2014;20:28–38.
Becker JT, Kingsley L, Mullen J, Cohen B, Martin E, Miller EN, et al. Vascular risk factors, HIV serostatus, and cognitive dysfunction in gay and bisexual men. Neurology. 2009;73:1292–9.
Price JL, Davis PB, Morris JC, White DL. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer’s disease. Neurobiol Aging. 1991;12:295–312.
Brew BJ, Pemberton L, Blennow K, Wallin A, Hagberg L. CSF amyloid beta42 and tau levels correlate with AIDS dementia complex. Neurology. 2005;65:1490–2.
Gisslen M, Krut J, Andreasson U, Blennow K, Cinque P, Brew BJ, et al. Amyloid and tau cerebrospinal fluid biomarkers in HIV infection. BMC Neurol. 2009;9:63.
Marra CM, Zhao Y, Clifford DB, Letendre S, Evans S, Henry K, et al. Impact of combination antiretroviral therapy on cerebrospinal fluid HIV RNA and neurocognitive performance. AIDS. 2009;23:1359–66.
Peluso MJ, Meyerhoff DJ, Price RW, Peterson J, Lee E, Young AC, et al. Cerebrospinal fluid and neuroimaging biomarker abnormalities suggest early neurological injury in a subset of individuals during primary HIV infection. J Infect Dis. 2013;207:1703–12.
Banks WA, Ercal N, Price TO. The blood–brain barrier in neuroAIDS. Curr HIV Res. 2006;4:259–66.
Anderson E, Zink W, Xiong H, Gendelman HE. HIV-1-associated dementia: a metabolic encephalopathy perpetrated by virus-infected and immune-competent mononuclear phagocytes. J Acquir Immune Defic Syndr. 2002;31:S43–54.
Andras IE, Toborek M. Amyloid beta accumulation in HIV-1-infected brain: the role of the blood brain barrier. IUBMB Life. 2013;65:43–9.
Andras IE, Eum SY, Huang W, Zhong Y, Hennig B, Toborek M. HIV-1-induced amyloid beta accumulation in brain endothelial cells is attenuated by simvastatin. Mol Cell Neurosci. 2010;43:232–43.
Erickson MA, Banks WA. Blood–brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J Cereb Blood Flow Metab. 2013;33:1500–13.
Raber J, Huang Y, Ashford JW. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol Aging. 2004;25:641–50.
Chang L, Andres M, Sadino J, Jiang CS, Nakama H, Miller E, et al. Impact of apolipoprotein E epsilon4 and HIV on cognition and brain atrophy: antagonistic pleiotropy and premature brain aging. Neuroimage. 2011;58:1017–27.
Burt TD, Agan BK, Marconi VC, He W, Kulkarni H, Mold JE, et al. Apolipoprotein (apo) E4 enhances HIV-1 cell entry in vitro, and the APOE epsilon4/epsilon4 genotype accelerates HIV disease progression. Proc Natl Acad Sci U S A. 2008;105:8718–23.
Morgan EE, Woods SP, Letendre SL, Franklin DR, Bloss C, Goate A, et al. Apolipoprotein E4 genotype does not increase risk of HIV-associated neurocognitive disorders. J Neurovirol. 2013;19:150–6.
Desplats P, Dumaop W, Smith D, Adame A, Everall I, Letendre S, et al. Molecular and pathologic insights from latent HIV-1 infection in the human brain. Neurology. 2013;80:1415–23.
Minghetti L, Visentin S, Patrizio M, Franchini L, Ajmone-Cat MA, Levi G. Multiple actions of the human immunodeficiency virus type-1 Tat protein on microglial cell functions. Neurochem Res. 2004;29:965–78.
Mollace V, Nottet HS, Clayette P, Turco MC, Muscoli C, Salvemini D, et al. Oxidative stress and neuroAIDS: triggers, modulators and novel antioxidants. Trends Neurosci. 2001;24:411–6.
Harman D, Hendricks S, Eddy DE, Seibold J. Free radical theory of aging: effect of dietary fat on central nervous system function. J Am Geriatr Soc. 1976;24:301–7.
Mattson MP, Haughey NJ, Nath A. Cell death in HIV dementia. Cell Death Differ. 2005;12:893–904.
Tian C, Sun L, Jia B, Ma K, Curthoys N, Ding J, et al. Mitochondrial glutaminase release contributes to glutamate-mediated neurotoxicity during human immunodeficiency virus-1 infection. J Neuroimmune Pharmacol. 2012;7:619–28.
Shi Q, Gibson GE. Oxidative stress and transcriptional regulation in Alzheimer disease. Alzheimer Dis Assoc Disord. 2007;21:276–91.
Nakamura T, Watanabe A, Fujino T, Hosono T, Michikawa M. Apolipoprotein E4 (1–272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol Neurodegener. 2009;4:35.
Garvey LJ, Pavese N, Politis M, Ramlackhansingh A, Brooks DJ, Taylor-Robinson SD, et al. Increased microglia activation in neurologically asymptomatic HIV-infected patients receiving effective ART. AIDS. 2014;28:67–72.
Zhang Y, Wang M, Li H, Zhang H, Shi Y, Wei F, et al. Accumulation of nuclear and mitochondrial DNA damage in the frontal cortex cells of patients with HIV-associated neurocognitive disorders. Brain Res. 2012;1458:1–11.
Coughlin JM, Wang Y, Ma S, Yue C, Kim PK, Adams AV, et al. Regional brain distribution of translocator protein using [C]DPA-713 PET in individuals infected with HIV. J Neurovirol. 2014;20:219–32.
Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, et al. Microglia, amyloid, and cognition in Alzheimer’s disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32:412–9.
Kim J, Yoon JH, Kim YS. HIV-1 Tat interacts with and regulates the localization and processing of amyloid precursor protein. PLoS One. 2013;8:e77972.
Wojda U, Salinska E, Kuznicki J. Calcium ions in neuronal degeneration. IUBMB Life. 2008;60:575–90.
Vidal F, Gutierrez F, Gutierrez M, Olona M, Sanchez V, Mateo G, et al. Pharmacogenetics of adverse effects due to antiretroviral drugs. AIDS Rev. 2010;12:15–30.
Letendre SL, McCutchan JA, Childers ME, Woods SP, Lazzaretto D, Heaton RK, et al. Enhancing antiretroviral therapy for human immunodeficiency virus cognitive disorders. Ann Neurol. 2004;56:416–23.
Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–71.
Lyn-Cook Jr LE, Lawton M, Tong M, Silbermann E, Longato L, Jiao P, et al. Hepatic ceramide may mediate brain insulin resistance and neurodegeneration in type 2 diabetes and non-alcoholic steatohepatitis. J Alzheimers Dis. 2009;16:715–29.
Grassi MP, Clerici F, Perin C, Zocchetti C, Borella M, Cargnel A, et al. HIV infection and drug use: influence on cognitive function. AIDS. 1995;9:165–70.
Nix LM, Tien PC. Metabolic syndrome, diabetes, and cardiovascular risk in HIV. Curr HIV/AIDS Rep. 2014;11:271–8.
El-Sadr WM, Mullin CM, Carr A, Gibert C, Rappoport C, Visnegarwala F, et al. Effects of HIV disease on lipid, glucose and insulin levels: results from a large antiretroviral-naive cohort. HIV Med. 2005;6:114–21.
S Roriz-Filho J, Sá-Roriz TM, Rosset I, Camozzato AL, Santos AC, Chaves ML, et al. (Pre)diabetes, brain aging, and cognition. Biochim Biophys Acta. 2009;1792:432–43.
Yaffe K. Metabolic syndrome and cognitive disorders: is the sum greater than its parts? Alzheimer Dis Assoc Disord. 2007;21:167–71.
Cohen RA, Poppas A, Forman DE, Hoth KF, Haley AP, Gunstad J, et al. Vascular and cognitive functions associated with cardiovascular disease in the elderly. J Clin Exp Neuropsychol. 2009;31:96–110.
Correia SC, Santos RX, Carvalho C, Cardoso S, Candeias E, Santos MS, et al. Insulin signaling, glucose metabolism and mitochondria: major players in Alzheimer’s disease and diabetes interrelation. Brain Res. 2012;1441:64–78.
Izycka-Swieszewska E, Zoltowska A, Rzepko R, Gross M, Borowska-Lehman J. Vasculopathy and amyloid beta reactivity in brains of patients with acquired immune deficiency (AIDS). Folia Neuropathol. 2000;38:175–82.
Nelson L, Gard P, Tabet N. Hypertension and inflammation in Alzheimer’s disease: close partners in disease development and progression! J Alzheimers Dis. 2014;41:331–43.
Acknowledgments
This work was supported by the National Institutes of Health (Grant R01 MH074368 and Grant P01 AA019072) and the National Institute of Neurological Disorders and Stroke (Grant R01 NS080655).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Cohen, R.A., Seider, T.R. & Navia, B. HIV effects on age-associated neurocognitive dysfunction: premature cognitive aging or neurodegenerative disease?. Alz Res Therapy 7, 37 (2015). https://doi.org/10.1186/s13195-015-0123-4
Published:
DOI: https://doi.org/10.1186/s13195-015-0123-4