
Abstract
The molecular mechanisms underlying HIV-induced inflammation, which persists even during effective long-term treatment, remain incompletely defined. Here, we studied pathogenic and nonpathogenic simian immunodeficiency virus (SIV) infections in macaques and African green monkeys, respectively. We longitudinally analyzed genome-wide DNA methylation changes in CD4 + T cells from lymph node and blood, using arrays. DNA methylation changes after SIV infection were more pronounced in lymph nodes than blood and already detected in primary infection. Differentially methylated genes in pathogenic SIV infection were enriched for Th1-signaling (e.g., RUNX3, STAT4, NFKB1) and metabolic pathways (e.g., PRKCZ). In contrast, nonpathogenic SIVagm infection induced DNA methylation in genes coding for regulatory proteins such as LAG-3, arginase-2, interleukin-21 and interleukin-31. Between 15 and 18% of genes with DNA methylation changes were differentially expressed in CD4 + T cells in vivo. Selected identified sites were validated using bisulfite pyrosequencing in an independent cohort of uninfected, viremic and SIV controller macaques. Altered DNA methylation was confirmed in blood and lymph node CD4 + T cells in viremic macaques but was notably absent from SIV controller macaques. Our study identified key genes differentially methylated already in primary infection and in tissues that could contribute to the persisting metabolic disorders and inflammation in HIV-infected individuals despite effective treatment.
Similar content being viewed by others

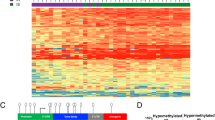
Introduction
DNA methylation, in concert with other epigenetic mechanisms, has multifaceted effects on the immune system, including T helper lineage fate determination [1], differentiation of memory cells [2], resolution of inflammation [3] and control of cellular proliferation [4]. DNA methylation can influence immune activation and inflammation as demonstrated by the observation that aberrant DNA methylation occurs in autoimmune and inflammatory diseases, such as systemic lupus erythematosus and rheumatoid arthritis [5,6,7].
HIV and SIV infections in humans and macaques (MAC), respectively, induce a chronic state of immune activation, which is the driving force behind CD4 + T cell depletion and AIDS [8]. Under efficient anti-retroviral treatment (ART), residual inflammation persists in most individuals, increasing the risk of non-AIDS comorbidity and mortality [9]. Despite intense efforts, the molecular mechanisms underlying the persistent inflammation in virologically suppressed people living with HIV (PLWH) remain incompletely characterized [10]. HIV persists in cellular and anatomical reservoirs in virologically suppressed PLWH. Recent studies have elucidated that HIV targets preferentially CD4 + T cells that display a particular metabolic profile with strong glycolytic activity [11, 12].
HIV infection of CD4 + T cells in vitro induces changes in DNA methylation of genes that regulate immune responses (interferon gamma (IFNG), interleukin-2 (IL2), forkhead box P3 (FOXP3) and programmed cell death 1 (PDCD1, the gene encoding PD-1)) [13,14,15,16]. Two studies in adults and perinatally HIV-infected children demonstrated that HIV infection is associated with changes in DNA methylation, including genes on the HLA locus in chromosome 6 [17, 18]. Another study that compared a pair of twins with discordant HIV infection status showed that several other genes are also differentially methylated, including insulin-like growth factor binding protein 6 (IGFBP6) [19]. Previous studies on HIV infection have generally been performed cross-sectionally and analyzed blood but not tissues. A genome-wide, longitudinal analysis of how HIV affects the host methylome in tissue CD4 + T cells is lacking. The study of CD4 + T cells is important because not only they are the main target cell of the virus but also because of their abnormal activation during HIV infection; most CD4 + T cells that are abnormally activated during HIV infection are not infected. Moreover, under current ART regimens, CD4 + T cells show a diminished capacity to restore their normal metabolic function compared to CD8 + T cells, B cells and NK cells [20]. Initiation of ART during acute HIV-1 infection leads to a better control of immune activation than initiation in chronic infection [10]. However, initiation of ART, even at very early stages of infection, does not fully restore all HIV-associated disorders. For instance, ART started in Fiebig stage III of HIV-1 acute infection restores Th17 cell numbers but not their polyfunctionality [21].
African nonhuman primates are the animal reservoir of HIV. These natural hosts of SIV, such as African green monkeys (AGM) and sooty mangabeys, display similar high levels of viral load in blood and gut as untreated PLWH [10, 22,23,24,25]. However, natural hosts of SIV do not display chronic immune activation or progression to AIDS [22, 24]. After mounting an acute immune response to SIV during primary infection, natural hosts downregulate the SIV-induced immune activation [24]. This control of immune activation is maintained for their entire life span, suggesting an active, efficient and long-lasting mechanism. Moreover, unlike human or MAC, natural hosts maintain a normal T helper subset balance [10, 26, 27]. Given the robust and long-lasting control of immune activation in the natural hosts, we raise the hypothesis that specific epigenetic modifications contribute to maintaining this long-term control.
In the current study, we compared changes in DNA methylation induced by pathogenic and nonpathogenic SIV infection. We performed a longitudinal analysis to identify DNA methylation modifications induced in response to SIV infection in the two distinct animal models. The DNA methylation patterns were analyzed in a genome-wide manner from purified CD4 + T cells. We analyzed CD4 + T cells not only in blood, but also in a major organ for the education of immune responses and site of viral replication, the lymph nodes (LN). Herein, we searched for differences in DNA methylation profiles between acute and chronic infection. In order to analyze which DNA methylation profiles are associated with uncontrolled viremia and immune activation, we compared viremic MAC to two non-progressive animal models: SIV-infected AGM and SIV controller MAC. SIV controller MAC mimic humans who can spontaneously control HIV [28, 29]. In these three non-human primate models, we investigated whether SIV infection-induced changes in DNA methylation occurred in genes associated with (1) immune activation and regulation; (2) T cell differentiation; (3) anti-viral immune responses such as viral restriction factors; (4) antigen-presentation; (5) cell metabolism; and (6) cell proliferation. The study revealed novel genes regulated by DNA methylation in CD4 + T cells and specific genes differentially methylated during pathogenic and nonpathogenic SIV infection related to modulation of immune response, metabolism and tissue integrity.
Results
SIV infections course
Five AGM and five Chinese rhesus MAC were infected with SIVagm.sab92018 and SIVmac251, respectively, and followed longitudinally (Additional file 2: Table 1). The viremia levels were similar in all animals except in one MAC. Already before SIV infection, this animal did not express CD28 on its peripheral CD4 + T cells (Additional file 11 Figure 1A). CD28 is important for immune activation through the T cell receptor [30]. This animal did not control post-acute viremia, did not seroconvert or display T cell activation in blood or lymph nodes and quickly lost CD4 + T cells in the blood (Additional file 1: Figure 1B–G). We therefore excluded this animal from the epigenetic study.

Effect of SIV infection on genome-wide DNA methylation of CD4 + T cells. CD4 + T cell subsets were defined in LN based on CD95 and CD28 expression and divided into central memory (Tcm), effector memory (Tem) and naïve T cells for a AGM and b MAC. Individual samples are depicted using stacked bar charts. Circular representation of the c AGM and d MAC genomes, with differentially methylated sites for each of the conditions indicated by dots. Blue and orange dots indicate a hypomethylation and hypermethylation post-infection compared to baseline, respectively. For AGM, the tracks correspond to LN day + 14, LN chronic phase and blood chronic phase from the outside inwards. For MAC, the tracks correspond to LN day + 14, LN chronic phase, blood day + 14 and blood chronic phase from the outside inwards. The numbers of e DMP and f DMG for AGM (blue) and MAC (red) are depicted. Bars depicting LN are filled with dark red and blue, while bars depicting blood are dotted and filled with light red and blue. Hypomethylated sites are shown as negative numbers and hypermethylated as positive numbers. g The percentage of gene-associated and intergenic DMP is shown for AGM and MAC. h The distribution of DMP among gene regions is shown. LN lymph node, DMP differentially methylated probes, DMG differentially methylated genes, TSS transcription start site, UTR untranslated region, AGM African green monkeys (sabaeus), MAC Chinese rhesus macaque. The analyses included for LN and blood, respectively, 5 and 7 MAC at baseline, 4 and 3 MAC in acute infection and 13 and 10 MAC in chronic infection, as well as AGM 5 and 10 AGM at baseline, 4 and 0 in acute infection, and 8 and 9 in chronic infection
For all other animals, from both species, the infection profile was typical [25, 31, 32]. AGM and MAC showed a median viremia peak at day 9–14 post-infection (p.i.) of 8.6 × 107 and 3.7 × 106 RNA copies/mL blood, followed by a reduction in viremia in the chronic phase to 4.7 × 104 and 2.5 × 104 RNA copies/mL blood, respectively (Additional file 1: Figure 2A). The levels of viral DNA in LN were higher in AGM CD4 + T cells than in MAC CD4 + T cells in the acute phase (Additional file 1: Figure 2B). At 65 days p.i., the DNA viral load had decreased 38-fold in AGM LN compared to day 14 p.i., which corresponds to the reported viral control in LN of AGM [25, 27, 33, 34]. In contrast, MAC displayed only a 4.4-fold decrease compared to day 14 post-infection. MAC partially recovered CD4 + T cell counts (median fold-change of 0.90 at day 62 p.i. compared to baseline) after the acute phase, while AGM had a complete CD4 + T cell recovery (Additional file 1: Figure 2C). AGM showed a lower level of T cell activation in blood and LN than MAC, as expected (Additional file 1: Figure 2D–F). SIV infection led to similar increased CD4 + central memory T cells in lymph nodes in both species, which became in some animals already apparent during acute phase, but was more pronounced during the chronic phase (Fig. 1a, b).

Time and tissue-specific changes in DNA methylation of CD4 + T cells following SIV infection. a Heatmaps show methylation levels for each of the differentially methylated probes (DMP) in the rows and individual animals in the columns for MAC LN and blood. Blue indicates highly methylated and yellow indicates low levels of methylation after z scale normalization per probe. LN lymph node. a Acute phase (day14). The height of each of the heatmaps is proportional to the number of differentially methylated probes. b Venn diagram showing the number (and percentages in parentheses) of overlapping DMG in the different conditions for MAC. c Heatmaps showing methylation levels for AGM as described for MAC above. d Venn diagram showing the overlap in DMG for AGM as described for MAC above. e Venn diagram showing the overlap in DMG found in both species. AGM African green monkeys (sabaeus), MAC Chinese rhesus macaque. The number of animals included is the same as in Fig. 1
In summary, the MAC and AGM included in the epigenetic study showed virologic and immunologic parameters similar to those previously reported [25, 31, 32].
Effect of SIV infection on DNA methylation patterns in CD4 + T cells in vivo
Genome-wide changes in DNA methylation in CD4 + T cells of LN and blood were analyzed longitudinally in the 5 AGM and 4 MAC described above. DNA methylation was measured at three timepoints: before infection, during acute (day 14 p.i.) and early chronic infection (day 62–65 p.i.). In addition, we analyzed pre-infection and chronic infection samples (after day 200 p.i.) from another fourteen AGM and twelve rhesus MAC previously described (Additional file 1: Figure 3 and Additional file 2: Table 1) [32, 35,36,37].

Effect of SIV infection on the Th1 pathway using ingenuity pathway analysis. a The canonical Th1 pathway is depicted, with genes within this pathway indicated as symbols and lines indicating regulation. Genes of the canonical Th1 pathway that were differentially methylated (DMG) in MAC are shown in grey; other genes are shown in white. Genes that were differentially expressed (DEG) have a purple border. Some genes were both differentially methylated and differentially expressed following infection (DMG and DEG) and are shown in grey with a purple border. b Heatmaps of gene expression of Th1-related genes during SIV infection. Columns correspond to conditions (species, tissue, phase of infection) and rows to individual genes. The mean log2 fold-changes to baseline are indicated, with blue corresponding to increased expression and yellow to decreased expression. Six AGM (sabaeus) and six Chinese rhesus macaques were analyzed for gene expression during acute (day + 14) and chronic infection (day + 65) from blood, and six and five animals per species were analyzed for acute and chronic infection, respectively, in LN. MB = MAC blood, AB = AGM blood, ML = MAC lymph node, AL = AGM lymph node, A = acute, C = chronic
Using the Infinium HumanMethylation450K BeadChip (Illumina) by restricting the analysis to a subset of probes that target CpGs at a unique location on the MAC and AGM genomes, we were able to study methylation at 157,164 and 154,108 CpG sites for AGM and MAC, respectively [38]. As expected, genome-wide methylation pattern showed a typical bimodal distribution (Additional file 1: Figure 4A) and a strong correlation was observed when all probes were compared between MAC and AGM (Additional file 1: Figure 4B). The overall distribution within the genome of the differentially methylated sites was therefore similar between AGM and MAC.

Analysis of the changes in DNA methylation of metabolic genes and of insulin sensitivity before and after SIVmac infection in macaques. a Heatmap showing median changes in methylation levels of insulin-signaling and insulin growth factor 1/2-signaling genes during SIVmac infection. Columns correspond to individual animals (rhesus macaques) and rows to probes. Blue indicates increased methylation and yellow decreased methylation compared to baseline, defined as the median of expressions of the pre-infection samples (blood: n = 5 pre-infection, 3 acute infection, 10 chronic infection; lymph node: n = 7 pre-infection, 4 acute infection, 13 chronic infection). Probes with a statistically significant change as compared to baseline are marked by a red box. b Insulin sensitivity is shown for MAC (red, n = 6) and AGM (blue, n = 11). Individual animals are shown in light color and median in dark color. **p < 0.01 compared to baseline using Friedman test and Dunn’s multiple comparison. c Correlation between change in insulin sensitivity following SIV infection and CD4 + T cell proliferation in LN is shown. Individual animals (MAC, n = 6 in red and AGM, n = 5 in blue) and linear regression results are depicted (for all animals combined). Pearson rho and p value are shown for either MAC alone or for MAC and AGM combined
For AGM LN CD4 + T cells, we identified 143 differentially methylated probes (DMP, 79 hypermethylated and 64 hypomethylated) during the acute phase and 521 DMP (203 hypermethylated and 318 hypomethylated) during the chronic phase of SIV infection when compared to baseline (Fig. 1c, e). In MAC LN CD4 + T cells, 630 DMP (449 hypermethylated and 181 hypomethylated) were identified in the acute phase, while 214 DMP (96 hypermethylated and 118 hypomethylated) were identified in the chronic phase (Fig. 1d, e). In blood CD4 + T cells from AGM, 86 DMP were identified in the chronic phase (40 hypermethylated and 46 hypomethylated). In MAC, we identified 336 DMP in blood in the acute phase (163 hypermethylated and 173 hypomethylated) and 49 in the chronic phase (24 hypermethylated and 25 hypomethylated).
The majority of the DMP were associated with genes (DMG): 62% for MAC and 60% for AGM (Fig. 1f, g). Gene-associated DMP mapped most frequently to gene promoters and gene bodies: 30.9% and 25.2% mapped to promoters for AGM and MAC, respectively, while 54% and 60.8% of DMP mapped to gene bodies (Fig. 1h). The remainder of gene-associated DMP mapped to the first exon or 5′ UTR or 3′ UTR (Fig. 1h). The complete list of DMP and associated genes is shown in Additional file 3: Table 2.
Thus, this analysis indicates that SIV infection modifies DNA methylation pattern in a substantial number of genes within CD4 + cells. MAC and AGM both showed more changes in LN than in blood, MAC mostly during acute infection and AGM predominantly during chronic infection.
Changes in DNA methylation dependent on tissue and stage of SIV infection
We next compared the tissue-dependent changes in DNA methylation for AGM and MAC (Fig. 2a–d). Only few genes were similarly affected in blood and LN CD4 + T cells, suggesting that changes were tissue-specific. For MAC, 25 out of 750 (3.3%) DMG were found in both LN and blood. These included genes encoding cytokines and transcription factors, such as LTA (lymphotoxin alpha), FOXP1 (forkhead box protein 1) and TRIM26 (tripartite motif containing 26) (Additional file 3: Table 2). In AGM, only 2 out of 464 DMG were found in both tissues (0.4%): CDHR5 (cadherin-related family member 5) and LRRC57 (leucine-rich repeat containing 57).
We then analyzed time-dependent DNA methylation changes in blood and LN. The DNA methylation profiles in acute and chronic infection were closer to each other than to the profiles before infection in both blood and LN and for both AGM and MAC (Fig. 2). DNA methylation profiles still considerably differed between acute phase and chronic infection in both species (Fig. 2a, c). In MAC LN, 20 out of 520 DMG were found in common between acute and chronic infection (3.8%). These DMG included genes encoding proteins with known immunological roles such as KIT ligand (KITLG), Runt-related transcription factor 3 (RUNX3) and Protein kinase C zeta (PRKCZ). In MAC blood, 4 out of 260 DMG were found in common between acute and chronic infection (1.5%). These genes included the immune genes encoding major histocompatibility complex, class II, DM alpha (HLA-DMA) and interleukin-5 receptor subunit alpha (IL5RA). Similarly, in AGM LN, 23 out of 414 (5.6%) of DMG were common between both timepoints. Four out of these were small nucleolar RNAs (snoRNA): SNORA1, SNORA32, SNORA8 and snoZ40. In total, 698 (93.0%) and 439 (94.6%) of all DMG were unique to a timepoint and tissue for MAC and AGM, respectively (Fig. 2b, d).
Thirty-five DMG (3%) were identified common in both species (Fig. 2e). Genes that were common between the two species included TRIM26 and growth factor receptor-bound protein 2 (GRB2).
Taken together, the changes in DNA methylation detected following SIV infection were most often different between blood and LN, dynamic over time and species-specific. This emphasizes the need of analyzing tissues during infection. The analysis revealed differential methylation in genes, such as FOXP1, TRIM26, LTA, RUNX3 and HLA upon SIV infection.
DNA methylation changes in pathways related to Th1 in SIVmac infection and to tissue integrity in SIVagm infection
We investigated among the DMG whose pathways were specifically enriched in MAC or AGM. In the first approach, we used all genes that were identified as differentially methylated following SIV infection, combining both tissues and timepoints. The most significantly increased pathways in MAC were that of colorectal cancer metastasis signaling (p = 10–4.4), Th1 pathway (p = 10–3.2), Wnt/B-catenin signaling (p = 10–3.2) and retinoic acid receptor (RAR) activation (p = 102.7) (Table 1). These pathways included many genes related to cell proliferation and inflammation, such as the E2F transcription factor 4 (E2F4), mitogen-activated protein kinase MAP2K1, guanine nucleotide binding protein (G Protein), alpha stimulating (GNAS), Nuclear Factor Kappa B Subunit 1 (NFKB1), BCL2 Like 1 (BCL2L1) and tumor necrosis factor alpha (TNF) and also to T cell differentiation. A total of 13 genes of the Th1 and TFH pathways were differentially methylated, including signal transducer and activator of transcription 4 (STAT4), NOTCH4, inducible T cell costimulator (ICOS), nuclear factor of activated T cells (NFATC2) and major histocompatibility complex class II DO alpha (HLA-DOA) (Fig. 3).
The most significantly increased pathways altered by SIV infection in AGM corresponded for instance to neuregulin signaling (p = 10–3.3), nitric oxide signaling (p = 10–3.3) and caveolar-mediated endocytosis signaling (p = 10–3.1). These pathways included genes related to signaling and cell proliferation (protein kinase C, epidermal growth factor receptor), immunosuppression (arginase), protein folding (heat shock protein 90), endocytosis and gap junctions (Table 2).
Collectively, in SIV-infected MAC, significant alterations were found in cell cycling and Th1 genes, while in SIV-infected AGM the alterations were rather linked to cell growth, immune regulation and tissue integrity.
SIVmac infection induces DNA methylation changes in metabolic pathways in lymph nodes
Given the deleterious impact of HIV persistence and chronic immune activation on the immune responses and the relevance of the virus–host interactions within LN, we performed an enrichment analysis focusing on the LN from MAC. DMG observed during the chronic phase were compared to baseline (Table 3). Three of the most significantly enriched pathways were related to metabolism, such as phosphatase and tensin homolog (PTEN) signaling, phosphatidylinositol 3-kinase (PI3K)/AKT signaling and insulin-like growth factor 1 (IGF-1) signaling. DMG associated with these pathways included MAP2K1, PRKCZ, growth factor receptor-bound protein 2 (GRB2) and RPS6KB2 (ribosomal protein S6 kinase B2).
We further investigated the observed changes in DNA methylation in MAC regarding insulin-signaling- and IGH-signaling-related genes by looking at the 13 corresponding probes within these pathways (Fig. 4a). The majority of differential methylation was observed in LN (10 genes in LN and 1 gene in blood). Three insulin-signaling- and IGH-signaling-related genes were differentially methylated during the acute phase of SIVmac infection and 9 during chronic infection. This enrichment during the chronic phase was even more notable given that the number of total genome-wide differentially methylated probes in LN was higher during the acute than chronic SIVmac infection (Fig. 1b, c).
To test whether the changes in DNA methylation were associated with functional alterations in metabolism, we investigated insulin sensitivity before and during SIVagm and SIVmac infection. Levels of glucose and insulin were measured in both AGM and MAC, and insulin sensitivity was calculated using the Quicki index (Fig. 4b) [39]. Only MAC displayed changes in insulin sensitivity in chronic infection (p < 0.01), while AGM did not. The magnitude of this increase in insulin sensitivity correlated with levels of CD4 + T cell proliferation in LN (Fig. 4c).
To better understand the exquisite control of viral load and inflammation in AGM LN during chronic infection, we performed pathway enrichment analysis similar as described above. In contrast to MAC, we did not detect any significantly enriched pathways for AGM LN.
Thus, these findings link changes detected in DNA methylation together with functional changes in metabolism and immune activation during SIV infection. They highlight the rapid and profound metabolic changes in acute SIVmac infection in LN, contrasting with the paucity in LN during SIVagm infection.
Relation between the detected changes in DNA methylation and gene expressions
To further test the relevance of the changes in DNA methylation that we have observed during SIV infection, we analyzed if the genes that were differentially methylated also display differential expression levels. For doing so, we used transcriptome data of CD4 + T cells. These data were from a different cohort of six AGM and MAC that we previously reported [32]. The twelve animals were of the same species, infected with the same virus, same infectious dose, same infection route, in a similar environment, and CD4 + T cells from blood and LN were isolated in exactly the same way and at the same timepoints as the AGM and MAC in the current study. For AGM, a total of 2819 differentially expressed genes (DEG, adjusted p value < 0.05 and fold change > 1.5) were identified, combining all sites and infection stages. Of all AGM DMG, 77 DMG (18.4%) were in differentially expressed genes (Fig. 5a). Among these genes were IL-21, IL-31 and lymphocyte activating 3 (LAG3). For MAC, a total of 3340 differentially expressed genes were identified in total, and 105 DMG (15.2%) were in differentially expressed genes (Fig. 5b), including interleukin-1 receptor associated kinase 2 (IRAK2), protein inhibitor of activated STAT1 (PIAS1), PRKCZ, FOXP1, TNF alpha-induced protein 3 (TNFAIP3 or A20) and Wnt family member 3 (WNT3). The complete list of genes that were both differentially methylated and expressed is shown in Additional file 4: Table 3 and the methylation and expression profiles for three genes are exemplified in Fig. 5c–e.

Integration of changes in DNA methylation and changes in gene expression levels during SIV infection. a AGM and b MAC Venn diagrams showing the number of differentially methylated genes (DMG) and differentially expressed genes (DEG). Percentages within parentheses. Only genes that were present in both microarray types were included. Example plots showing methylation levels and expression levels for c AGM IL31 (cg09750599) (n = 4–10), d MAC FOXP1 (cg05384123) and e MAC SGMS1 (cg10631515) (n = 3–13). Methylation is indicated with blue (right axis) and expression with red (left axis). Mean levels are shown for lymph node (LN, dark) and blood (light). Baseline levels are denoted as day 0. *Indicates significant difference compared to pre-infection as described in the methods, with the color corresponding to its condition
To further test the relevance of the detected profiles, we also analyzed if there was a concordance between DNA methylation and gene expression changes in the Th1 pathway. Of the 71 genes included in the Th1 pathway (ingenuity pathway analysis), 33 genes were differentially expressed (DEG) following SIVmac infection (Fig. 3b). Three of these genes were both differentially methylated and differentially expressed: ICOS, NFATC2, HLA-DOA.
Overall, we observed an overlap between DNA methylation and gene expression in as much as 15–18% of the genes. The analysis reveals potential novel targets of DNA methylation during SIV infection, such as Runx3, IL21 and IL31.
Validation of DNA methylation changes in an independent animal cohort
We furthermore aimed to validate the identified differentially methylated sites using an orthogonal technology. Moreover, we investigated whether the changes in DNA methylation we observed were distinct between viremic and non-viremic SIVmac infections. Therefore, we developed bisulfite pyrosequencing assays to investigate probes that were differentially methylated during SIVmac infection. We selected 17 differentially methylated probes in 11 genes that were either: differentially methylated in more than one tissue or timepoint; and/or were also differentially expressed following infection; and/or possess a known important immunological or metabolic function. These selected genes were ICOS (affected by DNA methylation in blood during acute infection, DEG), FOXP1 (LN and blood chronic infection, DEG), LTA (LN and blood chronic infection), MAPK12 (blood acute and chronic infection), MAPK-associated protein 1 (MAPKAP1, blood chronic infection), nitrogen permease regulator 2-like protein (NPRL2) and RUNX3 (LN acute and chronic), PRKCZ (LN acute and chronic, DEG), NFKB1 and sphingomyelin synthase 2 (SGMS2, LN chronic infection). Levels of DNA methylation measured by pyrosequencing significantly correlated with those observed by microarray for 12 out of 17 probes when re-analyzing the same samples (Fig. 6).

Comparison of methylation levels by bisulfite pyro-sequencing and microarrays. Symbols denote individual animals. Forty-two samples from Chinese rhesus macaques were analyzed. Cells from LN and blood are depicted as circles and triangles, respectively. Symbol color corresponds to timepoint with pre-infection (blue), acute infection (red) and chronic infection (green) shown. Linear regression lines are shown, and spearman rho and p value s are indicated per probe
DNA methylation was then measured in CD4 + T cells isolated from LN and blood from an unrelated colony of 17 Indian rhesus macaques (Additional file 1: Figure 3). Animals were either uninfected with SIV (n = 4), or chronically infected with SIVmac239 or SIVmac251 (n = 13). Seven of the 13 SIV-infected animals controlled viremia below the limit of detection (37.5 copies/mL) and were defined as SIV controllers. The other six SIV-infected animals had a median viral load of 2.2 × 105 (range 5 × 104–3.6 × 106). FOXP1, LTA, MAPKAP, NFKB1 and RUNX3 were differentially methylated in LN between uninfected and infected viremic animals using pyrosequencing analysis, confirming our microarray results (Fig. 7a). MAPKAP, NFKB1, PRKCZ and RUNX3 methylation levels were significantly increased in viremic compared to SIV controller animals, with no significant differences between uninfected and SIV controller animals for any probe site. In blood CD4 + T cells, methylation levels in FOXP1, ICOS and LTA were significantly increased in viremic animals versus uninfected animals (Fig. 7b). Viremic animals also showed increased methylation in LTA and MAPK12 compared to SIV controller animals, with again no significant differences between uninfected and SIV controller animals for any probe site. Multidimensional scaling analysis on all methylation sites covered within the probes demonstrated that SIV-infected animals clustered separately from uninfected animals with little overlap between viremic and SIV controller animals (Fig. 7c, d).

Validation of methylation changes in an independent cohort of 17 Indian rhesus macaques. Comparison of DNA methylation levels at the 17 selected probe sites in the validation cohort by sequencing CD4 + T cells from a LN and b blood. *p < 0.05, Mann–Whitney, **p < 0.01, Mann–Whitney. c LN and d blood multidimensional scaling showing clustering of validation animals for all CpG included in the 16 bisulfite pyrosequencing assays. Symbols represent individual animals that are uninfected (red circles, n = 4), infected and viremic (green triangles, n = 6) or SIV controllers (blue squares, n = 7). Ellipses depict 50% confidence intervals. Kruskal stress is 0.14 and 0.13 for LN and blood, respectively
Taken together, the changes in DNA methylation identified using microarrays were confirmed using bisulfite sequencing in an independent animal cohort. This was the case for example for RUNX3, FOXP1, LTA and NFKB1. Moreover, the analysis also showed that in contrast to viremic macaques, the selected DNA methylation sites in blood and LN CD4 + T cells from SIV controller animals were not different from those of uninfected animals.
Discussion
In this study, we measured genome-wide DNA methylation changes following SIV infection in CD4 + T cells of two compartments (blood and LN) longitudinally at three timepoints (before, acute and chronic infection). Genes of interest were confirmed as being differentially methylated using both an independent cohort of MAC and an ortholog method (bisulfite pyrosequencing). Functional relevance of the identified changes in DNA methylation was further confirmed in vivo by gene expression analysis and blood insulin sensitivity analyses. In viremic MAC, differentially methylated genes were found to be enriched in Th1 differentiation and metabolic pathways, including IGF-1 signaling (Fig. 8). The analyses further showed that AGM display DNA methylation patterns different from MAC upon SIV infection with preferential changes in genes associated with immune regulation and tissue growth or repair. SIV controllers displayed a distinct profile that was similar to the non-infected animals. DNA methylation changes were identified already in primary infection and were more abundant in lymph node than blood CD4 + T cells.

Model depicting DNA print changes in pathogenic and nonpathogenic SIV infection. MAC macaque, AGM African green monkeys
One potential limitation of this study is that human microarrays were used to measure DNA methylation on non-human primate genomes. However, alignment and stringent quality control of the included probes to the AGM and MAC genomes revealed that still 162,053 and 158,970 probes could be confidently used covering 16,295 and 16,611 genes, respectively [38]. Of those, 157,164 and 154,108 probes were detected with high confidence in the current study. Moreover, we confirmed differential DNA methylation in genes such as FOXP1, LTA, MAPKAP, NFKB1, PRKCZ and RUNX3 by bisulfite pyrosequencing. In addition, some genes for which methylation patterns were particularly stable over time after infection (HLA, detected both in acute and in chronic in SIV-infected macaques) correspond to those also reported as most often differentially methylated in HIV-infected individuals in a study of whole blood during chronic infection [17]. Similarly, CpG associated with ICOS, FOXP1, HLA-II, NOTCH4 and TRIM were found differentially methylated in whole blood from HIV-infected children under anti-retroviral therapy compared to non-infected children [18]. The fact that our findings in part replicate earlier work and that we identified similar genes in independent animals gives further credence to this study and earlier reports.
Another limitation to our study was that DNA methylation and gene expression were not performed on the same samples, but on two separate cohorts. This prevented us from fully integrating changes in DNA methylation and gene expression on an individual animal level, and we therefore were restricted to comparing gene lists. Around 15–18% of the genes that were differentially methylated after SIV infection were also differentially expressed. The range was the same between AGM and MAC and also similar to those described in previous reports [40,41,42]. Depending on location, DNA methylation can activate or repress gene expression or lead to alternative splicing [43]. In MAC, DMG were enriched for Th1 differentiation, which was mirrored by changes in gene expression in the same pathway. In particular, three genes (ICOS, NFATC2 and HLA-DOA) were both differentially methylated and expressed. Moreover, several key transcription factors in this pathway were differentially methylated: STAT4, RUNX3 and NFKB1. This is in line with previous reports in the literature indicating a stronger Th1 response in MAC compared to natural hosts [23, 26]. These data also agree with the notion that during chronic HIV/SIVmac infection, increased levels of follicular T helper cells with Th1 characteristics (TFH1) are found, which could potentially contribute to driving the chronic interferon signature observed during pathogenic infections [44]. The observation that a gene such as RUNX3, which is important for Th1 function [45], is already differentially methylated during acute infection might be an indication that this mechanism could enforce cell fates and/or be actively involved in maintaining Th cell subset disbalance during HIV/SIV infection.
We cannot exclude that some of the alterations in DNA methylation we observed are due to dynamic changes over time in cell composition. However, DNA methylation changes were detected already during the acute phase of infection (day 14 p.i.), before we observed the largest differences in memory phenotype. Moreover, several of the genes observed as differentially methylated in our study have previously been found differentially methylated in inflammatory disorders and autoimmune diseases characterized by an interferon DNA methylation signature including systemic lupus erythematosus (LTA, RUNX3, NFATC2, PRKCZ) [46,47,48], Behçet’s disease (STAT4, RUNX3, PRKCZ) [49], multiple sclerosis (RUNX3, HLA-DOA, PRKCZ) [50, 51], primary Sjøgren’s syndrome (LTA, RUNX3, NFATC2, HLA-DOA, NFKB1, PRKCZ) [52,53,54] and rheumatoid arthritis (NFATC2, ICOS, NFKB1, PRKCZ) [55,56,57]. Furthermore, the differentially methylated genes NFATC2, STAT4, HLA-DOA, FOXP1, ICOS, LTA, MAPK12, NFKB1, PRKCZ and RUNX3 have all been identified as interferon regulated genes [58]. This indicates the existence of common activated pathways between viral-induced inflammation and non-infectious inflammatory disorders.
We show that SIV infection in MAC led to alterations in DNA methylation in metabolic pathways, including IGF-1 signaling. Previously, insulin signaling and type 2 diabetes have been found to be associated with changes in DNA methylation [59, 60]. A study in which a HIV-infected and uninfected twin were studied identified a differential DNA methylation in the gene IGFBP6, which inhibits IGF-2 signaling [19, 61]. Insulin signaling regulates the capacity of cells to take up glucose, which is the energy source for activated T cells that switch to aerobic glycolysis [62]. Indeed, insulin receptor signaling in T cells is associated with increased effector function [63]. Our study suggests an increase in insulin sensitivity during the very early chronic phase of SIVmac infection (day 62 p.i.). This contrasts with other studies showing an insulin resistance in HIV infection. However, increases in insulin sensitivity have previously been observed in HIV infection [64]. Furthermore, we studied insulin sensitivity only in the early phase of chronic SIV infection (day 63 p.i.). The alteration in CD4 + T cell DNA methylation in metabolic pathways was especially pronounced in LN. The lymphatic system plays a role in whole-body metabolism via lipid transport and is thought to be involved in the pathogenesis of obesity and type 2 diabetes [65]. In a more general perspective, our study indicates that some of the metabolic changes induced by SIV infection are epigenetically fixed already soon after infection. This might in part explain why ART generally does not revert all HIV-induced abnormalities in the CD4 + T cells or why it might take many years to fully revert.
Our study uncovered that DNA methylation patterns in CD4 + T cells of SIV controller MAC were more similar to uninfected than to viremic animals. Only a few studies have analyzed the transcriptional profile of the whole CD4 + T cell compartment in HIV controllers. Concordant with our pyrosequencing data showing no significant methylation changes in SIV controllers, gene expression profiles in HIV controllers were documented as being similar to that of uninfected subjects and different from viremic individuals [66, 67]. The different profiles in DNA methylation between SIV controller animals and viremic MAC could be either a direct consequence of SIV replication in the latter and/or its associated immune activation. However, AGMs also strongly control viral replication in LN and, despite this, showed more methylation changes than SIV controllers in LN. While DNA methylation changes were apparent in nonpathogenic SIVagm infection, these were most often not related to immunological anti-viral response pathways. Strikingly, DNA methylation changes in viremic AGM were different from those detected in viremic MAC. Pathways involved with tissue integrity and immune regulation were enriched in AGM. This could thus contribute to the lack of chronic inflammation and intact tissue structures (such as integrity of follicular dendritic cell network and lack of fibrosis in LN of AGM in contrast to MAC) [10, 68, 69]. It is in agreement with observations that AGMs are able to actively maintain integrity of the gut mucosal barrier throughout SIV infection [69,70,71]. The lack of tissue damage could help AGM to avoid disease progression as dextran sulfate sodium-induced colitis, and tissue damage during SIVagm infection was demonstrated to lead to increased viral load, microbial translocation and inflammation [72].
Another observation of interest in the AGM was the differential methylation of regulatory genes, such as LAG3, IL21 and IL31 in AGM. The IL-31 cytokine is known to be mainly secreted by activated CD4 + T cells, often Th2 cells, acting on a broad range of immune and non-immune cells. IL-31 is involved in a variety of fundamental physiological processes, such as cell growth, metabolism and immune regulation [73]. IL-21 is known to be secreted by TFH cells in LN and upregulated in Th17 and Th2 cells. IL-21 is reduced in HIV/SIV infections and used as immunotherapy in preclinical trials in MAC to restore Th17 cells and reduce inflammation in the gut during SIVmac infection [74, 75]. IL-21 is known to stimulate NK and CD8 + T cell anti-tumor response [76]. Therefore, the enrichment in favor of Th2 (and not Th1 as in MAC) concomitant with eventually stronger IL-21 expression compared to MAC might contribute in AGM to the lower inflammation and/or induction of lymphocytes with improved suppressive capacity upon SIVagm infection [77].
To our knowledge, this is the first time that such a genome-wide epigenetic study has been performed in CD4 + cells from tissues longitudinally from baseline throughout a viral infection. Our study revealed that in the animal model of HIV (macaques infected by SIVmac), the differentially methylated sites are more frequent in LN than in blood in both acute and chronic phase of infection. All over, the pattern we detected could contribute to explain why ART initiation even in primary HIV-1 infection does not fully revert the metabolic CD4 + T cells back to normal [20, 21, 78]. Moreover, this study identified targets that are associated with nonpathogenic SIV infection and exploring their potential role in tissue repair or resilience could ultimately contribute to identifying novel targets for host-directed therapies to reduce tissue damage and comorbidities. Finally, some modulated pathways identified in CD4 + T cells here have also been described in inflammatory disorders. For instance, CD4 + T cells in Lupus patients show epigenetic changes in the insulin pathway [79]. Thus, our study might point to pathways commonly impacted by HIV and CD4 + T cell-associated inflammatory disorders.
In conclusion, we show that the infection with SIV led to specific changes in DNA methylation in CD4 + T cells, in particular in the LN and already during primary infection. Moreover, epigenetic changes in genes associated with Th1 cell differentiation and metabolism could be linked to pathogenic infection and were absent in aviremic macaques and natural hosts. Nonpathogenic SIVagm infection was characterized by epigenetic modifications in genes associated with immunoregulation and tissue integrity. These genes could represent novel candidates for immunotherapies targeting pathways associated with non-AIDS-related comorbidity and/or abnormal immune activation of CD4 + T cells.
Methods
Sample collection and animal infections
For the DNA methylation microarray cohort, the samples have been collected from 19 AGM (Chlorocebus sabaeus) and 16 Chinese rhesus macaques (Macaca mulatta). The AGM and Chinese rhesus macaques were infected i.v. with 250 TCID50 of SIVagm.sab92018 or 50 AID50 SIVmac251, respectively, as described previously [32]. For the pyrosequencing validation cohort, we used samples collected from 17 Indian rhesus macaques, out of which 13 were infected with repeated intra-rectal low doses of 250 TCID50 SIVmac251, an oral dose of 6000 TCID50 SIVmac239 or an intra-venous dose of 100 or 500 TCID50 SIVmac239, resulting in 6 viremic and 7 SIV controllers (Additional file 2: Table 1).
Blood was collected by venipuncture on EDTA tubes and shipped to Institut Pasteur to follow up infection parameters. Blood samples were collected at days − 53, − 49, − 35, − 7, 2, 7, 9, 14, 28, 35, 52 and chronic infection (day 62–day 1787, Additional file 2: Table 1). Peripheral lymph nodes were collected at baseline, day 14 and chronic infection (day 62–day 850, Additional file 2: Table 1). Material from the pyrosequencing cohort was collected at necropsy and cryopreserved.
CD4 + T cell isolation and DNA extraction
CD4 + T cells from peripheral blood mononuclear cells (PBMC) and LN were purified as described previously [32]. Briefly, surgically removed LN were disrupted mechanically after which CD4 + cells were isolated using magnetic anti-CD4 beads (Miltenyi). CD4 + T cell purity after isolation was confirmed using flow cytometry (median 97%, IQR 93–98%, Additional file 1: Figure 5). DNA was extracted from CD4 + T cells using the DNeasy blood and tissue kit (Qiagen), according to manufacturer’s protocol.
SIV viremia and DNA quantification
Viral RNA was measured by qPCR as described previously [32]. Viral DNA was measured in CD4 + T cells by qPCR as previously reported, using primers and probes designed specifically for SIVagm.sab and SIVmac [25, 31]. Positive and negative controls were used to exclude sample contamination. Sample preparation, enzyme mix preparation and PCR setup were performed in three separate rooms also to avoid PCR contamination. Viral copy numbers were calculated using plasmids of SIVagm and SIVmac, respectively. Primers and plasmids for CCR5 were used to normalize the viral levels to the number of cells [80].
Flow cytometry
Whole blood and LN cells in suspension were stained with the following antibodies: CD95 (DX2), CD3 (SP34-2), CD4 (L200) (all BD Biosciences), CD28 (CD28.2), CD20 (2H7) (both eBioscience) and Ki67 (MIB-1, Dako). FcR Blocking Reagent (Miltenyi) was used to block unspecific antibody binding, and live/dead cell viability dye (Invitrogen) was used to exclude dead cells. Cells were labeled with surface-binding antibodies, then fixed with 4% paraformaldehyde and permeabilized using saponin prior to incubation with Ki-67. Events were collected on a BD LSR-II flow cytometer, running BD FACS Diva 6.0 software, and analyzed with FlowJo 9.4.10 (TreeStar). Anti-mouse compensation beads (BD Biosciences) and Arc Amine Reactive Compensation Bead Kit (Life Technologies) were used to define compensation levels.
Measurement of insulin sensitivity
Plasma from EDTA tubes collected after overnight fasting was used to determine levels of insulin (ELISA, Merck Millipore) and glucose (Abcam, enzymatic assay) following manufacturer’s instructions. Insulin sensitivity was calculated from these levels using the quantitative insulin sensitivity check index (QUICKI) score: 1/(log(fasting insulin [µU/mL]) + log(fasting glucose [mg/dL])) [81].
DNA methylation microarrays and DMP calling
DNA methylation was measured at three timepoints for MAC: before infection, during acute (day 14 p.i.) and during early chronic infection (day 62–65 p.i.) (Additional file 1: Figure 3 and Additional file 2: Table 1). For AGM, we measured changes in DNA methylation in CD4 + T cells at the same three timepoints in LN and at two timepoints in blood (before infection and early chronic infection, as not enough material was available during acute infection). Additional pre-infection and chronic infection samples were also analyzed (Additional file 2: Table 1).
Bisulfite treatment was performed on 1 µg of DNA using the EpiTect Fast DNA Bisulfite kit (Qiagen), and 200 ng of bisulfite-converted DNA was hybridized to the Infinium HumanMethylation450K BeadChip according to manufacturer’s instructions.
Since the 450K BeadChip is designed for the analysis of human samples, probes compatible with the Macaca mulatta and Chlorocebus sabaeus genomes were identified, yielding > 160,000 of the 450 K probes targeting functional CpGs in the respective genomes with a maximum of a single mismatch underlying the probe sequence with the exception of the last 5 bases from the 3′ end. This resulted in the ability to study 162,053 probes for AGM and 158,970 probes for MAC, covering 16,259 and 16,611 genes, respectively [38]. The recently developed annotations files [38] were used for each species, and data were filtered and normalized (AGM and macaques separately) based on the CpG island categories of the species-specific annotation files with the SQN Pipeline [82]. All probes with a detection p value above 0.01 were removed. Following this filter, 157,164 and 154,108 probes were detected with high confidence in the current study for AGM and MAC, respectively. A threshold of 90% validated probes compared to the annotation file was set as a sample quality control, leading to the removal of one sample. Intra-individual normalization was performed using a subset quantile normalization approach based on the new functional annotation of probes followed by quantile normalization between samples.
Differentially methylated probes (DMP) were identified for each tissue by first filtering probes with a 5% difference compared to baseline in paired samples, followed by a Mann–Whitney test between baseline and the acute or chronic phase taking into account both paired samples and additional unpaired samples. Probes with a nominal p value < 0.005 and a median methylation difference > 5% were considered significant.
Bisulfite pyrosequencing
Differentially methylated probes of interest were validated using bisulfite pyrosequencing. Primers for regions of interest were designed based on the rheMac8 (UCSC) genome build, and the 1 µg of genomic DNA was bisulfited with EpiTect Fast DNA Bisulfite kit (Qiagen) following manufacturer’s instructions. Bisulfited DNA (1 µl) was amplified by PCR (see Additional file 5: Table 4 for primers sequences) with either Hot Start Taq DNA Polymerase (Qiagen) or Phusion U Hot Start PCR Master Mix (ThermoFisher) at 95 °C for 15 min, followed by 50 cycles at 95 °C for 30 s, 10 s at the respective annealing temperature and 72 °C for 20 s and a final extension step at 72 °C for 5 min. Quantitative DNA methylation analysis was performed by pyrosequencing on a PyroMark Q96 MD (Qiagen) and analyzed using the Pyro-Q-CpG software (Qiagen).
Transcriptome analysis
Transcriptomic microarray data that we previously published using the same CD4 + T cell purification method, viral inoculations and obtained at the same timepoints were reanalyzed to integrate these data with our methylome data [32]. Probe expression values were extracted using R Bioconductor. Scale normalization was performed, and control probes were removed after normalization. Aggregation of probe expression values was then performed at the gene level using median values. Differentially expressed genes were identified using a two-tailed paired Student t test (q value < 0.05 and fold change > 1.5). p value s were corrected using the FDR method. Individual fold-change expression values were calculated based on their matched control samples. Heatmaps and multidimensional scaling (MDS) were generated using R. The Kruskal stress indicated in the MDS representations indicates the quality of the representation and are proportional to the amount of information lost during the dimensionality reduction process. Functional enrichment analyses were done using ingenuity pathway analysis (IPA, Qiagen).
Statistics
Statistical inference analyses were performed using Prism 5.0 (GraphPad) and R. The Spearman rank test was used to assess the correlations between variables. Two-tailed nonparametric tests were used throughout to compare between groups.
Availability of data and materials
DNA methylation microarray data including both raw and processed data (filtered and normalized) have been deposited in GEO (GSE143708). Gene expression data were previously described [32] and are deposited in the transcriptome data repository MACE (https://mace.ihes.fr), under accession numbers 3070984318 (AGM) and 2,932,572,286 (RM). All other underlying data are provided in the manuscript or available upon request.
Abbreviations
- SIV:
-
Simian immunodeficiency virus
- PLWH:
-
People living with HIV
- AGM:
-
African green monkeys
- MAC:
-
Macaques
- LN:
-
Lymph nodes
- PBMC:
-
Peripheral blood mononuclear cells
- DMP:
-
Differentially methylated probes
- DMG:
-
Differentially methylated genes
- DEG:
-
Differentially expressed genes
- p.i.:
-
Post-infection
- snoRNA:
-
Small nucleolar RNAs
References
Makar KW, Wilson CB. DNA methylation is a nonredundant repressor of the Th2 effector program. J Immunol. 2004;173(7):4402–6.
Hashimoto S, Ogoshi K, Sasaki A, Abe J, Qu W, Nakatani Y, et al. Coordinated changes in DNA methylation in antigen-specific memory CD4 T cells. J Immunol. 2013;190(8):4076–91.
El Gazzar M, Yoza BK, Chen X, Hu J, Hawkins GA, McCall CE. G9a and HP1 couple histone and DNA methylation to TNFalpha transcription silencing during endotoxin tolerance. J Biol Chem. 2008;283(47):32198–208.
Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25(3):338–42.
Fogel O, Richard-Miceli C, Tost J. Epigenetic Changes in Chronic Inflammatory Diseases. Adv Protein Chem Struct Biol. 2017;106:139–89.
Mazzone R, Zwergel C, Artico M, Taurone S, Ralli M, Greco A, et al. The emerging role of epigenetics in human autoimmune disorders. Clin Epigenetics. 2019;11(1):34.
Wu H, Chen Y, Zhu H, Zhao M, Lu Q. The pathogenic role of dysregulated epigenetic modifications in autoimmune diseases. Front Immunol. 2019;10:2305.
Giorgi JV, Hultin LE, McKeating JA, Johnson TD, Owens B, Jacobson LP, et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis. 1999;179(4):859–70.
Sandler NG, Sereti I. Can early therapy reduce inflammation? Curr Opin HIV AIDS. 2014;9(1):72–9.
Paiardini M, Muller-Trutwin M. HIV-associated chronic immune activation. Immunol Rev. 2013;254(1):78–101.
Angin M, Volant S, Passaes C, Lecuroux C, Monceaux V, Dillies MA, et al. Metabolic plasticity of HIV-specific CD8(+) T cells is associated with enhanced antiviral potential and natural control of HIV-1 infection. Nat Metab. 2019;1(7):704–16.
Valle-Casuso JC, Angin M, Volant S, Passaes C, Monceaux V, Mikhailova A, et al. Cellular metabolism is a major determinant of HIV-1 reservoir seeding in CD4(+) T cells and offers an opportunity to tackle infection. Cell Metab. 2019;29(3):611-626.e5.
Youngblood B, Noto A, Porichis F, Akondy RS, Ndhlovu ZM, Austin JW, et al. Cutting edge: Prolonged exposure to HIV reinforces a poised epigenetic program for PD-1 expression in virus-specific CD8 T cells. J Immunol. 2013;191(2):540–4.
Nakayama-Hosoya K, Ishida T, Youngblood B, Nakamura H, Hosoya N, Koga M, et al. Epigenetic repression of interleukin-2 expression in senescent CD4+ T cells during chronic human immunodeficiency virus type-1 infection. J Infect Dis. 2014;211:28–39.
Pion M, Jaramillo-Ruiz D, Martinez A, Munoz-Fernandez MA, Correa-Rocha R. HIV infection of human regulatory T cells downregulates Foxp3 expression by increasing DNMT3b levels and DNA methylation in the FOXP3 gene. AIDS. 2013;27(13):2019–29.
Mikovits JA, Young HA, Vertino P, Issa JP, Pitha PM, Turcoski-Corrales S, et al. Infection with human immunodeficiency virus type 1 upregulates DNA methyltransferase, resulting in de novo methylation of the gamma interferon (IFN-gamma) promoter and subsequent downregulation of IFN-gamma production. Mol Cell Biol. 1998;18(9):5166–77.
Gross AM, Jaeger PA, Kreisberg JF, Licon K, Jepsen KL, Khosroheidari M, et al. Methylome-wide analysis of chronic HIV infection reveals five-year increase in biological age and epigenetic targeting of HLA. Mol Cell. 2016;62(2):157–68.
Shiau S, Strehlau R, Wang S, Violari A, Do C, Patel F, et al. Distinct epigenetic profiles in children with perinatally-acquired HIV on antiretroviral therapy. Sci Rep. 2019;9(1):10495.
Zhang Y, Li SK, Yi Yang K, Liu M, Lee N, Tang X, et al. Whole genome methylation array reveals the down-regulation of IGFBP6 and SATB2 by HIV-1. Sci Rep. 2015;5:10806.
Korencak M, Byrne M, Richter E, Schultz BT, Juszczak P, Ake JA, et al. Effect of HIV infection and antiretroviral therapy on immune cellular functions. JCI Insight. 2019;4(12):e126675. https://doi.org/10.1172/jci.insight.126675.
Schuetz A, Deleage C, Sereti I, Rerknimitr R, Phanuphak N, Phuang-Ngern Y, et al. Initiation of ART during early acute HIV infection preserves mucosal Th17 function and reverses HIV-related immune activation. PLoS Pathog. 2014;10(12):e1004543.
Chahroudi A, Bosinger SE, Vanderford TH, Paiardini M, Silvestri G. Natural SIV hosts: showing AIDS the door. Science. 2012;335(6073):1188–93.
Silvestri G, Sodora DL, Koup RA, Paiardini M, O’Neil SP, McClure HM, et al. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity. 2003;18(3):441–52.
Sodora DL, Allan JS, Apetrei C, Brenchley JM, Douek DC, Else JG, et al. Toward an AIDS vaccine: lessons from natural simian immunodeficiency virus infections of African nonhuman primate hosts. Nat Med. 2009;15(8):861–5.
Gueye A, Diop OM, Ploquin MJ, Kornfeld C, Faye A, Cumont MC, et al. Viral load in tissues during the early and chronic phase of non-pathogenic SIVagm infection. J Med Primatol. 2004;33(2):83–97.
Ploquin MJ, Desoutter JF, Santos PR, Pandrea I, Diop OM, Hosmalin A, et al. Distinct expression profiles of TGF-beta1 signaling mediators in pathogenic SIVmac and non-pathogenic SIVagm infections. Retrovirology. 2006;3:37.
Favre D, Lederer S, Kanwar B, Ma ZM, Proll S, Kasakow Z, et al. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog. 2009;5(2):e1000295.
Bruel T, Hamimi C, Dereuddre-Bosquet N, Cosma A, Shin SY, Corneau A, et al. Long-term control of simian immunodeficiency virus (SIV) in cynomolgus macaques not associated with efficient SIV-specific CD8+ T-cell responses. J Virol. 2015;89(7):3542–56.
Daskalaki M, Neumann B, Klippert A, Matz-Rensing K, Kaup FJ, Stahl-Hennig C. Long-term efficient control of SIV infection in macaques is associated with an intact intestinal barrier. J Med Primatol. 2017;46(4):144–8.
Linsley PS, Ledbetter JA. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol. 1993;11:191–212.
Diop OM, Gueye A, Dias-Tavares M, Kornfeld C, Faye A, Ave P, et al. High levels of viral replication during primary simian immunodeficiency virus SIVagm infection are rapidly and strongly controlled in African green monkeys. J Virol. 2000;74(16):7538–47.
Jacquelin B, Mayau V, Targat B, Liovat AS, Kunkel D, Petitjean G, et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Invest. 2009;119(12):3544–55.
Beer B, Scherer J, zur Megede J, Norley S, Baier M, Kurth R. Lack of dichotomy between virus load of peripheral blood and lymph nodes during long-term simian immunodeficiency virus infection of African green monkeys. Virology. 1996;219(2):367–75.
Martinot AJ, Meythaler M, Pozzi LA, Dalecki Boisvert K, Knight H, Walsh D, et al. Acute SIV infection in sooty mangabey monkeys is characterized by rapid virus clearance from lymph nodes and absence of productive infection in germinal centers. PLoS ONE. 2013;8(3):e57785.
Jacquelin B, Petitjean G, Kunkel D, Liovat AS, Jochems SP, Rogers KA, et al. Innate immune responses and rapid control of inflammation in African green monkeys treated or not with interferon-alpha during primary SIVagm infection. PLoS Pathog. 2014;10(7):e1004241.
Parker R, Dutrieux J, Beq S, Lemercier B, Rozlan S, Fabre-Mersseman V, et al. Interleukin-7 treatment counteracts IFN-alpha therapy-induced lymphopenia and stimulates SIV-specific cytotoxic T lymphocyte responses in SIV-infected rhesus macaques. Blood. 2010;116(25):5589–99.
Dutrieux J, Fabre-Mersseman V, Charmeteau-De Muylder B, Rancez M, Ponte R, Rozlan S, et al. Modified interferon-alpha subtypes production and chemokine networks in the thymus during acute simian immunodeficiency virus infection, impact on thymopoiesis. Aids. 2014;28(8):1101–13.
Pichon F, Busato F, Jochems S, Jacquelin B, Le Grand R, Deleuze J-F, et al. Analysis and annotation of genome-wide DNA methylation patterns in two nonhuman primate species using the Infinium Human Methylation 450K and EPIC BeadChips. 2020:2020.05.09.081547.
Quon MJ. QUICKI is a useful and accurate index of insulin sensitivity. J Clin Endocrinol Metab. 2002;87(2):949–51.
Chen K, Zhao BS, He C. Nucleic acid modifications in regulation of gene expression. Cell Chem Biol. 2016;23(1):74–85.
Hannon E, Gorrie-Stone TJ, Smart MC, Burrage J, Hughes A, Bao Y, et al. Leveraging DNA-methylation quantitative-trait loci to characterize the relationship between methylomic variation, gene expression, and complex traits. Am J Hum Genet. 2018;103(5):654–65.
Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356(6337):eaaj2239. https://doi.org/10.1126/science.aaj2239.
Tirado-Magallanes R, Rebbani K, Lim R, Pradhan S, Benoukraf T. Whole genome DNA methylation: beyond genes silencing. Oncotarget. 2017;8(3):5629–37.
Velu V, Mylvaganam G, Ibegbu C, Amara RR. Tfh1 cells in germinal centers during chronic HIV/SIV infection. Front Immunol. 2018;9:1272.
Kohu K, Ohmori H, Wong WF, Onda D, Wakoh T, Kon S, et al. The Runx3 transcription factor augments Th1 and down-modulates Th2 phenotypes by interacting with and attenuating GATA3. J Immunol. 2009;183(12):7817–24.
Imgenberg-Kreuz J, Carlsson Almlof J, Leonard D, Alexsson A, Nordmark G, Eloranta ML, et al. DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann Rheum Dis. 2018;77(5):736–43.
Joseph S, George NI, Green-Knox B, Treadwell EL, Word B, Yim S, et al. Epigenome-wide association study of peripheral blood mononuclear cells in systemic lupus erythematosus: Identifying DNA methylation signatures associated with interferon-related genes based on ethnicity and SLEDAI. J Autoimmun. 2019;96:147–57.
Ulff-Moller CJ, Asmar F, Liu Y, Svendsen AJ, Busato F, Gronbaek K, et al. Twin DNA methylation profiling reveals flare-dependent interferon signature and b cell promoter hypermethylation in systemic lupus erythematosus. Arthritis Rheumatol. 2018;70(6):878–90.
Yu H, Du L, Yi S, Wang Q, Zhu Y, Qiu Y, et al. Epigenome-wide association study identifies Behcet’s disease-associated methylation loci in Han Chinese. Rheumatology (Oxford). 2019;58(9):1574–84.
Huynh JL, Garg P, Thin TH, Yoo S, Dutta R, Trapp BD, et al. Epigenome-wide differences in pathology-free regions of multiple sclerosis-affected brains. Nat Neurosci. 2014;17(1):121–30.
Maltby VE, Lea RA, Graves MC, Sanders KA, Benton MC, Tajouri L, et al. Genome-wide DNA methylation changes in CD19(+) B cells from relapsing-remitting multiple sclerosis patients. Sci Rep. 2018;8(1):17418.
Altorok N, Coit P, Hughes T, Koelsch KA, Stone DU, Rasmussen A, et al. Genome-wide DNA methylation patterns in naive CD4+ T cells from patients with primary Sjogren’s syndrome. Arthritis Rheumatol. 2014;66(3):731–9.
Braekke Norheim K, Imgenberg-Kreuz J, Jonsdottir K, Janssen EA, Syvanen AC, Sandling JK, et al. Epigenome-wide DNA methylation patterns associated with fatigue in primary Sjogren’s syndrome. Rheumatology (Oxford). 2016;55(6):1074–82.
Miceli-Richard C, Wang-Renault SF, Boudaoud S, Busato F, Lallemand C, Bethune K, et al. Overlap between differentially methylated DNA regions in blood B lymphocytes and genetic at-risk loci in primary Sjogren’s syndrome. Ann Rheum Dis. 2016;75(5):933–40.
Glossop JR, Emes RD, Nixon NB, Packham JC, Fryer AA, Mattey DL, et al. Genome-wide profiling in treatment-naive early rheumatoid arthritis reveals DNA methylome changes in T and B lymphocytes. Epigenomics. 2016;8(2):209–24.
Guo S, Zhu Q, Jiang T, Wang R, Shen Y, Zhu X, et al. Genome-wide DNA methylation patterns in CD4+ T cells from Chinese Han patients with rheumatoid arthritis. Mod Rheumatol. 2017;27(3):441–7.
Webster AP, Plant D, Ecker S, Zufferey F, Bell JT, Feber A, et al. Increased DNA methylation variability in rheumatoid arthritis-discordant monozygotic twins. Genome Med. 2018;10(1):64.
Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, et al. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013;41(Database issue):D1040–6.
Zhou Z, Sun B, Li X, Zhu C. DNA methylation landscapes in the pathogenesis of type 2 diabetes mellitus. Nutr Metab (Lond). 2018;15:47.
Zhao J, Goldberg J, Bremner JD, Vaccarino V. Global DNA methylation is associated with insulin resistance: a monozygotic twin study. Diabetes. 2012;61(2):542–6.
Bach LA. Recent insights into the actions of IGFBP-6. J Cell Commun Signal. 2015;9(2):189–200.
Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol. 2015;6:1.
Tsai S, Clemente-Casares X, Zhou AC, Lei H, Ahn JJ, Chan YT, et al. Insulin receptor-mediated stimulation boosts T cell immunity during inflammation and infection. Cell Metab. 2018;28(6):922-34 e4.
Hommes MJ, Romijn JA, Endert E, Eeftinck Schattenkerk JK, Sauerwein HP. Insulin sensitivity and insulin clearance in human immunodeficiency virus-infected men. Metabolism. 1991;40(6):651–6.
Jiang XG, Tian W, Nicolls MR, Rockson SG. The lymphatic system in obesity, insulin resistance, and cardiovascular diseases. Front Physiol. 2019;10:1402.
Hyrcza MD, Kovacs C, Loutfy M, Halpenny R, Heisler L, Yang S, et al. Distinct transcriptional profiles in ex vivo CD4(+) and CD8(+) T cells are established early in human immunodeficiency virus type 1 infection and are characterized by a chronic interferon response as well as extensive transcriptional changes in CD8(+) T cells. J Virol. 2007;81(7):3477–86.
Wu JQ, Dwyer DE, Dyer WB, Yang YH, Wang B, Saksena NK. Genome-wide analysis of primary CD4+and CD8+T cell transcriptomes shows evidence for a network of enriched pathways associated with HIV disease. Retrovirology. 2011;8:18.
Huot N, Bosinger SE, Paiardini M, Reeves RK, Muller-Trutwin M. Lymph node cellular and viral dynamics in natural hosts and impact for HIV cure strategies. Front Immunol. 2018;9:780.
Barrenas F, Raehtz K, Xu CL, Law L, Green RR, Silvestri G, et al. Macrophage-associated wound healing contributes to African green monkey SIV pathogenesis control. Nat Commun. 2019;10:5101.
Raehtz KD, Barrenas F, Xu C, Busman-Sahay K, Valentine A, Law L, et al. African green monkeys avoid SIV disease progression by preventing intestinal dysfunction and maintaining mucosal barrier integrity. PLoS Pathog. 2020;16(3):e1008333.
He T, Brocca-Cofano E, Gillespie DG, Xu C, Stock JL, Ma D, et al. Critical role for the adenosine pathway in controlling simian immunodeficiency virus-related immune activation and inflammation in gut mucosal tissues. J Virol. 2015;89(18):9616–30.
Hao XP, Lucero CM, Turkbey B, Bernardo ML, Morcock DR, Deleage C, et al. Experimental colitis in SIV-uninfected rhesus macaques recapitulates important features of pathogenic SIV infection. Nat Commun. 2015;6:8020.
Zhang Q, Putheti P, Zhou Q, Liu Q, Gao W. Structures and biological functions of IL-31 and IL-31 receptors. Cytokine Growth Factor Rev. 2008;19(5–6):347–56.
Ortiz AM, Klase ZA, DiNapoli SR, Vujkovic-Cvijin I, Carmack K, Perkins MR, et al. IL-21 and probiotic therapy improve Th17 frequencies, microbial translocation, and microbiome in ARV-treated SIV-infected macaques. Mucosal Immunol. 2016;9(2):458–67.
Micci L, Ryan ES, Fromentin R, Bosinger SE, Harper JL, He T, et al. Interleukin-21 combined with ART reduces inflammation and viral reservoir in SIV-infected macaques. J Clin Invest. 2015;125(12):4497–513.
Hermans D, Gautam S, Garcia-Canaveras JC, Gromer D, Mitra S, Spolski R, et al. Lactate dehydrogenase inhibition synergizes with IL-21 to promote CD8(+) T cell stemness and antitumor immunity. Proc Natl Acad Sci U S A. 2020;117:6047–55.
Huot N, Jacquelin B, Garcia-Tellez T, Rascle P, Ploquin MJ, Madec Y, et al. Natural killer cells migrate into and control simian immunodeficiency virus replication in lymph node follicles in African green monkeys. Nat Med. 2017;23(11):1277–86.
Hocqueloux L, Avettand-Fenoel V, Jacquot S, Prazuck T, Legac E, Melard A, et al. Long-term antiretroviral therapy initiated during primary HIV-1 infection is key to achieving both low HIV reservoirs and normal T cell counts. J Antimicrob Chemother. 2013;68(5):1169–78.
Jeffries MA, Dozmorov M, Tang YH, Merrill JT, Wren JD, Sawalha AH. Genome-wide DNA methylation patterns in CD4(+) T cells from patients with systemic lupus erythematosus. Epigenetics-Us. 2011;6(5):593–601.
Kornfeld C, Ploquin MJ, Pandrea I, Faye A, Onanga R, Apetrei C, et al. Antiinflammatory profiles during primary SIV infection in African green monkeys are associated with protection against AIDS. J Clin Invest. 2005;115(4):1082–91.
Katz A, Nambi SS, Mather K, Baron AD, Follmann DA, Sullivan G, et al. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab. 2000;85(7):2402–10.
Touleimat N, Tost J. Complete pipeline for Infinium((R)) Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics. 2012;4(3):325–41.
Acknowledgments
We would like to thank all the animal care workers and veterinarians at the IDMIT Center for their excellent work.
Funding
SJ was supported by a fellowship from the University Paris Diderot, Sorbonne Paris Cité. NH was supported by the Fondation Beytout. MP was the recipient of a fellowship from Sidaction and NT by a fellowship from the ANRS (French Agency for Research on HIV and Hepatitis). MMT received a grant from the French Agency of AIDS Research, ANRS (AO 2012–2, 13390). The Infectious Disease Models and Innovative Therapies (IDMIT) center in Fontenay-aux-Roses, France, is funded by the French government’s Investissements d’Avenir program for infrastructures (PIA) under grant ANR-11-INBS-0008 and the PIA grant ANR-10-EQPX-02-01. The funding bodies had no role in the design of the study and collection, analysis, or interpretation of data or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
SJ, BJ, JT and MMT designed the studies. SJ, BJ, FB, ER, NH and MP conducted the experiments. NT, FP, YL and SJ performed the bioinformatic analyses. BJ, NDB and RLG coordinated the animal follow-ups. CSH and RC provided samples. SJ, JT and MMT wrote the manuscript. MMT obtained the funding and supervised the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The animals of the microarray-based DNA methylation study were housed at the Commissariat à l'Energie Atomique (CEA, Fontenay-aux-Roses, France, permit number: A 92–032-02). The CEA complies with Standards for Human Care and Use of Laboratory of the Office for Laboratory Animal Welfare (OLAW, USA) under OLAW Assurance number #A5826-01. The Central Committee for Animals at Institut Pasteur or Ethical Committee of Animal Experimentation (CETEA-DSV, IDF, France) approved all animal experimental procedures (Notification numbers: 10-051b, 12-006b). All experimental procedures were conducted in strict accordance with the international European guidelines 2010/63/UE on protection of animals used for experimentation and other scientific purposes (French decree 2013–118). For the pyrosequencing validation, the rhesus monkeys used in this study were cared for by experienced staff at the German Primate Center (DPZ) and housed according to the German Animal Welfare Act which complies with the European Union guidelines on the use of non-human primates for biomedical research and the Weatherall report. The study was approved by the Lower Saxony State Office for Consumer Protection and Food Safety and performed with the project licences 33.14.42502-04-119/09, 33.19-4502-05-10A041, 33.14-42,502-04-10/0037, 33.19-42,502-04-12/0820, 33.19-42,502-04-15/2001, and 33.19-42,502-04-17/2500. In line with § 11 of the German Animal Welfare act, the DPZ has the permission to breed and house non-human primates under license number 392001/7 granted by the local veterinary office.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Supplementary figures.
Additional file 2.
Supplementary Table 1. Description of animals used for characterization of DNA methylation during SIV infection.
Additional file 3.
Supplementary Table 2. List of all identified differentially methylated probes (DMP) as defined by the genome-wide array.
Additional file 4.
Supplementary Table 3. List of genes that were both differentially methylated and differentially expressed in AGM and MAC as defined by the genome-wide array.
Additional file 5.
Supplementary Table 4. List of primers used for pyrosequencing.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jochems, S.P., Jacquelin, B., Tchitchek, N. et al. DNA methylation changes in metabolic and immune-regulatory pathways in blood and lymph node CD4 + T cells in response to SIV infections. Clin Epigenet 12, 188 (2020). https://doi.org/10.1186/s13148-020-00971-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-020-00971-w