
Abstract
Background and aims
Stroke is the leading cause of adult-onset disability. Although clinical factors influence stroke outcome, there is a significant variability among individuals that may be attributed to genetics and epigenetics, including DNA methylation (DNAm). We aimed to study the association between DNAm and stroke prognosis.
Methods and results
To that aim, we conducted a two-phase study (discovery-replication and meta-analysis) in Caucasian patients with ischemic stroke from two independent centers (BasicMar [discovery, N = 316] and St. Pau [replication, N = 92]). Functional outcome was assessed using the modified Rankin Scale (mRS) at three months after stroke, being poor outcome defined as mRS > 2. DNAm was determined using the 450K and EPIC BeadChips in whole-blood samples collected within the first 24 h. We searched for differentially methylated positions (DMPs) in 370,344 CpGs, and candidates below p-value < 10–5 were subsequently tested in the replication cohort. We then meta-analyzed DMP results from both cohorts and used them to identify differentially methylated regions (DMRs).
After doing the epigenome-wide association study, we found 29 DMPs at p-value < 10–5 and one of them was replicated: cg24391982, annotated to thrombospondin-2 (THBS2) gene (p-valuediscovery = 1.54·10–6; p-valuereplication = 9.17·10–4; p-valuemeta-analysis = 6.39·10–9). Besides, four DMRs were identified in patients with poor outcome annotated to zinc finger protein 57 homolog (ZFP57), Arachidonate 12-Lipoxygenase 12S Type (ALOX12), ABI Family Member 3 (ABI3) and Allantoicase (ALLC) genes (p-value < 1·10–9 in all cases).
Discussion
Patients with poor outcome showed a DMP at THBS2 and four DMRs annotated to ZFP57, ALOX12, ABI3 and ALLC genes. This suggests an association between stroke outcome and DNAm, which may help identify new stroke recovery mechanisms.
Similar content being viewed by others


Introduction and background
Ischemic stroke (IS) is the leading cause of adult-onset disability and the second cause of death according to current epidemiological data [1]. Specifically, one out of four individuals will experience a stroke during their life, and one-third of stroke survivors will have some degree of long-term disability [2]. Although stroke incidence has decreased during the last few decades thanks to the improvement in secondary prevention strategies, its prevalence is higher due to the increase in life expectancy [3], causing significant direct and indirect economic costs for the healthcare system and reducing the quality of life of patients and caregivers [4].
After the acute phase, there is a huge variation in stroke outcome among patients. A significant proportion of this variation can be explained considering clinical variables such as history of vascular risk factors, clinical management, previous cerebrovascular burden, among others [5]. However, even after considering these factors, there is still a considerable degree of variability between individuals with similar clinical conditions, which is thought to be caused by genetics [6]. Previous research indeed found 89 loci associated with stroke risk [7]. Role of genetics in stroke recovery, on the other hand, has been less studied, and current knowledge is mostly restricted to two genome-wide association studies (GWAS) [8, 9], which provided insight on the biological mechanisms involved in stroke outcome and proposed potential therapeutical targets.
Moreover, during the last decade there has been a growing interest in stroke epigenetics, which are hereditable and modifiable factors that regulate gene expression without altering DNA sequence [10]. DNA methylation (DNAm), as the addition of a methyl group in a cytosine-phosphate-guanine context (CpG), is related to gene silencing or expression, and it represents the most studied epigenetic mechanism in stroke research. A recent article indeed confirmed specific DNAm signatures in patients with stroke as compared to individuals without [11]. However, epigenetics of stroke recovery has been rarely studied, and only one previous project interrogated the role of DNAm in early neurological evolution, as the difference between initial and at discharge National Institute of Health Stroke Scale (NIHSS). This study found that differences in the methylation levels in the EXOC4 gene yielded a worse neurological course after stroke [12]. On the other hand, to our better knowledge, no study has reported a relationship between DNAm and 3-month outcome, as measured by the modified Rankin Scale (mRS), which might provide important insight into the epigenetic regulation of stroke recovery after the acute phase. Therefore, our main objective was to analyze the relation between DNAm and stroke outcome to achieve a better understanding on the role of epigenetics in stroke recovery.
Methods
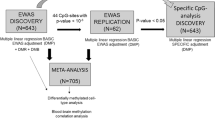
A two-phase EWAS was conducted in stroke patients to identify differentially methylated positions (DMPs) associated with stroke outcome. The study consisted of a discovery-replication phases followed by a meta-analysis. Subsequently, we explored the differentially methylated regions (DMRs) and the enriched biological pathways. We show a visual representation of the study design in Fig. 1. Full methods are available in supplemental material.

Study design diagram. Discovery sample was conformed by two cohorts of patients with stroke, BasicMar-1 (450K Illumina Chip) and BasicMar-2 (EPIC Illumina Chip). Replication phase consisted in two cohorts as well (St. Pau-1, 450 K Illumina Chip; and St. Pau-2, EPIC Illumina Chip). In both cases we filtered cases based on strict stringent criteria defined in a previous study (supplemental methods). We then searched Differentially Methylated Positions (DMPs) in the discovery cohort, and those CpG candidates having a p-value < 10–5 were tested for significance in the replication phase. Both stages were meta-analyzed to search for differentially methylated regions (DMRs) and biological pathways. Gene expression analyses were ran for the replicated DMPs. *In the replication phase, patients could not be rule out according to previous functional status and stroke location, as this cohort did not collect these data
Setting and participants
Discovery sample
European-ancestry IS patients were selected from 2 cohorts nested in the BasicMar register (BasicMar-1 [N = 619] & BasicMar-2 [N = 380]) following strict inclusion criteria [9]: (1) acute anterior ischemic stroke patients; (2) modified Rankin Scale (mRS) < 3; (3) initial NIHSS > 2. The exclusion criteria were: (1) lacunar stroke etiology and (2) recurrence of stroke during the follow-up period. For more details consult supplementary methods. A total of 323 subjects met these inclusion and exclusion criteria (Fig. 1, discovery sample).
Replication sample
Replication sample consisted in a subset of 92 patients who belong to the St. Pau cohort (St. Pau-1 [N = 29] & St. Pau-2 [N = 63]). Selection criteria were the same as discovery sample except for the following data, which was not available in the replication cohort: (1) previous functional status; (2) whether the infarct was posterior or anterior. Therefore, patients were not rule-out based on these criteria.
Clinical severity and functional outcome
Stroke severity and functional evaluation were assessed by stroke neurologists. Clinical severity was evaluated using the NIHSS [13] upon arrival at the hospital, at 24 h and at 3 months after stroke onset. Previous stroke functional outcome was scored according to the mRS [14]. The primary endpoint was the functional outcome after 3 months. Poor outcome was defined as a mRS score from 3 to 6. Etiological stroke subtypes were classified according to TOAST criteria [15].
DNA methylation quantification
DNA was extracted from whole peripheral blood collected in 10-mL EDTA tubes during the first 24 h after stroke onset. For the discovery study, BasicMar-1 (N = 252), DNAm was analyzed using the Human Methylation 450 K Beadchip (Illumina, Netherlands, Eindhoven; more than 450,000 CpGs) in two different technical runs, while for BasicMar-2 (N = 71) we used the Infinium Methylation EPIC Beadchip (Illumina, Netherlands, Eindhoven; more than 850,000 CpGs) in one technical run. Therefore, a total of 323 subjects were included in the discovery phase before quality controls (Fig. 1). For the replication study, we used the 450 K Beadchip for St. Pau-1 (N = 29) and the EPIC Beadchip for St. Pau-2 (N = 63).
DNA methylation quality controls
Intensity files from both studies were loaded using the Minfi library [16]. We then calculated β values, which range from 0 (completely unmethylated CpG) to 1 (completely methylated). We continued applying a series of quality controls (QCs) at the sample and probe level. Briefly, we excluded not detected probes (detection p-values > 0.05 or beadcount lower than 5 in 5% of samples) or that were located at allosomal, multihit or polymorphic positions. Regarding samples, we excluded those showing sex mismatch or a low call rate (less than 98%). For each batch we normalized the β matrix using the beta-mixture quantile normalization method and merged these batches into two strata: discovery and replication samples [17]. For each stratum we corrected the batch effect and adjusted for the estimated cellular counts (see more details in supplementary methods). After this set of QCs, we ended having a discovery sample composed of 316 individuals and 370,344 CpGs (supplementary Table S1), while in replication phase we had 92 individuals and 358,834 CpGs.
Gene expression quantification
Eighteen individuals from the BasicMar cohort, fulfilling the same stringent selection criteria as stated previously, were assessed for whole transcriptome expression in peripheral blood samples collected in PAXgene tubes (Qiagen) at 6 h, 24 h and 3 month post-stroke and stored at -80ºC until further use. Briefly, total RNA was isolated using the PAXgene Blood RNA extraction Kit (Qiagen) and analyzed with the GeneChip Human Gene 2.0 ST (Affymetrix) at the Microarray Analysis Service of Hospital del Mar Research Institute with Ovation WB Solution commercial kit (NuGEN). After quantity and quality check, mRNA samples were converted to complementary DNA (cDNA), labeled, hybridized, washed and scanned to generate CEL data files, following standard protocols. Importantly, in seven of these patients we also had DNAm data.
Statistical analysis and bioinformatics
Descriptive analyses
Data were expressed as mean (± standard deviation), median (interquartile range) or count (percentage) according to the type and distribution of each variable. Main demographic, clinical and outcome variables were compared between patients with good (mRS 0 to 2) and poor prognosis (mRS 3 to 6) using t-, U Mann–Whitney or χ2 tests, as appropriate. To know which of these factors were independently associated with stroke outcome we built a logistic regression model using a forward-stepwise algorithm based on Akaike information criterion (AIC).
Differentially methylation positions
We firstly explored whether patients with poor stroke outcome presented DMPs. To that aim, we first build linear models in the discovery sample in which methylation at the CpG level was the dependent variable and dichotomized mRS was the independent variable of interest. This set of models were additionally adjusted for: age, sex, smoking habit, type 2 diabetes (DM2), hypertension (HT), dyslipidemia, NIHSS at 24 h and previous mRS. These covariables were selected based on previous analyses (see Sect. "Descriptive analyses": previous mRS, NIHSS at 24 h and age) or previous literature reporting associations between vascular risk factors and DNAm (smoking, HT, DM2 and dyslipidemia) [18, 19]. We used 24-h NIHSS instead of initial NIHSS to account for the effect of treatment on stroke prognosis. We set α value at 0.05 and epigenome-wide significance at 10–7 (Bonferroni adjustment). Nominal significance was set at 10–5, and those CpGs under this cutoff were considered as candidates to be replicated. Besides, we conducted a bootstrap to test the robustness of our results in the discovery study and a Bayesian method to correct p-values for statistic inflation (Bacon library, see the supplementary methods) [20]. By applying these adjustments, we accounted for two of the main sources of bias in EWAS: influential cases and test-statistic inflation. Finally, we checked whether the relationship between stroke outcome and DNAm was moderated by relevant variables such as blood cell fractions and stroke subtype (see the supplementary methods).
Candidates of interest (p-value < 10–5) were then tested in the replication cohort. Models were constructed adjusting for the same set of variables as in the discovery study. All results were adjusted for multiple testing at this stage (false discovery rate, Benjamini & Hochberg method). Finally, all candidates were annotated using the Illumina manifest data and the GREAT software [21]. Additionally, we checked the methylation levels by mRS levels (from 0 to 5–6) of CpGs that were nominally significant at both stages to infer whether these DMPs were ordinally associated with mRS.
We additionally combined full epigenome-wide results coming from both cohorts using the METAL software [22]. We used a fixed effects model weighted by the number of subjects from each cohort. Epigenome-wide and nominal significance levels were set using the same cutoffs as described above. We considered a candidate as replicated when Q-value was lower than 0.05 in the replication phase and meta-analysis p-value was significant at the epigenome-wide level (10–7) [12].
Post-EWAS analyses
We compared the longitudinal expression profiles (at 6 h, 24 h and 3 months) between patients with good and poor outcomes for the genes annotated to the replicated CpGs. This analysis aimed to confirm whether DMPs corresponded to changes in gene expression levels. See the supplementary methods for additional details on the statistical analysis.
In addition, using the meta-analyzed p-values, we searched for differentially methylated regions and biological pathways in patients with poor outcome as described in the supplemental methods.
Results
Descriptive analysis
Discovery sample
We included 316 patients in the discovery study (Fig. 1). Patients had a median age of 77.5 years (Q1-Q3 = 69.0 to 83.0) and 166 (52.5%) had a poor stroke outcome (mRS > 2) at 3 months after stroke onset, as summarized in Table 1. In the univariate analyses, patients with poor prognosis were older, more likely to be men and had a higher initial and 24-h stroke severity (Table 1). When we built a multivariate logistic regression model, NIHSS at 24 h, previous mRS and age remained as independent factors significantly associated with poor prognosis (supplementary Table S2).
Replication sample
We recruited 92 participants for the replication stage (Fig. 1). Main characteristics of these subjects can be found in supplementary Table S3. There were 55 (59.8%) participants with a poor stroke outcome, and average age was 76.5 (Q1-Q3 = 69.0 to 81.0). Variables associated with poor stroke outcome were similar as those observed in the discovery phase (Table 1 & supplementary Table S3). When we compared both cohorts, we only observed that patients in the discovery sample had a higher prevalence of hypertension (discovery vs replication: 236 [74.7%] vs 54 [58.7%]), diabetes mellitus (129 [40.8%] vs 15 [16.3%]) and were more likely to be active smokers (85 [26.9%] vs 12 [13%]). We observed no differences between cohorts in terms of stroke prognosis, demographic variables, other vascular risk factors and stroke severity (p-value > 0.05 in all cases).
Differentially methylated positions
Discovery stage
After applying quality controls, we tested the association between 370,344 CpGs and stroke outcome (good vs poor outcome) in the discovery sample (supplementary Table S1). We found 29 nominally significant CpGs (p-value < 10–5), which were considered as candidates to be replicated (Fig. 2 & supplementary Table S4), most of them being hypomethylated in patients with poor outcome (Fig. 3A). Moreover, nominally significant CpGs were more frequently located within the gene body compared to non-significant CpGs (Fig. 3B). Only one of these CpGs, annotated at the promotor region of Gamma-Aminobutyric Acid Type A receptor subunit beta 3 gene (GABRB3), was significant at the genome-wide level (p-value < 10–7, Fig. 2 & supplementary Table S4). Differences in methylation between patients with good and poor prognosis in this set of 29 nominally significant CpGs are displayed in supplementary Figure S1.

Epigenome-wide association study of stroke outcome. Manhattan plots. The blue and pink Manhattan plots correspond to the discovery and meta-analysis stages, respectively. Red solid line represents the cutoff for epigenome-wide significance, while the green dashed line for nominal significance (10–5). The annotated hit (THBS2) was validated in the replication study (p-value < 0.05)

Differentially methylated positions. A Bar plots showing the proportion of hyper- and hypomethylated CpGs in patients with poor outcome by CpG statistical significance (10–5). B Localization of CpGs by statistical significance (10–5). C Results obtained in the replication phase in the subset of nominally significant CpG. Dots correspond to β-coefficients and error bars to 95% confidence interval. Blue CpGs are those significant candidates after correcting for multiple testing. D Dots represent marginal means of multivariate models interrogating the association between methylation at these sites and stroke outcome for both discovery and replication studies. Error bars correspond to 99% confidence interval
To correct for the influence of outlier observations, we ran a bootstrap in nominally associated CpGs (N = 29) and all of them remained significant (supplementary Figure S2). On the other hand, when we corrected our results by test-statistic inflation we found that only 6 out of 29 CpGs remained nominally significant (p-value < 10–5) after the adjustment (supplementary Table S4 & supplementary Figure S3).
We also checked whether the relationship between methylation at these 29 CpG sites and stroke outcome was moderated by blood cell factions, finding that only 2 CpGs (cg14092276 & cg06710648) were differentially hypomethylated only in granulocyte cells (supplementary Table S5). Finally, none of these 29 candidates CpGs showed a significant interaction with stroke subtype (supplementary Table S6), which suggests that differences in DNA methylation between patients with good and poor prognosis were constant across stroke etiologies.
Replication stage
The 29 candidate DMPs from the discovery stage were tested for association with poor stroke outcome in the replication sample. Two of these CpGs, cg24391982 (thrombospondin-2 [THBS2] gene) and cg21900495 (insulin promoter factor 1 [PDX1]), were nominally significant (p-value < 0.05), and the direction of the effect was consistent with that observed in the discovery stage (Fig. 3C & supplementary Table S4). However, only cg24391982 (THBS2) was significant after correcting for multiple testing in the replication stage (false discovery rate, Q-value < 0.05; Fig. 3C & supplementary Table S4). Both candidates were hypomethylated in patients with poor outcome and were located at the gene body according to the Illumina manifest. Differences in methylation signal between patients with good and poor prognosis for both candidates and both stages are represented in Fig. 3D. In supplementary Figure S4 we show the methylation signal at these CpGs for each value of the mRS, observing similar results.
Meta-analysis
Discovery and replication results were finally meta-analyzed in a fixed effect model, and we found that cg24391982 (THBS2) was significant at the genome-wide level (p-value = 6.39·10–9), while cg21900495 was only nominally significant (p-value = 4.0·10–7, Fig. 2 & supplementary Table S4). We also found 10 DMPs at nominal significance (p-value < 10–5; supplementary Table S7). One of these DMPs (cg16805094) was also annotated to THBS2 gene, and in supplementary Figure S5 we show the meta-analyzed p-values obtained in the full region of this gene.
Expression analyses
We also studied the longitudinal expression of THBS2 in a small sample with gene expression data comprising 13 and 5 patients with good (72.2%) and poor prognosis (28.8%), respectively (N = 18, supplementary Table S8). There was no significant main effect of time on THBS2 expression (p-value = 0.068). Similarly, the interaction between time and stroke outcome was not statistically significant (p-value = 0.753), suggesting that patients with poor and good outcomes followed similar trajectories over time. However, patients with poor outcomes showed a higher expression of THBS2 at 24 h and 3 months post-stroke (p-value < 0.05, Fig. 4A). We also conducted this analysis for PDX1, which was only nominally significant, finding no significant results (Fig. 4A).

Gene expression analysis. Longitudinal expression of genes annotated to CpGs that were replicated. A THBS1 and PDX1 expression levels at 6 h, 24 h and 3 months post-stroke in patients with good (blue, N = 13) and poor (red, N = 5) outcome. Models were adjusted for age and sex. B Correlation between DNAm and gene expression (N = 7). *p-value ≤ 0.05
In seven individuals, we had both DNAm and gene expression data. Figure 4B shows the correlation between DNAm at cg24391982 and the expression of the THBS2 gene. Although the correlation was not statistically significant, we observed a trend toward significance, indicating that hypermethylation was associated with THBS2 silencing (r = -0.75, p-value = 0.087). Additionally, we analyzed the correlation between DNAm at cg21900495 and the expression of the PDX1 gene, finding no significant results (Fig. 4B).
Differentially methylated regions
We explored whether existed differentially methylated regions (DMRs) using the meta-analyzed p-values as input. We show the significant DMRs in supplementary Table S9. After correcting for multiple testing, we found 4 DMRs that were annotated to the following genes: zinc finger protein 57 homolog (ZFP57, 19 CpGs), Arachidonate 12-Lipoxygenase 12S Type (ALOX12, 11 CpGs), ABI Family Member 3 (ABI3, 6 CpGs) and Allantoicase (ALLC, 5 CpGs). We also found one DMR that was marginally significant (Q-value = 0.052), which was annotated to the gene Homeobox A5 (HOXA5, 8 CpGs). In Fig. 5 we show the locus plots for these significant DMRs (Q-value < 0.05). As expected, CpGs that conformed each region were positively correlated. DMRs annotated to ZFP57, ALOX12 and ABI3 were located at the gene body, while ALLC was an intergenic region.

Differentially methylated regions in patients with poor stroke outcome. Region plots showing the four significant DMRs associated with stroke outcome in the meta-analysis (Q-value < 0.05). Each dot represents one CpG conforming the region. The Y-axis corresponds to the -log10 p-value and the X-axis to the CpG location. The dot color corresponds to the correlation of each CpG with the most significant CpG of each region (red indicates a positive correlation, while blue an inverse correlation). At the bottom of each plot there is the correlation matrix of the methylation levels of those CpGs conforming the region
Gene set enrichment analysis
We observed no significant gene set enrichment in the meta-analyses results after adjusting for multiple testing. In supplementary Figure S6 we show top candidate gene sets for Gene-Ontologies and Reactome (Q-value < 0.25). Among top candidates of Reactome database, we found interleukin-27 signaling (p-value = 1.0·10–4) and presynaptic function of Kainate receptors (p-value = 4.7·10–4), but none of these was significant after adjusting for multiple testing (supplementary Figure S6).
Discussion
This is the first study that explored how whole blood DNAm measured during the acute phase conditions the functional status at 3 months after stroke onset. The study consisted of a two-phase discovery and replication studies and a meta-analysis. It reports that hypomethylation at CpG cg24391982 (THBS2) is associated with poor stroke outcome (mRS > 2). Besides, our study also identified several DMRs annotated to the following genes: ZFP57, ALOX12, ABI3, ALLC.
The most consistent finding in our study is the association between methylation at THBS2 gene and stroke outcome. THBS2, or alternatively TSP-2, is a member of the thrombospondin subgroup A family, together with thrombospondin-1 (THBS1), which are angiostatic factors involved in angiogenesis inhibition, but also in synaptogenesis and cell–matrix interactions [23]. Previous studies in humans reported that this protein shows specific temporal profiles within the acute phase, being upregulated in patients with stroke as compared to controls [24]. Other studies conducted in patients at increased cardiovascular risk found that higher THBS2 expression was related to cardiovascular mortality and adverse cardiovascular events [25]. Similarly, levels of THBS1 have been linked to adverse outcomes and complications after aneurysmal subarachnoid hemorrhage [26]. In our study we observed that patients with a poor stroke outcome showed lower methylation levels as compared to patients with good prognosis. In general terms, hypomethylation is associated with increased gene expression, but this might depend on the localization of the CpG respective to the gene, so we cannot draw a causal conclusion [10]. When we studied gene expression, we found that patients with poor outcome showed a higher THBS2 expression at 24 h and 3 months after stroke onset, in line with the previous literature. However, our gene expression data were limited to a small group of participants. Further larger studies tracking patients throughout the entire acute phase and collecting data on both DNA methylation and THBS2 expression will provide better insight into the role of this gene in stroke prognosis and its potential as a therapeutic target.
Another interesting DMP was annotated to PDX1 gene, which is a transcriptional activator at several genes, including insulin, somatostatin and glucose transporter type 2 [27]. Variants in this gene have been associated with increased risk of diabetes mellitus and hypertension [27, 28]. Both hyperglycemia and elevated blood pressure have been associated with a worse recovery after stroke, which might explain the link between methylation at PDX1 gene and poor stroke outcome in our study [29, 30]. However, results about this locus should be interpreted with caution because this DMP was only nominally significant. Moreover, we observed no differences in PDX1 expression between patients with poor and good outcome.
We also found several DMRs significantly associated with stroke outcome. One of them was annotated to the ALOX12 gene, a member of the lipoxygenase enzymes family, which are implicated in both pro- and anti-atherogenic processes [31]. Kim et al. (2020) found that ALOX12 displayed higher methylation levels in plaques than in non-plaque intima [32], in line with results from Portilla-Fernández and collaborators (2020), who reported a DMR at ALOX12 gene associated with carotid intima media thickness [33]. Carotid intima media thickness, in turn, is known to predict 3-month outcome as measured by mRS [34], linking our results with those relating atherogenesis to ALOX12 activity.
We also observed 3 additional DMRs annotated to ZFP57, ALLC, ABI3, and all of them have been described to be involved in Alzheimer’s disease (AD) [35,36,37,38]. For instance, hypermethylation at ALLC has been associated with advanced Braak stages [35, 36]. Similarly, Li et al. (2021) described a DMR at gene ZFP57 in patients with AD showing a clinical progression within a follow-up. Finally, rare coding variants in ABI3, a gene highly expressed in microglia, were associated with increased risk of AD in a case–control study [38]. Cognitive impairment is one of the main consequences of stroke, such that 20 to 30% of stroke survivors show some degree of cognitive impairment [39]. Our results might suggest that methylation at these specific sites might be contributing to a worse stroke outcome via a decline in cognitive function.
As strengths of the study, it is worth highlighting first its robust design, based on two-phases, discovery and replication studies followed by a meta-analysis. Secondly, the solid selection criteria in the discovery sample are nested in a cohort of well-phenotyped stroke participants [9]. It has been also considered step-by-step several sources of bias in epigenomic studies, such as the influence by outlier observations, results inflation or the effect of confounding variables, and finally, the inclusion of several epigenetic approaches such as studying DMPs, DMRs and functional enrichment. Moreover, we studied the gene expression of genes annotated to replicated DMPs in a small group of participants, offering further insight into the biological significance of these DMPs.
As limitations, we could not exclude patients based on previous mRS or stroke location in the replication study, which might have added noise to the replication results, even if these participants seemed to be healthier as compared to the discovery sample. Despite the fact that it is the largest study so far to address this issue with such rigorous phenotyping and selection criteria, the sample size could be insufficient for detecting other less intense associations, especially at the replication stage. However, these two limitations could prevent finding other new associations that require higher statistical power, but do not diminish the significance or robustness of the reported associations. Moreover, our study reports associations but does not establish causality. Further experimental studies will be needed to investigate this aspect. Another recurrent limitation in epigenetic studies is the use of whole-blood samples to estimate DNAm. Although it has been reported a good correlation between whole-blood and cerebral DNA methylation in some biological processes, it is true that some specific brain-tissue mechanisms may not be detected in blood. However, there are no practical means to study brain-tissue samples during the acute phase of patients who survive a stroke event. Finally, this study involved only Caucasian individuals, and our results might not generalize to other ethnicities. Future international collaborative efforts are needed to conduct a trans-ethnic EWAS of stroke outcome.
Conclusions
Methylation at THBS2 gene, involved in angiogenesis, is associated with poor stroke outcome at 3 months. Furthermore, the regions analysis revealed four DMRs annotated to genes previously related to atherogenesis and cognitive impairment. These findings suggest an association between DNAm and stroke outcome, which might help to identify new stroke recovery mechanisms.
Availability of data
Data will be shared upon reasonable request from qualified researchers.
References
Feigin VL, Brainin M, Norrving B, Martins S, Sacco RL, Hacke W, et al. World Stroke Organization (WSO): global stroke fact sheet 2022. Int J Stroke. 2022;17:18–29.
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics-2015 update : A report from the American Heart Association. Circulation. 2015.
Feigin V, Krishnamurthi R, Parmar P, Norrving B, Mensah G, Bennett D, et al. Update on the global burden of ischaemic and. Neuroepidemiology. 2016;45:161–76.
Alvarez-Sabín J, Quintana M, Masjuan J, Oliva-Moreno J, Mar J, Gonzalez-Rojas N, et al. Economic impact of patients admitted to stroke units in Spain. Eur J Health Econ. 2017;18:449–58.
Jimenez-Conde J, Biffi A, Rahman R, Kanakis A, Butler C, Sonni S, et al. Hyperlipidemia and reduced white matter hyperintensity volume in patients with ischemic stroke. Stroke. 2010;41:437–42.
Dichgans M, Pulit SL, Rosand J. Stroke genetics: discovery, biology, and clinical applications. Lancet Neurol. 2019;18:587–99.
Mishra A, Malik R, Hachiya T, Jürgenson T, Namba S, Posner DC, et al. Stroke genetics informs drug discovery and risk prediction across ancestries. Nature. 2022;611:115–23.
Söderholm M, Pedersen A, Lorentzen E, Stanne TM, Bevan S, Olsson M, et al. Genome-wide association meta-analysis of functional outcome after ischemic stroke. Neurology. 2019;92:E1271–83.
Mola-Caminal M, Carrera C, Soriano-Tárraga C, Giralt-Steinhauer E, Díaz-Navarro RM, Tur S, et al. PATJ low frequency variants are associated with worse ischemic stroke functional outcome. Circ Res. 2019;124:114–20.
Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–68.
Soriano-Tárraga C, Lazcano U, Giralt-Steinhauer E, Avellaneda-Gómez C, Ois Á, Rodríguez-Campello A, et al. Identification of 20 novel loci associated with ischaemic stroke. Epigenome Wide Assoc Study Epigenet. 2020;15:988–97.
Cullell N, Soriano-Tárraga C, Gallego-Fábrega C, Cárcel-Márquez J, Muiño E, Llucià-Carol L, et al. Altered methylation pattern in EXOC4 is associated with stroke outcome: an epigenome-wide association study. Clin Epigenetics. 2022;14:1–17.
Lyden PD, Lu M, Levine SR, Brott TG, Broderick J. A modified national institutes of health stroke scale for use in stroke clinical trials: preliminary reliability and validity. Stroke. 2001;32:1310–6.
Farrell B, Godwin J, Richards S, Warlow C. The United Kingdom transient ischaemic attack (UK-TIA) aspirin trial: final results. J Neurol Neurosurg Psychiat. 1991;54:1044–54.
Adams HP Jr, Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL, Marsh EE 3rd. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in acute stroke treatment. Stroke. 1993;24(1):35–41.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9.
Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29:189–96.
Soriano-Tárraga C, Jiménez-Conde J, Giralt-Steinhauer E, Mola-Caminal M, Vivanco-Hidalgo RM, Ois A, et al. Epigenome-wide association study identifies TXNIP gene associated with type 2 diabetes mellitus and sustained hyperglycemia. Hum Mol Genet. 2016;25:609–19.
Carreras-Gallo N, Dwaraka VB, Cáceres A, Smith R, Mendez TL, Went H, et al. Impact of tobacco, alcohol, and marijuana on genome-wide DNA methylation and its relationship with hypertension. Epigenetics. 2023;18:2214392.
van Iterson M, van Zwet EW, Heijmans BT, Hoen PAC, van Meurs J, Jansen R, et al. Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol. 2017;18:1–13.
McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501.
Willer CJ, Li Y, Abecasis GR. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1.
Liauw J, Hoang S, Choi M, Eroglu C, Choi M, Sun GH, et al. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab. 2008;28:1722–32.
Navarro-Sobrino M, Rosell A, Hernández-Guillamon M, Penalba A, Boada C, Domingues-Montanari S, et al. A large screening of angiogenesis biomarkers and their association with neurological outcome after ischemic stroke. Atherosclerosis. 2011;216:205–11.
Golledge J, Clancy P, Hankey GJ, Norman PE. Relation between serum thrombospondin-2 and cardiovascular mortality in older men screened for abdominal aortic aneurysm. Am J Cardiol. 2013;111:1800–4.
Chen Q, Ye ZN, Liu JP, Zhang ZH, Zhou CH, Wang Y, et al. Elevated cerebrospinal fluid levels of thrombospondin-1 correlate with adverse clinical outcome in patients with aneurysmal subarachnoid hemorrhage. J Neurol Sci. 2016;369:126–30.
Yamada Y, Matsui K, Takeuchi I, Oguri M, Fujimaki T. Association of genetic variants with hypertension in a longitudinal population-based genetic epidemiological study. Int J Mol Med. 2015;35:1189–98.
Steinthorsdottir V, Thorleifsson G, Sulem P, Helgason H, Grarup N, Sigurdsson A, et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat Genet. 2014;46:294–8.
Maiër B, Kubis N. Hypertension and its impact on stroke recovery: from a vascular to a parenchymal overview. Neural Plast. 2019;2019.
Dziedzic T, Pera J, Trabka-Janik E, Szczudlik A, Slowik A. The impact of postadmission glycemia on stroke outcome: glucose normalisation is associated with better survival. Atherosclerosis. 2010;211:584–8.
Gertow K, Nobili E, Folkersen L, Newman JW, Pedersen TL, Ekstrand J, et al. 12- and 15-lipoxygenases in human carotid atherosclerotic lesions: Associations with cerebrovascular symptoms. Atherosclerosis. 2011;215:411–6.
Kim JY, Choi BG, Jelinek J, Kim DH, Lee SH, Cho K, et al. Promoter methylation changes in ALOX12 and AIRE1: novel epigenetic markers for atherosclerosis. Clin Epigenet. 2020;12:1–13.
Portilla-Fernández E, Hwang SJ, Wilson R, Maddock J, Hill WD, Teumer A, et al. Meta-analysis of epigenome-wide association studies of carotid intima-media thickness. Eur J Epidemiol. 2021;36:1143–55.
Lehmann ALCF, Alfieri DF, de Araújo MCM, Trevisani ER, Nagao MR, Pesente FS, et al. Carotid intima media thickness measurements coupled with stroke severity strongly predict short-term outcome in patients with acute ischemic stroke: a machine learning study. Metab Brain Dis. 2021;36:1747–61.
Zhang L, Silva TC, Young JI, Gomez L, Schmidt MA, Hamilton-Nelson KL, et al. Epigenome-wide meta-analysis of DNA methylation differences in prefrontal cortex implicates the immune processes in Alzheimer’s disease. Nat Commun. 2020;11.
Smith AR, Smith RG, Pishva E, Hannon E, Roubroeks JAY, Burrage J, et al. Parallel profiling of DNA methylation and hydroxymethylation highlights neuropathology-associated epigenetic variation in Alzheimer’s disease. Clin Epigenet. 2019;11:1–13.
Li QS, Vasanthakumar A, Davis JW, Idler KB, Nho K, Waring JF, et al. Association of peripheral blood DNA methylation level with Alzheimer’s disease progression. Clin Epigenet. 2021;13:1–16.
Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. 2017;49:1373–84.
Kalaria RN, Akinyemi R, Ihara M. Stroke injury, cognitive impairment and vascular dementia. Biochim Biophys Acta Mol Basis Dis. 2016;1862:915–25.
Acknowledgements
We want to show our gratitude to patients and their families, who collaborated in this study with their time and efforts.
Funding
This work was supported by grants from the Spanish Ministry of Science and Innovation, Instituto de Salud Carlos III with the grants “Registro BASICMAR” Funding for Research in Health (PI051737), Fondos de Investigación Sanitaria ISC III (PI12/01238), (PI15/00451), (PI18/00022), (PI21/00593); Sara Borrell program, funded by Instituto de Salud Carlos III (CD22/00001, J.J.-B.); and Fondos FEDER/EDRF Spanish stroke research network INVICTUS + (RD16/0019/0002) and Grant “RICORS-ICTUS” (RD21/0006/0021) funded by Instituto de Salud Carlos III (ISCIII), and by the European Union NextGenerationEU, Mecanismo para la Recuperación y la Resiliencia (MRR). Additional support provided by the Fundació la Marató TV3 with the grant “GOD’s project. Genestroke Consortium” (76/C/2011) and Recercaixa’13 (JJ086116). Fundings were received from National Institute of Health, SiGN study, The NINDS Stroke Genetics Network Study (U01NS069208) and CaNVAS (1R01NS114045-01).
Author information
Authors and Affiliations
Contributions
J.J.B, I.F.P & CGF conducted the analyses. J.J.B. built the figures and wrote the first draft of the manuscript. J.J.C. and I.F.C. participated in the conceptualization of the study, acquired the funding and supervised the project. A.R.C., E.C.G. and E.G.S. participated in the data collection. A.S.P. and A.M.G. collaborated in the data curation and harmonization. L.R. and C.S.T conducted the DNA extractions and processed the DNA methylation data. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval
We declare that all the cohorts and samples involved in the study followed the national and international guidelines (Deontological Code. Declaration of Helsinki) and complied with the current personal data protection regulations, The Regulation (EU) 2016/679 of the European Parliament, and Ley Orgánica 3/2018 on protection of digital rights (LOPDPGDD). Local Institutional Review Boards (IRB) approved all study aspects.
Consent for publication
Not applicable.
Competing interests
Authors have nothing to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jiménez-Balado, J., Fernández-Pérez, I., Gallego-Fábrega, C. et al. DNA methylation and stroke prognosis: an epigenome-wide association study. Clin Epigenet 16, 75 (2024). https://doi.org/10.1186/s13148-024-01690-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-024-01690-2