
Abstract
Adaptive nanopore sequencing as a diagnostic method for imprinting disorders and episignature analysis revealed an intragenic duplication of Exon 6 and 7 in UBE3A (NM_000462.5) in a patient with relatively mild Angelman-like syndrome. In an all-in-one nanopore sequencing analysis DNA hypomethylation of the SNURF:TSS-DMR, known contributing deletions on the maternal allele and point mutations in UBE3A could be ruled out as disease drivers. In contrast, breakpoints and orientation of the tandem duplication could clearly be defined. Segregation analysis in the family showed that the duplication derived de novo in the maternal grandfather. Our study shows the benefits of an all-in-one nanopore sequencing approach for the diagnostics of Angelman syndrome and other imprinting disorders.
Similar content being viewed by others

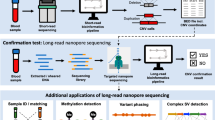

Introduction
Angelman syndrome (AS, OMIM #105,830) patients were first described by Angelman [1] and are clinically characterized by severe intellectual disability and speech impairment as well as ataxia, epilepsy and distinct behavioral profiles [21]. It is caused by a loss of function of the maternal copy of the gene encoding the ubiquitin ligase E3A (UBE3A, OMIM * 601,623) and other genes on chromosome 15q11–13 [21]. Known as a genomic imprinting disorder, AS is caused by genetic or epigenetic defects, that lead to the disruption of imprinted genes. Human genomic imprinting in turn is defined by epigenetic modifications of a small set of human genes based on their parental origin, that lead to their monoallelic expression [24]. AS is caused by four possible molecular pathomechanisms: (1) interstitial deletions of 5–7 Mb spanning the imprinted region on 15q11.2q13 on the maternal allele (70–75% of AS cases), (2) maternally inherited UBE3A point mutations (5–10%), (3) imprinting defects causing aberrant DNA methylation within chromosome 15q11–q13 that disrupt the expression of maternally inherited UBE3A (3–5%) and (4) uniparental disomy (UPD) of the paternal chromosome 15q11–q13 (2–3%) [5, 7]. Maternally derived duplications affecting several Mb in the 15q11-q13 region and their genotype–phenotype correlation have been previously reported [2]. However, they seem to cause entities distinctive from AS. The smallest duplication in the 15q11-q13 region as part of a detailed case report, encompassed the complete UBE3A gene and was found in patients with developmental delay and neuropsychiatric symptoms [14]. The ClinVar-Database contains seven entries of partial heterozygous UBE3A duplications (> 50 bp, Supp. Table 1), ranging from 59 bp to 70 kb size.
Technical and bioinformatical developments over the last 12 months have led to a significant improvement in sequencing accuracy of Oxford Nanopore Technologies (ONT) sequencing platforms thereby providing a long-read sequencing tool which, together with its capability for copy number change and DNA methylation detection as well as phasing of parental alleles for homozygosity analysis, can cover virtually all putative molecular mechanisms of AS in an all-in-one approach [22]. One exception with regard to AS is thought to be the discrimination between an epimutation at the SNURF:TSS-DMR and a paternal heterodisomy without genotyping closely related family members [9]. Theoretically, adaptive sampled (or WGS) Nanopore sequencing methylation data on several other DMRs on the same chromosome could be used to solve the majority of such cases. In view of the above, Nanopore sequencing represents a versatile first-tier diagnostic tool for imprinting disorders and episignature analysis replacing several conventional techniques such as (MS)-MLPA, Bisulfite Pyro- and DNA Sanger Sequencing that were part of a traditional stepwise diagnostic process. Furthermore, the technology offers the unique possibility to bypass wet lab enrichments by a bioinformatic approach that uses the possibility of reversing the voltage across pores to enable selection of fragments for sequencing based on real-time assessment of a small initial part of a read. Known as adaptive sampling [12], this feature can be used to enrich regions of interest or deplete unwanted fragments.
Here we report on a family with an intragenic tandem duplication of Exon 6 and 7 in UBE3A. The index patient was diagnosed with AS-like syndrome. The maternally inherited copy number change was uncovered by adaptive nanopore sequencing, with which duplication breakpoints and orientation of the duplication could be clearly defined. In line with the expected segregation of Angelman syndrome in the family, the de novo duplication in the unaffected mother of the index case was located on the allele inherited from her father.
Material and methods
Case presentation
Our patient is the first and only child of nonconsanguinous parents. The pregnancy was the result of IVF (couple sterility of unexplained cause) and was completely unremarkable (no gestational diabetes, no medication, and no indication of infections or fetal growth disorders). Due to a pathological CTG and a labor arrest cesarean section at 39 + 2 gestational week (GW) was carried out. While, birth weight (2960 g, 10th percentile) and length (51 cm, 32nd percentile) were normal, microcephaly was diagnosed (head circumference 32 cm, below 1st percentile, − 2.6 z).
Early on, the parents had the impression that their son was not developing appropriately. A first presentation in the neuropediatric outpatient clinic took place at the age of 14 months, when a clear developmental delay (no free sitting, no crawling) with dystrophy and microcephaly was diagnosed. EEG examination revealed pathological findings in terms of beta disturbance and epileptic potentials. At the age of 17 months EEG was found unchanged. No seizures were reported by the parents. MRI examination of the brain revealed unremarkable findings, as did echocardiography, ultrasonography of the abdomen, X-ray examination of the thorax, and basic metabolic screening. Endocrinologic causes for the dystrophy were excluded.
Initial presentation to our genetic counseling center was at 19 months of age. The patient presented with a friendly demeanor, delayed developmental milestones (no sitting, no walking without support, and no words) and reduced (< 1st percentile) weight (8500 g, − 2.4 z), height (77.1 cm, − 2.1 z), and head circumference (43.9 cm, − 4.1 z). He showed subtle dysmorphic features such as large mouth, small chin, prominent nose and mildly deep set ears.
EEG at 33 months of age was unremarkable. Due to pronounced sleep disturbances, melatonin was administered in phases.
At 34 months of age, our patient developed Kawasaki syndrome with coronary ectasia.
At the time of the last clinical reevaluation, the patient was 40 months old. Head circumference (46 cm, − 3.88 z), length (88 cm, − 2.7 z), and weight (11 kg, − 2.6 z) were below the first percentile. Seizures had not occurred. He could walk a few steps but was very insecure and he still explored a lot with his mouth. Salivation was increased. He was vocalizing but did not speak. He has a very friendly disposition and laughs a lot.
A typical but comparatively mild course of Angelman syndrome with dystrophy, short stature, microcephaly and global developmental delay with severe speech delay was diagnosed. EEG examinations initially showed abnormal findings, the last EEG had been inconspicuous and seizures had not occurred.
The family history was unremarkable.
Ethical dimension
This clinical case is reported under the premises of broad consent, that is the consent of patients and/or their legal proxies that clinical data and biological samples collected in the course of diagnostics and treatment are used for research purposes. This concept of broad consent has been acknowledge as a standard procedure for the Johannes Gutenberg University Medical Center by the regulatory authority, the Ethics Commission of the Chamber of Physicians in Rhineland-Palatinate. This regulatory board, however, is responsible for the ethical approval of clinical studies with sufficient numbers of participants to generate statistical power. Healing attempts in single cases like off-label use of cancer medication e.g. for childhood cancer or compassionate release of beneficial study drugs for single patients outside a study are controlled by the clinical ethics committee of the Johannes Gutenberg University Medical Center. In the specific case reported here, we informed the family about the use of diagnostic data and subsequent findings in biological material collected during diagnostics and treatment in the setting of case reports and publications. The clinical ethics committee agreed to include the following statement in our paper: This case report is a relevant contribution to clinically relevant research. Informed consent was granted by broad and individual consent. This includes information about the fact that both, the nature of research and the nature of the clinical case may lead to a situation in which a willing and technically able third party may be able to relate data and reported findings to an individual person. Consent was given under these premises and thus the clinical ethics committee of the Johannes Gutenberg University Medical Center in Mainz, Germany, has (a) no ethical concerns regarding the study in principle; (b) concludes that the ethical questions does not need to be addressed in the framework of clinical studies and thus by the Ethics Commission of the Chamber of Physicians based on the fact, that a single case is reported; (c) weighs the informed consent and expressed autonomy of the patient and/or parents or legal proxies against the risk of relating data and findings to an individual person and thus comes to the conclusion that the underlying research is ethically sound and the presented research including data and findings can justifiably be published.
DNA isolation
Genomic DNA from peripheral blood was extracted by Gentra Puregene Blood Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions, followed by quality (NanoDrop) and quantity (Qubit) assessment. Extracted DNA was stored at − 20 °C until further use.
Nanopore sequencing and bioinformatic processing
Native barcoding sequencing libraries were prepared from 400 ng genomic DNA using the Native Barcoding Sequencing kit SQK-NBD114.24 (ONT, Oxford Nanopore Technologies Ltd., Oxford, UK) following the manufacturer’s protocol. The clean-up step after adapter ligation was intended to size-select fragments and was done with Long Fragment Buffer. The barcoded patient library, among others, was loaded on a single R10.4.1 (FLO-PRO114M) flow cell and sequenced on a PromethION 24 device within 72 h. MinKNOW (v23.06.04) was used to supervise the initial sequencing run, including adaptive sampling with enrichment of intended genomic regions by setting human genome build hg19 as input reference. Genomic regions subject to medically relevant parent-of-origin methylation or Epi-variants as potential contributors to hereditary conditions [3, 8, and 16] were set as the target regions in the BED format file. The entire genomic and 5 kb flanking sequence of genes associated with the criteria above was also subjected to adaptive sampling. The total size of the target regions was 24,431,679 bps. Information on target regions will be available upon request. Base-calling and alignment to the human reference genome (hg19) via Minimap2 [11] was performed using Dorado software from ONT (v0.3.4) with a super accuracy model with base modifications (dna_r10.4.1_e8.2_400bps_sup@v4.2.0_5mCG_5hmCG@v2). Haplotype-aware small variant calling was accomplished using DeepVariant (v1.5, model “ONT_R104”) [15]. Phasing of reads was performed with whatsapp (v2.0) which uses nanopore long reads to link adjacent single nucleotide variants and then phases the mapped reads to infer the haplotypes [13]. The structural variations were called using Sniffles (version 2.0.3) [23]. Copy numbers were analyzed from the aligned files utilizing the CNVpytor software (v1.3.1), which discovers and analyses copy number variations and alterations based on read depth [17].
MLPA
100 ng of DNA were used together with the SALSA MLPA Probemix P336 UBE3A-B1 (MRC Holland, Amsterdam, NL) and used for DNA copy number quantification according to the manufacturer’s instructions. Copy number analysis was performed using GeneMarker software v.3.0.1.
Pyrosequencing
500 ng of DNA were bisulfite treated by the EZ DNA Methylation-Direct Kit (Zymo, Irvine, USA). 100 ng of bisulfite treated DNA were PCR amplified (Table 1) by FastStart Taq DNA Polymerase (Roche, Basel, CH) according to the manufacturer’s instructions. Afterwards quantification of DNA methylation was carried out by Pyrosequencing (Qiagen, Hilden, GER) which offers better resolution than MS-MLPA.
Segregation analysis to elucidate the parental origin of the copy number change that occurred de novo in the unaffected mother was performed by genotype analysis. Therefore, 100 ng of genomic DNA from the mother and her parents were PCR amplified (Table 1) by FastStart Taq DNA Polymerase according to the manufacturer’s instructions. Afterwards absolute quantification of allele frequency was carried out by Pyrosequencing (v.2.5.10).
Sanger sequencing of duplication breakpoint
100 ng of DNA were PCR amplified (Table 1) and sequenced on a SeqStudio Flex Genetic Analyzer (ThermoFisher Scientific, Waltham, USA) according to the manufacturer’s instructions. Sequence analysis was performed using Mutation Surveyor software v.5.1.
Results
Copy number analysis
A large interstitial deletion of 5–7 Mb in the imprinted region 15q11.2q13 on the maternal allele was ruled out based on DNA methylation quantification of the SNURF:TSS-DMR by nanopore and pyrosequencing (Fig. 1A) as well as copy number analysis of chromosome 15q11.2q13 using nanopore data (Supp. Figure 2).

A Integrative genomics viewer (IGV) screen capture images of bam files showing the SNURF:TSS-DMR (primary DMR). Both haplotypes and their corresponding DNA methylation are shown (top: maternal haplotype with methylated CpGs in red, bottom: paternal haplotype with unmethylated CpGs in blue) B Quantification of DNA methylation of the SNURF:TSS-DMR by Bisulfite Pyrosequencing of 3 CpGs. Mean methylation plus standard deviation of three technical replicates per sample are shown. From left for each CpG: index patient, healthy controls 1-3 and a sample of a known Angelman syndrome patient (maternal class I deletion on Chr.15q11.2q13) serving as an assay quality control
Instead, an intragenic heterozygous duplication of Exon 6 and 7 (NM_000462.5) was identified by nanopore sequencing and confirmed by MLPA copy number analysis (Fig. 2A and B). Size (23,340 b), localization and orientation of the tandem duplication in UBE3A was resolved by nanopore sequencing (Fig. 2A). MLPA analysis confirmed the duplication in the mother of the index patient but not her parents, indicating a de novo event on one of her parental alleles. The duplication breakpoints could be validated by breakpoint-PCR (Fig. 2C) plus subsequent Sanger Sequencing (Fig. 2D) and resulted in the following karyotype of the index case (ISCN 2020): seq[GRCh38] NC_000015.10:g. 25364087-25387427dup mat.

A IGV screen capture images showing breakpoints of the UBE3A duplication of Exon 6 and 7. B Peak Ratio Plot MLPA C Breakpoint-PCR between Intron 7 and Intron 5. Sample 1: mother of the index patient, 2: father of the index patient, 3: index patient, 4: healthy proband, NTC: no template control. D Sanger Sequencing of the Breakpoint-PCR product. Artificial Reference Sequence: bioinformatically combined sequence of intron 5 and intron 7 at the breakpoint detected by Nanopore Sequencing. Inserting two “NN‘s” at the breakpoint for better breakpoint visibility
The copy number change is predicted to be an out-of-frame duplication by the Reading Frame Checker of the Leiden Open Variation Database (LOVD v.3.0) on UBE3A transcript NM_000462.3.
Analysis of UBE3A genomic sequence
Nanopore sequencing revealed no SNVs in UBE3A that would lead to the observed phenotype of the index patient. Sequencing data of UBE3A showed 100% horizontal coverage of the entire gene with a mean vertical coverage of > 10 x.
Quantification of DNA methylation
Phased nanopore reads revealed no aberrant DNA methylation signature in the SNURF:TSS-DMR of the index patient (Fig. 1A). Quantification of DNA methylation in the SNURF:TSS-DMR by Bisulfite Pyrosequencing showed no differences compared to three unaffected control samples (Fig. 1B).
Additional to the SNURF:TSS-DMR, other DMRs on Chromosome 15 were analyzed in detail based on methylation status calculated from nanopore data. Quantification of DNA methylation in our index patient revealed no aberrant patterns in the NDN:TSS-DMR (Locus Reference Genomic identifier: LRG_1047), IGF1R:Int2-DMR (LRG_1055) and MKRN3:upstream enhancer region (LRG_1045) (Supp Fig. 1).
Segregation analysis
Based on nanopore sequencing data of the index case, allele frequency determination of several heterozygous SNPs throughout the duplicated region pointed towards a skewed ratio and thus potentially allow for further segregation analysis throughout the family. Pyrosequencing of rs77329250 that is located in the duplicated region of UBE3A confirmed 1/3:2/3 ratios of G:A in the index case and his mother, indicating that Adenine is associated with the heterozygous duplication (Fig. 3). The maternal grandmother showed homozygosity for Guanine at rs77329250, thus Adenine must be inherited by the maternal grandfather. This constellation is in line with a de novo event on the paternal inherited allele in the unaffected mother of the index case.

Segregation analysis by Pyrosequencing of rs77329250. A index case, B mother of index case, C maternal grandmother of the index case, D maternal grandfather of the index case
Discussion
To our knowledge, this is the first case report on a family with an intragenic tandem duplication of Exon 6 and 7 in UBE3A causing Angelman syndrome. The maternally inherited copy number change was uncovered by adaptive nanopore sequencing. Consistent with the inheritance pattern in Angelman syndrome, we have been able to detect the de novo duplication on the paternally inherited allele in the unaffected mother of the index case.
The detailed characterization of the copy number change (breakpoints and orientation) was done using nanopore sequencing data showing that the heterozygous duplication was placed in a tandem and non-inverted orientation within the UBE3A gene. Previous studies on duplications in 15q11-q13 [14] used chromosomal microarray analysis (CMA) and clinical testing often involves (MS)-MLPA to analyze copy number changes [6], thus making an unambiguous statement about the location and orientation of a duplication impossible. Based on the provided karyotype, accessible ClinVar entries with regard to partial duplications of UBE3A also suggest that CMA or Exome sequencing was performed. Nanopore sequencing assisted phasing of parental alleles allows for detection of uniparental isodisomies by analyzing loss of heterozygosity, thus allows for the discrimination between this type of UPD and an epimutation at the SNURF:TSS-DMR as the molecular cause of AS. Heterodisomies and epimutations at imprinted loci are difficult to distinguish if parental DNA is not available for segregation analysis [9]. However, adaptive nanopore sequencing could overcome this problem by the enrichment of further primary or secondary DMRs on the same chromosome. Assuming that the chosen regions underlie genomic imprinting in the starting material to be analyzed and are not known for multilocus imprinting disturbances (MLID) [10], they should show unaffected methylation patterns if an epimutation is the cause of an imprinting syndrome, while a complete heterodisomy of the affected chromosome is expected to show aberrant methylation patterns of the additional DMRs as well. As anticipated in a case of AS caused by an intragenic UBE3A copy number change, DNA methylation quantification revealed no aberrant patterns in the NDN:TSS-DMR, IGF1R:Int2-DMR as well as in the MKRN3:upstream enhancer region on Chromosome 15. Further studies on patients affected by imprinting disorders that are known to be caused by uniparental heterodisomies could prove our proposed scenario.
Since nanopore long-read sequencing technology can also detect DNA modifications without any additional wet lab effort, nanopore sequencing is a first-tier diagnostic method for elucidating all of the molecular causes of AS and other imprinting disorders in an all-in-one approach. Adaptive sampling is a unique feature of this sequencing technology that does not need wet lab-intensive enrichment of target regions and allows for multiplexing samples with sufficient vertical coverage on the same flowcell [12]. Currently the application still has the drawback that relatively large target regions have to be selected in order for the whole process to run effectively. Currently, further developments in bioinformatics are addressing this problem [18, 19] and significant efficiency improvements can be expected in the near future as a result.
Availability of data and materials
The data that support the findings of this study are not openly available due to reasons of sensitivity and to preserve individuals’ privacy under the European General Data Protection Regulation, but are available from the corresponding author upon reasonable request.
References
Angelman H. ‘Puppet’ children a report on three cases. Dev Med Child Neurol. 1965;7:681–8.
Al Ageeli E, Drunat S, Delanoë C, Perrin L, Baumann C, Capri Y, Fabre-Teste J, Aboura A, Dupont C, Auvin S, El Khattabi L, Chantereau D, Moncla A, Tabet AC, Verloes A. Duplication of the 15q11-q13 region: clinical and genetic study of 30 new cases. Eur J Med Genet. 2014;57(1):5–14.
Aref-Eshghi E, Bend EG, Colaiacovo S, Caudle M, Chakrabarti R, Napier M, Brick L, Brady L, Carere DA, Levy MA, Kerkhof J, Stuart A, Saleh M, Beaudet AL, Li C, Kozenko M, Karp N, Prasad C, Siu VM, Tarnopolsky MA, Ainsworth PJ, Lin H, Rodenhiser DI, Krantz ID, Deardorff MA, Schwartz CE, Sadikovic B. Diagnostic utility of genome-wide DNA methylation testing in genetically unsolved individuals with suspected hereditary conditions. Am J Hum Genet. 2019;104(4):685–700.
Back K. An investigation of regulation of MKRN3 monoallelic expression. Master's Thesis. Boston University. School of medicine. 2020. https://hdl.handle.net/2144/42082
Bird LM. Angelman syndrome: review of clinical and molecular aspects. Appl Clin Genet. 2014;16(7):93–104.
Beygo J, Buiting K, Ramsden SC, Ellis R, Clayton-Smith J, Kanber D. Update of the EMQN/ACGS best practice guidelines for molecular analysis of Prader-Willi and Angelman syndromes. Eur J Hum Genet. 2019;27(9):1326–40.
Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010;154C(3):365–76.
Court F, Tayama C, Romanelli V, Martin-Trujillo A, Iglesias-Platas I, Okamura K, Sugahara N, Simón C, Moore H, Harness JV, Keirstead H, Sanchez-Mut JV, Kaneki E, Lapunzina P, Soejima H, Wake N, Esteller M, Ogata T, Hata K, Nakabayashi K, Monk D. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014;24(4):554–69.
Deest M, Brändl B, Rohrandt C, Eberlein C, Bleich S, Müller F-J, Frieling H. Long-read nanopore sequencing reveals novel common genetic structural variants in Prader-Willi syndrome and associated psychosis. medRxiv. 2022 Available at: https://www.medrxiv.org/content/https://doi.org/10.1101/2022.07.18.22277235v1 (Accessed November 27, 2023).
Eggermann T, Yapici E, Bliek J, Pereda A, Begemann M, Russo S, Tannorella P, Calzari L, de Nanclares GP, Lombardi P, Temple IK, Mackay D, Riccio A, Kagami M, Ogata T, Lapunzina P, Monk D, Maher ER, Tümer Z. Trans-acting genetic variants causing multilocus imprinting disturbance (MLID): common mechanisms and consequences. Clin Epigenetics. 2022;14(1):41.
Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–100.
Loose M, Malla S, Stout M. Real-time selective sequencing using nanopore technology. Nat Methods. 2016;13:751–4.
Martin M, Patterson M, Garg S, Fischer SO, Pisanti N, Klau GW, Schoenhuth A, Marschall T. WhatsHap: fast and accurate read-based phasing. bioRxiv. Available at: https://www.biorxiv.org/content/https://doi.org/10.1101/085050v2 (Accessed December 01, 2023).
Noor A, Dupuis L, Mittal K, Lionel AC, Marshall CR, Scherer SW, Stockley T, Vincent JB, Mendoza-Londono R, Stavropoulos DJ. 15q11.2 duplication encompassing only the UBE3A gene is associated with developmental delay and neuropsychiatric phenotypes. Hum Mutat. 2015;36(7):689–93.
Poplin R, Chang PC, Alexander D, Schwartz S, Colthurst T, Ku A, Newburger D, Dijamco J, Nguyen N, Afshar PT, Gross SS, Dorfman L, McLean CY, DePristo MA. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol. 2018;36(10):983–7.
Sadikovic B, Levy MA, Kerkhof J, Aref-Eshghi E, Schenkel L, Stuart A, McConkey H, Henneman P, Venema A, Schwartz CE, Stevenson RE, Skinner SA, DuPont BR, Fletcher RS, Balci TB, Siu VM, Granadillo JL, Masters J, Kadour M, Friez MJ, van Haelst MM, Mannens MMAM, Louie RJ, Lee JA, Tedder ML, Alders M. Clinical epigenomics: genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet Med. 2021;23(6):1065–74.
Suvakov M, Panda A, Diesh C, Holmes I, Abyzov A. CNVpytor: a tool for copy number variation detection and analysis from read depth and allele imbalance in whole-genome sequencing. Gigascience. 2021;10(11):1074.
Ulrich JU, Lutfi A, Rutzen K, Renard BY. ReadBouncer: precise and scalable adaptive sampling for nanopore sequencing. Bioinformatics. 2022;38(Suppl 1):i153–60.
Weilguny L, De Maio N, Munro R, Manser C, Birney E, Loose M, Goldman N. Dynamic, adaptive sampling during nanopore sequencing using Bayesian experimental design. Nat Biotechnol. 2023;41(7):1018–25.
White HE, Durston VJ, Harvey JF, Cross NC. Quantitative analysis of SNRPN(correction of SRNPN) gene methylation by pyrosequencing as a diagnostic test for Prader-Willi syndrome and Angelman syndrome. Clin Chem. 2006;52(6):1005–13.
Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genet Med. 2010;12(7):385–95.
Yamada M, Okuno H, Okamoto N, Suzuki H, Miya F, Takenouchi T, Kosaki K. Diagnosis of prader-willi syndrome and angelman syndrome by targeted nanopore long-read sequencing. Eur J Med Genet. 2023;66(2): 104690.
Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, von Haeseler A, Schatz MC. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods. 2018;15(6):461–8.
Monk D, Mackay DJG, Eggermann T, Maher ER, Riccio A. Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat Rev Genet. 2019;20(4):235–48.
Acknowledgements
We thank the parents of our patient who kindly agreed to be part of this study. We thank Denise Seyler for her technical assistance.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
LH, SD, and ML were involved in conceptualization, formal analysis, and writing the main manuscript text. LH, VH and SuS were responsible for the clinical examinations. SD, LL, CH, SG and ML were responsible for methodology. NWP was evaluating the ethical perspective for publication of the present case report. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
See paragraph ethical dimension in Material and Methods section.
Consent for publication
Written informed consent was obtained from the parents for publication of the medical data.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Holthöfer, L., Diederich, S., Haug, V. et al. A case of an Angelman-syndrome caused by an intragenic duplication of UBE3A uncovered by adaptive nanopore sequencing. Clin Epigenet 16, 101 (2024). https://doi.org/10.1186/s13148-024-01711-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-024-01711-0