
Abstract
Background
Recent studies demonstrated that changes in DNA methylation (DNAm) and inactivation of two imprinted genes (MKRN3 and DLK1) alter the onset of female puberty. We aimed to investigate the association of DNAm profiling with the timing of human puberty analyzing the genome-wide DNAm patterns of peripheral blood leukocytes from ten female patients with central precocious puberty (CPP) and 33 healthy girls (15 pre- and 18 post-pubertal). For this purpose, we performed comparisons between the groups: pre- versus post-pubertal, CPP versus pre-pubertal, and CPP versus post-pubertal.
Results
Analyzing the methylome changes associated with normal puberty, we identified 120 differentially methylated regions (DMRs) when comparing pre- and post-pubertal healthy girls. Most of these DMRs were hypermethylated in the pubertal group (99%) and located on the X chromosome (74%). Only one genomic region, containing the promoter of ZFP57, was hypomethylated in the pubertal group. ZFP57 is a transcriptional repressor required for both methylation and imprinting of multiple genomic loci. ZFP57 expression in the hypothalamus of female rhesus monkeys increased during peripubertal development, suggesting enhanced repression of downstream ZFP57 target genes. Fourteen other zinc finger (ZNF) genes were related to the hypermethylated DMRs at normal puberty. Analyzing the methylome changes associated with CPP, we demonstrated that the patients with CPP exhibited more hypermethylated CpG sites compared to both pre-pubertal (81%) and pubertal (89%) controls. Forty-eight ZNF genes were identified as having hypermethylated CpG sites in CPP.
Conclusion
Methylome profiling of girls at normal and precocious puberty revealed a widespread pattern of DNA hypermethylation, indicating that the pubertal process in humans is associated with specific changes in epigenetically driven regulatory control. Moreover, changes in methylation of several ZNF genes appear to be a distinct epigenetic modification underlying the initiation of human puberty.
Similar content being viewed by others

Background
The onset of puberty is heralded by an increased pulsatile secretion of gonadotropin-releasing hormone (GnRH), which upon reaching the anterior pituitary activates the pituitary–gonadal axis. Epidemiological studies have provided evidence supporting a genetic influence on pubertal timing [1,2,3]. However, the age at normal puberty varies greatly among girls (8–13 years) and the genetic basis for such a variability remains largely unknown [4]. A potential underlying mechanism is the modulation of gene activity by epigenetic factors, which may be important for the broad regulation of pubertal timing [5]. In fact, it appears that up to 20% of the variance of puberty initiation involves environmental factors, such as nutrition, stress, exposure to endocrine-disrupting chemicals, and intrauterine conditions [5, 6].
Epigenetics refers to the alterations in gene expression that are not caused by changes in DNA sequence itself [7]. DNA methylation (DNAm) is one of the best studied epigenetic mechanisms involved in modulating gene activity [8, 9]. It consists of the covalent addition of a methyl (-CH3) group to the fifth position of the pyrimide base of DNA, cytosine, and occurs mostly in cytosine–phosphate–guanine (CpG) dinucleotides [8].
Epigenetics has been implicated as a regulatory system underlying GnRH secretion [10, 11]. The study of DNAm in the medial basal hypothalamus of male rhesus monkeys revealed a decrease in methylation status of the GnRH gene’s 5′ CpG island that paralleled an increase in GnRH mRNA levels across puberty [12]. Indeed, increased DNAm of gene promoters is commonly associated with gene silencing [13, 14]. Recently, silencers of the Polycomb group were identified as major drivers of an epigenetic mechanism of transcriptional repression that is lifted at the beginning of female puberty in rats, allowing the pubertal process to proceed unimpeded [10]. Importantly, manipulations of DNAm in animal models were shown to alter the onset of puberty. Thus, inhibiting DNAm resulted in pubertal failure, whereas inducing DNA hypermethylation led to earlier onset of puberty [10, 15]. DNAm also plays an essential role in genomic imprinting, an epigenetic phenomenon recently implicated in the regulation of puberty. Initial evidence for this concept came from studies showing that common variants located at the loci of three imprinted genes (MKRN3, DLK1, and KCNK9) were associated with the age at menarche in a large European women cohort [16]. More direct evidence was provided by the demonstration that central precocious puberty (CPP) due to loss-of-function mutations in the paternally expressed imprinted genes MKRN3 (makorin ring finger 3) and DLK1 (delta-like 1 homolog) is an imprinting disorder [17,18,19].
In the present study, we used peripheral blood leukocytes to investigate the relationship that may exist between DNAm patterns and pubertal timing in healthy and CPP-affected girls. We compared pre- with post-pubertal control subjects to interrogate changes in the methylome profile that occurs during physiological pubertal development. We also compared CPP patients with healthy girls to analyze the DNAm changes in CPP.
Results
Description of the analyzed human groups
We studied ten female patients with familial CPP (index cases) who were referred for clinical and/or genetic evaluation to the Endocrinology Unit at Clinical Hospital, Sao Paulo, Brazil. CPP in girls was diagnosed based on the presence of breast Tanner stage 2 (B2) before the age of 8 years, pubertal basal and/or GnRH-stimulated LH levels, and bone age advanced more than 1 year (Greulich and Pyle atlas). Clinical and hormonal features of the patients with CPP are described in Table 1. The mean age at pubertal onset of these girls was 6.4 years (ranging from 3 to 8 years). At the time of the first evaluation (mean age of 7.7 years), Tanner B3 was observed in 50% of the girls and Tanner B4 in the remaining 50%. The mean Δ [bone age − chronological age] was 2.2 ± 1.1 years. None of the patients were obese (mean body mass index (BMI) Z-score = 0.7). Mean basal LH levels were 1.4 ± 1.4 IU/L, and mean LH levels after GnRH stimulation were 18.9 ± 14 IU/L. Mean basal FSH levels were 3.4 ± 1.7 IU/L. Median estradiol (E2) levels were 13 pg/mL. All CPP patients had normal brain magnetic resonance imaging.
Familial CPP was defined by the presence of more than one affected member in a family [2]. The pedigrees of the ten families are illustrated in Fig. 1. Only female members were affected in all families. Regarding the mode of inheritance, CPP was maternally inherited in four families (pedigrees 2, 3, 6, 9), paternally inherited in four families (pedigrees 1, 7, 8, 10), and undetermined in two families (pedigrees 4 and 5). A MKRN3 inactivating mutation (p.R328H) was detected in family 10 by Sanger sequencing. Whole-genome sequencing revealed a complex defect in DLK1 (∼ 14 kb deletion and 269 bp duplication) in family 1 [18]. The remaining families (pedigrees 2 to 9) were previously studied by whole-exome sequencing without identifying a genetic mutation associated with CPP phenotype [17].

Pedigrees of the families with CPP. Squares indicate male members, circles female members, black symbols clinically affected members, and arrows the probands
The control group was composed of 33 healthy Brazilian girls. Pubertal stage was characterized by physical signs (Tanner criteria) and hormonal evaluation (Tables 2 and 3). Fifteen of these girls were at pre-pubertal stage with mean chronological age of 6.7 years, ranging from 2.6 to 9 years. All of them exhibited Tanner B1 upon physical medical evaluation and pre-pubertal basal LH levels (< 0.1 IU/L). The mean BMI Z-score was 0.2 ± 0.6. The remaining 18 girls were at pubertal stage, with mean chronological age of 13.1 years, ranging from 9.5 to 16.3 years. The majority of them (61%) had Tanner B4. In the pubertal group, mean basal LH levels were 5.2 ± 2.4 IU/L, mean basal FSH levels were 5.4 ± 1.8 IU/L, and mean E2 levels were 67.6 ± 42.7 pg/mL. The mean BMI Z-score was 0.1 ± 0.7.
Changes in DNA methylation associated with normal puberty
Comparison between pre-pubertal and pubertal healthy girls revealed the presence of 120 differentially methylated regions (DMRs) (false discovery rate (FDR) < 0.05 and methylation differences > 5%), with all but one (99%) being hypermethylated in the pubertal group (Table 4). Most of the DMRs (89 DMRs, 74%) were located on the X chromosome, none of them mapped to the pseudoautosomal regions (PAR) of this chromosome. The 120 DMRs harbored the promoter regions of 127 genes, whose functions were enriched for various biological processes, such as intracellular receptor signaling pathway, messenger RNA (mRNA) transcription, histone modification, and genetic imprinting (Additional file 1).
The single hypomethylated genomic region identified in pubertal girls encompassed the promoter region of ZFP57 (zinc finger protein 57), a KRAB domain-containing transcriptional repressor involved in both imprinting and methylation of multiple genomic loci [20, 21]. The DMR with the largest methylation difference (10.5%) contained the promoter region of RP11-313 J2.1, the zinc finger protein 91 pseudogene. In addition, the promoter regions of 13 other zinc finger (ZNF) genes (ZNF597, ZSCAN12P1, ZNF707, ZNF331, ZC4H2, ZNF182, ZNF41, ZIC3, RNF113A, ZDHHC9, PRDM8, PRDM9, RBM10) were related to these 120 DMRs.
We applied the Gene Set Enrichment Analysis (GSEA) to search for enriched transcription factors that could be targeting the identified DMRs. We detected enrichment for 20 transcription factors, and the ten most relevant are listed in Additional file 2. Importantly, one of them is the estrogen receptor (ER). Seven differentially methylated genes (SMARCA1, POU5F1, HTATSF1, ELF4, HMGB3, KLHL34, FGFR2) had ER binding sites in the region spanning up to 4 kb around their transcription start site (TSS).
Changes in DNA methylation associated with CPP
A unique DMR was detected between CPP cases and pre-pubertal controls (FDR < 0.05), and it was slightly more methylated in the CPP group (mean beta difference of 0.003391969). This genomic region (chr6: 33385679-33385786) harbored the promoter region of CUTA (homolog of Escherichia coli CutA), a gene ubiquitously expressed, including brain. Comparison between CPP cases and pubertal girls revealed the absence of DMRs (FDR < 0.05). Because of this, we explored the methylation levels at isolated CpG sites.
Comparison between CPP cases and pre-pubertal controls revealed 417 differentially methylated CpG sites (DMSs) (FDR < 0.05 and methylation differences > 10%), with the majority of them (338 DMSs, 81%) being hypermethylated in CPP patients (Fig. 2, Additional files 3 and 4). In silico functional analyses of the 199 known genes related to these 338 DMSs demonstrated enrichment for signaling pathways involved in cell communication (70 genes), regulation of response to stimuli (40 genes), and metabolism (10 genes). When comparing CPP cases with pubertal controls, we identified 605 DMSs (FDR < 0.05 and methylation differences > 10%), with the majority of them (539 DMSs, 89%) being hypermethylated in the CPP group (Fig. 3, Additional files 5 and 6). The functional characterization of 308 known genes related to these 539 DMSs revealed enrichment for metabolic pathways (13 genes), transport vesicles (10 genes), association with endocrine system diseases (10 genes), and carcinomas (13 genes). Forty-eight genes harboring hypermethylated CpG sites in CPP were ZNFs (Additional files 4 and 6).

Heatmap based on the methylation levels of the 417 DMSs identified between CPP cases and pre-pubertal controls. Cases are indicated in red and controls in green. Each column represents an individual, and each row represents a CpG site. Methylation levels are displayed in Z-score at the up left corner of the heatmap. Hierarchical clustering was applied to the CpG sites (rows) using Euclidian distance with average linkage

Heatmap based on the methylation levels of the 605 DMSs identified between CPP cases and post-pubertal controls. Cases are indicated in red and controls in green. Each column represents an individual, and each row represents a CpG site. Methylation levels are displayed in Z-score at the up left corner of the heatmap. Hierarchical clustering was applied to the CpG sites (rows) using Euclidian distance with average linkage
Normal methylation of the MKRN3 and DLK1 genes
Methylation status of the MKRN3 and DLK1 genes and their regulatory regions using two distinct methods revealed no differences between CPP patients and controls. The methylation analyses of MKRN3:TSS-DMR and MEG3/DLK1:IG-DMR (IG = intergenic) are showed in Additional file 7.
Hypothalamic expression of ZFP57 increases at puberty in nonhuman primates
To determine if hypomethylation of ZFP57 in peripheral blood leukocytes is accompanied by increased ZFP57 expression in the hypothalamus at the time of puberty, we measured ZFP57 mRNA levels in the medial basal hypothalamus (MBH) of pre- and peripubertal female rhesus monkeys. We observed that ZFP57 mRNA levels began to increase during late juvenile development and became significantly elevated at puberty, coinciding with the increase in GnRH and KISS1 expression that occurs at this time (Fig. 4a, b). To determine if a decrease in expression of ZNFs that become hypermethylated at puberty also occurs at puberty in the monkey hypothalamus, we selected five of these genes for mRNA measurement. Interestingly, expression of all five genes showed a tendency to decrease at puberty (Fig. 4b), with the change in ZNF597 being statistically significant. To further evaluate these results, we performed a correlation analysis of the changes in ZFP57 and ZNF597 expression that occurred with the advent of puberty and found the existence of a significant (p = 0.01) inverse correlation between the pubertal increase in ZFP57 mRNA levels and the decrease in ZNF597 expression (Fig. 4c). We also used the MBH of female rhesus monkeys to quantitate the hypothalamic expression of four ZNFs that become hypermethylated in CPP and found that their mRNA levels either increase at normal puberty (ZNF251) or showed no change (RNF113A, ZDBF2, and ZDHHC9) (Additional file 8a). This result is in keeping with the finding that ZNFs hypermethylated at CPP are not the same as those ZNFs that become hypermethylated at normal puberty.

Changes in expression of a GnRH, KISS1, and TAC3 and b selected ZNFs in the female monkey hypothalamus during pubertal development. One of the ZNFs examined (ZFP57) was hypomethylated at puberty in peripheral blood cells. The other five were hypermethylated. c Correlation analysis shows that a loss in ZNF597 expression observed at the time of monkey puberty is negatively correlated to the increase in ZFP57 mRNA levels detected at this time. The mRNA levels obtained were expressed as fold-change with regard to the values observed in the EJ group. Bars represent mean ± s.e.m. (n = 4–7/group) (*p < 0.05; vs EJ group; one-way ANOVA-SNK test). EJ early juvenile, LJ late juvenile, PUB peripubertal
In addition to defining the methylation status of MKRN3 and DLK1 in peripheral blood leukocytes of CPP patients and control subjects undergoing normal puberty, we examined the changes in MKRN3 and DLK1 mRNA levels that occur in the MBH of female monkeys at the time of puberty. No changes in expression for either gene were detected between the early juvenile and the pubertal phases of monkey puberty (Additional file 8b).
Figure 5 summarizes the main results of the present study.

Flow chart summarizing the main results of the present study
Discussion
In the last few years, it has become increasingly clear that epigenetic mechanisms contribute significantly to the regulation of pubertal timing. From a clinical perspective, it would be desirable to have minimally invasive methods for the identification and monitoring of at least some of these mechanisms. Here, we report the use of white blood cells to assess patterns of DNAm that occur in association with puberty in girls. Our findings reveal the existence of a broad pattern of DNA hypermethylation taking place in these cells at the time of both normal and central precocious puberty. These findings are consistent with an earlier report showing an increase in DNAm levels of peripheral blood cells during the puberty transition in girls [22].
Most of the changes we observed consisted of hypermethylation of either DMRs or DMSs, with ZNFs standing out as a population of transcriptional repressors affected by these alterations. The sole exception to this pattern was ZFP57, a transcriptional repressor required for methylation of downstream genes and imprinting of several other genes [20, 23]. Contrary to other ZNFs, ZFP57 was hypomethylated at puberty, suggesting that its expression may increase—instead of decrease—in tissues relevant to the pubertal transition. Measurement of ZFP57 expression in the hypothalamus of female rhesus monkeys undergoing puberty proved this assumption to be correct, as a significant increase in ZFP57 mRNA levels was detected at the time of puberty.
The ZFP57 gene, located at chromosome 6p22.1, encodes a protein of 516 amino acids that contains seven zinc finger motifs [21, 24]. The interaction of these motifs with specific DNA sequences in regulatory regions of target genes is required for ZNF proteins to control gene expression [24]. Importantly, in the absence of ZFP57, genomic imprinting is lost [20]. This is illustrated by the loss of differential DNA methylation at the imprinted regions Dlk1-Dio3 and Snrpn in homozygous mutant embryos (maternal-zygotic) derived from Zfp57 null female mice [20]. Notably, directly relevant to the present findings, ZFP57 has been shown to be required for normal imprinting of genomic regions controlling the expression of MKRN3 and DLK1, two genes encoding repressors of the pubertal process, with DLK1 being one of the genes most strongly affected by the absence of ZFP57 [20]. The increase of ZFP57 expression observed in the hypothalamus of pubertal female monkeys suggests that an important function of ZFP57 in the neuroendocrine brain might be to repress the activity of transcriptional repressors of puberty, such as the Polycomb complex or other ZNFs [10, 25]. Furthermore, the inverse correlation detected between the increase in ZFP57 mRNA levels and the decrease in ZNF597 expression that occurred in the hypothalamus at the time of monkey puberty suggests that ZNF597 might be one of the transcriptional inhibitors of primate puberty repressed by ZFP57. Further studies are required to test the validity of this idea.
It is now known that pubertal timing requires repression of inhibitory factors and that DNA hypermethylation of gene promoters is associated with gene silencing [11, 26, 27]. In humans, genome-wide association studies revealed associations between single-nucleotide polymorphisms located near ZNF131, ZNF462, and ZNF483 and earlier age of menarche, suggesting that ZNF genes can impact human pubertal development [16, 25, 28]. Supporting this concept are the demonstrations that MKRN3, also known as ZNF127, inhibits the human pubertal process and that MKRN3 loss-of-function mutations are the most frequent cause of familial CPP [29,30,31]. More recently, the DLK1 gene was also proposed to play an inhibitory role in the regulation of puberty, since its deficiency was associated with a CPP phenotype in syndromic and nonsyndromic cases [18, 32]. Within this context, our results showing a broad pattern of DNA hypermethylation at puberty suggest that—if hypermethylation of ZNFs also occurs in neuroendocrine cells controlling reproductive development—ZNF expression would decrease, and downstream target genes would escape from ZNF inhibitory control at the time of puberty. A specific example of this epigenetic interaction was recently provided by the demonstration that expression of several ZNFs decreases at puberty in the hypothalamus of female nonhuman primates and that preventing this change delayed pubertal timing [25]. In the present study, we measured the mRNA levels of five of these hypermethylated genes (ZNF597, ZNF41, ZNF707, ZNF331, and PRDM8) in de hypothalamus of developing female monkeys and found that all of them showed a tendency to decrease expression at the end of pubertal development, with the changes in ZNF597 being significant.
As indicated above, there were 14 ZNFs hypermethylated at normal puberty. Intriguingly, the largest methylation difference (10.5%) was related to the zinc finger protein 91 pseudogene. At present, we do not know if it is involved in the hypothalamic control of puberty, but such a role remains possible, especially considering that 2–20% of human pseudogenes are transcribed, with some being transcribed in a tissue-specific manner maintained over the years [33]. It is therefore plausible that pseudogenes have a functional role in specific cell populations, an idea supported by the finding that noncoding RNAs produced from pseudogenes can regulate gene expression [33].
The search for transcription factors targeting the 120 DMRs associated with the pubertal process revealed an enrichment for ER. We identified seven differentially methylated genes as ER target genes, suggesting the existence of a functional relationship between them. This relationship appears to be particularly relevant to the neuronal regulation of the pubertal process, as neuronal ERα is involved in the temporal coordination of GnRH secretion, and an inhibitory ER-mediated influence on kisspeptin neurons has been shown to keep puberty in check in female mice [34]. Recently, the methylome study of 30 girls identified changes in DNAm across puberty related to estrogen-responsive genes, suggesting that differential DNAm at puberty may in part result from exposure to pubertal levels of estradiol [35].
An intriguing finding of this study was the striking prevalence of X-linked DMRs related to puberty. All the 89 X-linked DMRs were mapped outside the PAR regions of the X chromosome. PAR are short regions of homology between the mammalian X and Y chromosomes which are located at the tips of the short arm (PAR1) and long arm (PAR2) of the chromosome, and they harbor genes that escape X-inactivation [36]. Most of X-linked genes are subject to X-inactivation in females to ensure dosage compensation [36]. However, 15–20% of X-chromosomal genes escape from inactivation, and 80% of them lie on the short arm [37, 38]. Moreover, these escaping genes can have different expression levels between tissues and between females [38, 39]. Of note, some of the X-linked DMRs related to puberty identified in our study affect genes that escape X-inactivation, as MSL3, NR0B1, RBM3 e HS6ST2, and others that are heterogeneous in escape, as FGF13 e o SLC25A5 [38]. The potential contribution of genes that escape X-inactivation to the timing of puberty was previously noticed. A case in point are the reports of girls with trisomy X (47, XXX karyotype) manifesting precocious puberty [40, 41]. Early activation of the hypothalamic–pituitary–gonadal axis in these girls was attributed to the extra X chromosome and more specifically to the expression of genes that escape X-inactivation. Early puberty was also described in females with Xp.11.22-p11.23 duplication [42]. In these patients, the duplicated X is preferentially activated, probably contributing to their clinical phenotype. Our results now demonstrate that changes in methylation of genes that escape X-inactivation occur in puberty.
A Danish study identified the methylation of a region on chromosome 7, which contains the promoter of TRIP6 (thyroid hormone receptor interactor 6), to be associated with human pubertal development [43]. Our results support and extend these earlier findings by demonstrating an overlap of ten DMRs with the reported data, with seven presenting methylation changes in the same direction, including the DMR containing the TRIP6 promoter (Table 5).
Our study unveils a genome-wide DNA hypermethylation in CPP, which is in accordance with animal studies [15]. To our knowledge, this is the first study describing changes in the methylome patterns of girls with CPP. Hypermethylated CpGs in 63 genes were identified in CPP patients, including 48 ZNF genes. We speculate that these genes can either contribute to CPP or represent epigenetic modifications resulting from functional changes affecting the complex genetic network underlying the CPP disease. Although we cannot distinguish between these two possibilities, we notice the absence of significant DMRs between pubertal healthy girls and CPP patients. This suggests that the main epigenetic modifications that modulate gene expression during puberty, either normal or precocious, are similar. However, this conclusion is tempered by the finding that differentially methylated genes in CPP are different from those differentially methylated in normal puberty. It might be that in CPP there are different genomic regions that become differentially methylated and that these regions remain epigenetically silent in normal puberty. In fact, only one DMR containing the promoter of CUTA was found to be more methylated in CPP patients than in pre-pubertal controls. This gene, mapped to chromosome 6p21.32, encodes a protein of 136 amino acids that plays a role in anchoring of acetylcholinesterase to neuronal membranes in the human brain [44]. The protein CUTA seems to be also involved in promoting proliferation and survival of glial cells [45]. The CUTA gene has not been implicated before in the regulation of pubertal development.
Changes in methylation of the two precocious puberty imprinted genes, MKRN3 and DLK1, could represent an interesting causal mechanism of sporadic and familial CPP. However, our results showing a normal methylation status of both genes exclude this potential mechanism as an underlying cause of CPP in our patients. It remains possible that the study of a much larger population of girls with CPP may provide evidence for such a relationship, or that the identification of novel mutations able to alter gene methylation patterns proved to be a causative factor [46,47,48,49].
Conclusion
By demonstrating a widespread pattern of DNA hypermethylation associated with normal and precocious puberty in girls, our results suggest that an epigenetic mechanism involving a chemical change in DNA architecture contributes to regulating pubertal timing in humans. Because these hypermethylation patterns involve several genes, the compelling possibility emerges that the net outcome of these alterations is a modified output from networks controlling the pubertal process. The overrepresentation of ZNFs among genes affected by differential methylation and the recent demonstration of an involvement of ZNFs in the central control of female puberty in monkeys suggest that ZNFs may provide a major regulatory pathway linking DNA methylation to the control of human puberty.
Methods
Hormone assays
Serum LH, FSH, and E2 levels were measured by ultrasensitive methods, immunofluorometric assay—IFMA (AutoDELFIA, Turku, Finland), or electrochemiluminometric assay—ECLIA (Cobas e601, Roche Diagnostics, USA), with good correlation among them. The inter-assay and intra-assay coefficients of variation were 5% or less. The hormonal profile was assessed by IFMA (patients) or ECLIA (control group). For the acute GnRH stimulation test, serum LH was measured at − 15, 0, 15, 30, 45, and 60 min after i.v. administration of 100 μg of GnRH. Basal LH levels > 0.6 IU/L (IFMA) or 0.2 IU/L (ECLIA) were considered as pubertal levels, and a GnRH-stimulated LH peak > 6.9 IU/L (IFMA) or 5.0 IU/L (ECLIA) were considered as a pubertal response [50, 51]. The LH level measured 2 h after the first administration of leuprorrelin depot 3.75 mg > 10 IU/L (IFMA) or 5.0 IU/L (ECLIA) was considered pubertal [51, 52].
Sample preparation and quality control
Genomic DNA was extracted from peripheral blood leukocytes using standard procedures. DNA quality and quantity were assessed by NanoDrop (Thermo Fisher Scientific), Qubit (Thermo Fisher Scientific), and electrophoresis on 1% agarose gel. The bisulfite-converted DNA (EZ DNA Methylation kit, Zymo Research) was hybridized in the Human Methylation 450 BeadChip microarray (HM450K, Illumina), following the Illumina Infinium HD methylation protocol. We used RnBeads tools to evaluate the quality of our data, and all samples provided high-quality data [53]. Briefly, experimental quality control was performed using the microarray positive and negative control probes for staining, hybridization, extension, target removal, bisulfite conversion, specificity, and non-polymorphic sites.
Data were extracted by the iScan SQ scanner (Illumina) using GenomeStudio software (v.2011.1), with the methylation module v.1.9.0, into IDAT files.
Probes were annotated using GRCh37/hg19 coordinates from UCSC regarding genomic positions and features (FDb.InfiniumMethylation.hg19 package), with additional annotations to identify probes that exhibit multiple alignments in the genome for posterior exclusion.
Methylation levels of the CpG sites were calculated as beta values, which range continuously from 0 (unmethylated) to 1 (fully methylated) (http://www.illumina.com).
Differential methylation analyses
These analyses were performed in the R environment using Bioconductor packages (http://www.bioconductor.org).
The RnBeads package was applied to the dataset [53]. Non-specific probes (n = 28,076) were removed due to the high likelihood of cross-hybridization. Background was corrected using the Noob method, which is based on a normal-exponential convolution using out-of-band probes [54]. Normalization of signal intensities values from probes types I and II was performed using SWAN method (Additional file 9), which adjusts the intensities based on a quantile approach [55].
Technical effects and cell blood composition were corrected using default parameters from RnBeads [56]. An expected association between surrogate variables and the age at the time of blood collection was identified, but correction was not applied because these variables are related to the study design (Additional file 10). The clinical treatment for CPP with GnRH analogue did not act as a co-variable.
After the pre-processing step, 443,042 CpG sites were analyzed in pairwise comparisons (pre-pubertal versus pubertal controls, familial CPP cases versus pre-pubertal controls, and familial CPP cases versus pubertal controls).
To identify DMSs, hierarchical linear models from the limma software package followed by a fitting based on the Bayes statistics was applied to M values (log of beta values) [57]. CpG sites presenting a FDR < 0.05 and methylation differences greater than 10% were considered as the most significant and selected for further analysis.
DMRcate was applied to identify DMRs, defined as a 300 nucleotides sequence with at least seven CpG sites presenting methylation changes in the same direction [58]. Genomic regions with FDR < 0.05 and mean methylation differences greater than 5% were considered the top ones.
In silico analyses
Functional enrichment analyses were performed on the Web-based Gene Set Analysis Toolket (WebGestalt) using the whole genome as background [59]. Features with adjusted p value < 0.05 provided by the Benjamini–Hochberg multiple test were considered significant. We also used the GSEA program to search for statistically significant associations between a defined set of genes and biological states [60].
Methylation analyses of MKRN3 and DLK1 loci
Bisulfite-converted DNA samples of all patients and controls were studied using the TaqMan Allele-Specific Methylated Multiplex Real-Time Quantitative Polymerase Chain Reaction to analyze the methylation status at MKRN3:TSS-DMR and MEG3/DLK1:IG-DMR [61].
Nonhuman primates
The MBH of female rhesus monkeys (Macaca mulatta) was obtained through the Oregon National Primate Research Center (ONPRC) Tissue Distribution Program. The animals were classified into different stages of pubertal development according to the criteria proposed by Watanabe and Terasawa [62]. Early juvenile (EJ) animals were 9 months to 1.8 years of age, late juvenile (LJ) were 2–2.9 years of age, and pubertal (Pub) were 3.1–4 years old. Plasma LH levels at these ages, measured using a different set of animals (n = 10/group), were 2.59 ± 0.97 (EJ), 3.88 ± 0.92 (LJ), and 6.48 ± 1.64 (Pub) ng/ml, respectively. The MBH was dissected by making a rostral cut along the posterior border of the optic chiasm, a caudal cut immediately in front of the mammillary bodies, and two lateral cuts half-way between the medial eminence and the hypothalamic sulci, as previously reported [63]. The tissue fragments were frozen in liquid nitrogen and stored at − 80 °C until RNA extraction.
RNA extraction and quantitative (q) PCR
Total RNA was extracted from the MBH of female rhesus monkeys using the RNeasy mini kit (Qiagen, Valencia, CA). DNA contamination was removed by on-column digestion with DNAse using the Qiagen RNase-free DNase kit (Qiagen, Valencia, CA). RNA concentrations were determined by spectrophotometric trace (Nanodrop, ThermoScientific, Wilmington, DE). Total RNA (500 ng) was transcribed into cDNA in a volume of 20 μl using 4 U of Omniscript reverse transcriptase (Qiagen, Valencia, CA). mRNA was measured using the SYBR GreenER™ qPCR SuperMix system (Invitrogen, Carlsbad, CA). Amplification primers were designed using the PrimerSelect tool of DNASTAR 14 software (Madison, WI) on the NCBI online Primer-Blast program (Additional file 11). PCR reactions were performed in a volume of 10 μl (1 μl of diluted cDNA, 5 μl of SYBR GreenER™ qPCR SuperMix, and 4 μl of primers mix; 1 μM of each gene-specific primer). The PCR conditions used were 5 min at 95 °C, 40 cycles of 15 s at 95 °C, and 60 s at 60 °C. Formation of a single SYBR Green-labeled PCR amplicon was confirmed by subjecting each PCR reaction to a three-step melting curve analysis (15 s at 95 °C, 1 min at 60 °C, ramping up to 95 °C at 0.5 °C/s, detecting every 0.5 s, and ending with 15 s at 95 °C). The qPCR reactions were performed using a QuantStudio 12 K Real-Time PCR system (Thermo Fisher, Waltham, MA), and a QuantStudio 12 K Flex software (Thermo Fisher, Waltham, MA) was used to detect threshold cycles (CTs). Standard curves were constructed by serially diluting (1/2 to 1/512) a pool of cDNAs derived from a mix of equal amounts of cDNA from each sample. The mRNA content of each sample was estimated by referring the corresponding CTs to the relative standard curve, and the values obtained were normalized for procedural losses using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA as the normalizing unit.
Abbreviations
- B:
-
Breast Tanner stage
- BMI:
-
Body mass index
- CpG:
-
Cytosine–phosphate–guanine
- CPP:
-
Central precocious puberty
- DMR:
-
Differentially methylated region
- DMS:
-
Differentially methylated site
- DNAm:
-
DNA methylation
- E2:
-
Estradiol
- ECLIA:
-
Electrochemiluminometric assay
- EJ:
-
Early juvenile
- ER:
-
Estrogen receptor
- FDR:
-
False discovery rate
- GnRH:
-
Gonadotropin-releasing hormone
- GSEA:
-
Gene Set Enrichment Analysis
- IFMA:
-
Immunofluorometric assay
- IG:
-
Intergenic
- LJ:
-
Late juvenile
- MBH:
-
Medial basal hypothalamus
- mRNA:
-
Messenger RNA
- PAR:
-
Pseudoautosomal region
- Pub:
-
Pubertal
- TSS:
-
Transcription start site
- ZNF:
-
Zinc finger
References
Palmert MR, Hirschhorn JN. Genetic approaches to stature, pubertal timing, and other complex traits. Mol Genet Metab. 2003;80:1–10.
de Vries L, Kauschansky A, Shohat M, Phillip M. Familial central precocious puberty suggests autosomal dominant inheritance. J Clin Endocrinol Metab. 2004;89:1794–800.
Fischbein S. Intra-pair similarity in physical growth of monozygotic and of dizygotic twins during puberty. Ann Hum Biol. 1977;4:417–30.
Palmert MR, Boepple PA. Variation in the timing of puberty: clinical spectrum and genetic investigation. J Clin Endocrinol Metab. 2001;86:2364–8.
Leka-Emiri S, Chrousos GP, Kanaka-Gantenbein C. The mystery of puberty initiation: genetics and epigenetics of idiopathic central precocious puberty (icpp). J Endocrinol Investig. 2017;40:789–802.
Latronico AC, Brito VN, Carel JC. Causes, diagnosis, and treatment of central precocious puberty. Lancet Diabetes Endocrinol. 2016. https://doi.org/10.1016/S2213-8587(15)00380-0.
Felsenfeld G. A brief history of epigenetics. Cold Spring Harb Perspect Biol. 2014;6(1):a018200.
Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–20.
Kanherkar RR, Bhatia-Dey N, Csoka AB. Epigenetics across the human lifespan. Front Cell Dev Biol. 2014;2:49.
Lomniczi A, Loche A, Castellano JM, Ronnekleiv OK, Bosch M, Kaidar G, Knoll JG, Wright H, Pfeifer GP, Ojeda SR. Epigenetic control of female puberty. Nat Neurosci. 2013;16:281–9.
Lomniczi A, Ojeda SR. The emerging role of epigenetics in the regulation of female puberty. Endocr Dev. 2016;29:1–16.
Kurian JR, Terasawa E. Epigenetic control of gonadotropin releasing hormone neurons. Front Endocrinol (Lausanne). 2013;4:61.
Smallwood SA, Kelsey G. De novo DNA methylation: a germ cell perspective. Trends Genet. 2012;28:33–42.
Deaton AM, Bird A. Cpg islands and the regulation of transcription. Genes Dev. 2011;25:1010–22.
Rzeczkowska PA, Hou H, Wilson MD, Palmert MR. Epigenetics: a new player in the regulation of mammalian puberty. Neuroendocrinology. 2014;99:139–55.
Perry JR, Day F, Elks CE, Sulem P, Thompson DJ, Ferreira T, He C, Chasman DI, Esko T, Thorleifsson G, Albrecht E, Ang WQ, Corre T, Cousminer DL, Feenstra B, Franceschini N, Ganna A, Johnson AD, Kjellqvist S, Lunetta KL, McMahon G, Nolte IM, Paternoster L, Porcu E, Smith AV, Stolk L, Teumer A, Tsernikova N, Tikkanen E, Ulivi S, Wagner EK, Amin N, Bierut LJ, Byrne EM, Hottenga JJ, Koller DL, Mangino M, Pers TH, Yerges-Armstrong LM, Hua Zhao J, Andrulis IL, Anton-Culver H, Atsma F, Bandinelli S, Beckmann MW, Benitez J, Blomqvist C, Bojesen SE, Bolla MK, Bonanni B, Brauch H, Brenner H, Buring JE, Chang-Claude J, Chanock S, Chen J, Chenevix-Trench G, Collee JM, Couch FJ, Couper D, Coviello AD, Cox A, Czene K, D’Adamo AP, Davey Smith G, De Vivo I, Demerath EW, Dennis J, Devilee P, Dieffenbach AK, Dunning AM, Eiriksdottir G, Eriksson JG, Fasching PA, Ferrucci L, Flesch-Janys D, Flyger H, Foroud T, Franke L, Garcia ME, Garcia-Closas M, Geller F, de Geus EE, Giles GG, Gudbjartsson DF, Gudnason V, Guenel P, Guo S, Hall P, Hamann U, Haring R, Hartman CA, Heath AC, Hofman A, Hooning MJ, Hopper JL, Hu FB, Hunter DJ, Karasik D, Kiel DP, Knight JA, Kosma VM, Kutalik Z, Lai S, Lambrechts D, Lindblom A, Magi R, Magnusson PK, Mannermaa A, Martin NG, Masson G, McArdle PF, McArdle WL, Melbye M, Michailidou K, Mihailov E, Milani L, Milne RL, Nevanlinna H, Neven P, Nohr EA, Oldehinkel AJ, Oostra BA, Palotie A, Peacock M, Pedersen NL, Peterlongo P, Peto J, Pharoah PD, Postma DS, Pouta A, Pylkas K, Radice P, Ring S, Rivadeneira F, Robino A, Rose LM, Rudolph A, Salomaa V, Sanna S, Schlessinger D, Schmidt MK, Southey MC, Sovio U, Stampfer MJ, Stockl D, Storniolo AM, Timpson NJ, Tyrer J, Visser JA, Vollenweider P, Volzke H, Waeber G, Waldenberger M, Wallaschofski H, Wang Q, Willemsen G, Winqvist R, Wolffenbuttel BH, Wright MJ, Boomsma DI, Econs MJ, Khaw KT, Loos RJ, McCarthy MI, Montgomery GW, Rice JP, Streeten EA, Thorsteinsdottir U, van Duijn CM, Alizadeh BZ, Bergmann S, Boerwinkle E, Boyd HA, Crisponi L, Gasparini P, Gieger C, Harris TB, Ingelsson E, Jarvelin MR, Kraft P, Lawlor D, Metspalu A, Pennell CE, Ridker PM, Snieder H, Sorensen TI, Spector TD, Strachan DP, Uitterlinden AG, Wareham NJ, Widen E, Zygmunt M, Murray A, Easton DF, Stefansson K, Murabito JM, Ong KK. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature. 2014;514:92–7.
Abreu AP, Dauber A, Macedo DB, Noel SD, Brito VN, Gill JC, Cukier P, Thompson IR, Navarro VM, Gagliardi PC, Rodrigues T, Kochi C, Longui CA, Beckers D, de Zegher F, Montenegro LR, Mendonca BB, Carroll RS, Hirschhorn JN, Latronico AC, Kaiser UB. Central precocious puberty caused by mutations in the imprinted gene mkrn3. N Engl J Med. 2013;368:2467–75.
Dauber A, Cunha-Silva M, Macedo DB, Brito VN, Abreu AP, Roberts SA, Montenegro LR, Andrew M, Kirby A, Weirauch MT, Labilloy G, Bessa DS, Carroll RS, Jacobs DC, Chappell PE, Mendonca BB, Haig D, Kaiser UB, Latronico AC. Paternally inherited dlk1 deletion associated with familial central precocious puberty. J Clin Endocrinol Metab. 2017;102:1557–67.
Eggermann T, Perez de Nanclares G, Maher ER, Temple IK, Tumer Z, Monk D, Mackay DJ, Gronskov K, Riccio A, Linglart A, Netchine I. Imprinting disorders: a group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin Epigenetics. 2015;7:123.
Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, Ferguson-Smith AC. A maternal-zygotic effect gene, zfp57, maintains both maternal and paternal imprints. Dev Cell. 2008;15:547–57.
Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, Dayanikli P, Firth HV, Goodship JA, Haemers AP, Hahnemann JM, Kordonouri O, Masoud AF, Oestergaard E, Storr J, Ellard S, Hattersley AT, Robinson DO, Temple IK. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in zfp57. Nat Genet. 2008;40:949–51.
Chen S, Mukherjee N, Janjanam VD, Arshad SH, Kurukulaaratchy RJ, Holloway JW, Zhang H, Karmaus W. Consistency and variability of DNA methylation in women during puberty, young adulthood, and pregnancy. Genet Epigenet. 2017;9:1179237X17721540.
Strogantsev R, Krueger F, Yamazawa K, Shi H, Gould P, Goldman-Roberts M, McEwen K, Sun B, Pedersen R, Ferguson-Smith AC. Allele-specific binding of zfp57 in the epigenetic regulation of imprinted and non-imprinted monoallelic expression. Genome Biol. 2015;16:112.
Lupo A, Cesaro E, Montano G, Zurlo D, Izzo P, Costanzo P. Krab-zinc finger proteins: a repressor family displaying multiple biological functions. Curr Genomics. 2013;14:268–78.
Lomniczi A, Wright H, Castellano JM, Matagne V, Toro CA, Ramaswamy S, Plant TM, Ojeda SR. Epigenetic regulation of puberty via zinc finger protein-mediated transcriptional repression. Nat Commun. 2015;6:10195.
Ojeda SR, Lomniczi A. Puberty in 2013: unravelling the mystery of puberty. Nat Rev Endocrinol. 2014;10:67–9.
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92.
Perry JR, Stolk L, Franceschini N, Lunetta KL, Zhai G, McArdle PF, Smith AV, Aspelund T, Bandinelli S, Boerwinkle E, Cherkas L, Eiriksdottir G, Estrada K, Ferrucci L, Folsom AR, Garcia M, Gudnason V, Hofman A, Karasik D, Kiel DP, Launer LJ, van Meurs J, Nalls MA, Rivadeneira F, Shuldiner AR, Singleton A, Soranzo N, Tanaka T, Visser JA, Weedon MN, Wilson SG, Zhuang V, Streeten EA, Harris TB, Murray A, Spector TD, Demerath EW, Uitterlinden AG, Murabito JM. Meta-analysis of genome-wide association data identifies two loci influencing age at menarche. Nat Genet. 2009;41:648–50.
Bulcao Macedo D, Nahime Brito V, Latronico AC. New causes of central precocious puberty: the role of genetic factors. Neuroendocrinology. 2014;100:1–8.
Simon D, Ba I, Mekhail N, Ecosse E, Paulsen A, Zenaty D, Houang M, Jesuran Perelroizen M, de Filippo GP, Salerno M, Simonin G, Reynaud R, Carel JC, Leger J, de Roux N. Mutations in the maternally imprinted gene mkrn3 are common in familial central precocious puberty. Eur J Endocrinol. 2015;174:1–8.
Abreu AP, Macedo DB, Brito VN, Kaiser UB, Latronico AC. A new pathway in the control of the initiation of puberty: the mkrn3 gene. J Mol Endocrinol. 2015;54:R131–9.
Geoffron S, Abi Habib W, Chantot-Bastaraud S, Dubern B, Steunou V, Azzi S, Afenjar A, Busa T, Pinheiro Canton A, Chalouhi C, Dufourg MN, Esteva B, Fradin M, Genevieve D, Heide S, Isidor B, Linglart A, Morice Picard F, Naud-Saudreau C, Oliver Petit I, Philip N, Pienkowski C, Rio M, Rossignol S, Tauber M, Thevenon J, Vu-Hong TA, Harbison MD, Salem J, Brioude F, Netchine I, Giabicani E. Chromosome 14q32.2 imprinted region disruption as an alternative molecular diagnosis of Silver-Russell syndrome. J Clin Endocrinol Metab. 2018;103:2436–46.
Pink RC, Wicks K, Caley DP, Punch EK, Jacobs L, Carter DR. Pseudogenes: pseudo-functional or key regulators in health and disease? RNA. 2011;17:792–8.
Mayer C, Acosta-Martinez M, Dubois SL, Wolfe A, Radovick S, Boehm U, Levine JE. Timing and completion of puberty in female mice depend on estrogen receptor alpha-signaling in kisspeptin neurons. Proc Natl Acad Sci U S A. 2010;107:22693–8.
Thompson EE, Nicodemus-Johnson J, Kim KW, Gern JE, Jackson DJ, Lemanske RF, Ober C. Global DNA methylation changes spanning puberty are near predicted estrogen-responsive genes and enriched for genes involved in endocrine and immune processes. Clin Epigenetics. 2018;10:62.
Helena Mangs A, Morris BJ. The human pseudoautosomal region (par): origin, function and future. Curr Genomics. 2007;8:129–36.
Spatz A, Borg C, Feunteun J. X-chromosome genetics and human cancer. Nat Rev Cancer. 2004;4:617–29.
Tukiainen T, Villani AC, Yen A, Rivas MA, Marshall JL, Satija R, Aguirre M, Gauthier L, Fleharty M, Kirby A, Cummings BB, Castel SE, Karczewski KJ, Aguet F, Byrnes A, Lappalainen T, Regev A, Ardlie KG, Hacohen N, MacArthur DG. Landscape of X chromosome inactivation across human tissues. Nature. 2017;550:244–8.
Zhang Y, Castillo-Morales A, Jiang M, Zhu Y, Hu L, Urrutia AO, Kong X, Hurst LD. Genes that escape X-inactivation in humans have high intraspecific variability in expression, are associated with mental impairment but are not slow evolving. Mol Biol Evol. 2013;30:2588–601.
Skordis N, Ferrari E, Antoniadou A, Phylactou LA, Fanis P, Neocleous V. GnRH-dependent precocious puberty manifested at the age of 14 months in a girl with 47, XXX karyotype. Hormones (Athens). 2017;16:209–11.
Grosso S, Anichini C, Berardi R, Balestri P, Pucci L, Morgese G. Central precocious puberty and abnormal chromosomal patterns. Endocr Pathol. 2000;11:69–75.
Giorda R, Bonaglia MC, Beri S, Fichera M, Novara F, Magini P, Urquhart J, Sharkey FH, Zucca C, Grasso R, Marelli S, Castiglia L, Di Benedetto D, Musumeci SA, Vitello GA, Failla P, Reitano S, Avola E, Bisulli F, Tinuper P, Mastrangelo M, Fiocchi I, Spaccini L, Torniero C, Fontana E, Lynch SA, Clayton-Smith J, Black G, Jonveaux P, Leheup B, Seri M, Romano C, dalla Bernardina B, Zuffardi O. Complex segmental duplications mediate a recurrent dup(X)(p11.22-p11.23) associated with mental retardation, speech delay, and EEG anomalies in males and females. Am J Hum Genet. 2009;85:394–400.
Almstrup K, Lindhardt Johansen M, Busch AS, Hagen CP, Nielsen JE, Petersen JH, Juul A. Pubertal development in healthy children is mirrored by DNA methylation patterns in peripheral blood. Sci Rep. 2016;6:28657.
Navaratnam DS, Fernando FS, Priddle JD, Giles K, Clegg SM, Pappin DJ, Craig I, Smith AD. Hydrophobic protein that copurifies with human brain acetylcholinesterase: amino acid sequence, genomic organization, and chromosomal localization. J Neurochem. 2000;74:2146–53.
Perrier AL, Cousin X, Boschetti N, Haas R, Chatel JM, Bon S, Roberts WL, Pickett SR, Massoulie J, Rosenberry TL, Krejci E. Two distinct proteins are associated with tetrameric acetylcholinesterase on the cell surface. J Biol Chem. 2000;275:34260–5.
Richards EJ. Inherited epigenetic variation--revisiting soft inheritance. Nat Rev Genet. 2006;7:395–401.
Berdasco M, Esteller M. Genetic syndromes caused by mutations in epigenetic genes. Hum Genet. 2013;132:359–83.
Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, Lee TY, Bodmer D, Hoenselaar E, Hendriks-Cornelissen SJ, Tsui WY, Kong CK, Brunner HG, van Kessel AG, Yuen ST, van Krieken JH, Leung SY, Hoogerbrugge N. Heritable somatic methylation and inactivation of msh2 in families with lynch syndrome due to deletion of the 3′ exons of tacstd1. Nat Genet. 2009;41:112–7.
Hou P, Liu D, Xing M. Genome-wide alterations in gene methylation by the BRAF v600e mutation in papillary thyroid cancer cells. Endocr Relat Cancer. 2011;18:687–97.
Brito VN, Batista MC, Borges MF, Latronico AC, Kohek MB, Thirone AC, Jorge BH, Arnhold IJ, Mendonca BB. Diagnostic value of fluorometric assays in the evaluation of precocious puberty. J Clin Endocrinol Metab. 1999;84:3539–44.
Macedo DB, Cukier P, Mendonca BB, Latronico AC, Brito VN. Advances in the etiology, diagnosis and treatment of central precocious puberty. Arq Bras Endocrinol Metabol. 2014;58:108–17.
Brito VN, Latronico AC, Arnhold IJ, Mendonca BB. A single luteinizing hormone determination 2 hours after depot leuprolide is useful for therapy monitoring of gonadotropin-dependent precocious puberty in girls. J Clin Endocrinol Metab. 2004;89:4338–42.
Assenov Y, Muller F, Lutsik P, Walter J, Lengauer T, Bock C. Comprehensive analysis of DNA methylation data with RnBeads. Nat Methods. 2014;11:1138–40.
Triche TJ Jr, Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD. Low-level processing of Illumina Infinium DNA methylation beadarrays. Nucleic Acids Res. 2013;41:e90.
Maksimovic J, Gordon L, Oshlack A. SWAN: subset-quantile within array normalization for Illumina Infinium Humanmethylation450 Beadchips. Genome Biol. 2012;13:R44.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, R VL, Clark SJ, Molloy PL. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6.
Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33:W741–8.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50.
Azzi S, Steunou V, Rousseau A, Rossignol S, Thibaud N, Danton F, Le Jule M, Gicquel C, Le Bouc Y, Netchine I. Allele-specific methylated multiplex real-time quantitative pcr (asmm rtq-pcr), a powerful method for diagnosing loss of imprinting of the 11p15 region in Russell Silver and Beckwith Wiedemann syndromes. Hum Mutat. 2011;32:249–58.
Watanabe G, Terasawa E. In vivo release of luteinizing hormone releasing hormone increases with puberty in the female rhesus monkey. Endocrinology. 1989;125:92–9.
Heger S, Mastronardi C, Dissen GA, Lomniczi A, Cabrera R, Roth CL, Jung H, Galimi F, Sippell W, Ojeda SR. Enhanced at puberty 1 (eap1) is a new transcriptional regulator of the female neuroendocrine reproductive axis. J Clin Invest. 2007;117:2145–54.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (to DSB); Fundação de Amparo à Pesquisa do Estado de São Paulo 2015/06281-7 (to MM), and 2013/03236-5 (to ACL); Conselho Nacional de Desenvolvimento Científico e Tecnológico 302849/2015-7 (to ACL); and National Institute of Health 1R01HD084542 and 8P51OD011092 for the operation of the Oregon National Primate Research Center (to SRO and AL).
Availability of data and materials
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
DSB collected and analyzed the human data and wrote the paper. ACL and ACVK designed the project, discussed the data, and wrote the manuscript. MM performed the bioinformatic analysis and wrote the manuscript. AL and SRO designed the monkey’s study, interpreted the results, and wrote the manuscript. CFA ran the qPCRs of the monkey’s hypothalamus. APMC and IN investigated the methylation status of MKRN3 and DLK1 loci. VNB, BBM, and ACL clinically evaluated the patients with CPP. DBM collected the data and studied the MKRN3 gene. MCS collected the data and studied the DLK1 gene. HMCP, EAMRR, and MFB clinically evaluated the control group. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the Clinical Hospital of the University of Sao Paulo Medical School (no. 45533215.9.0000.0068), and a written informed consent was obtained from control individuals, patients, and their parents.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Gene ontology of the genes related to the top DMRs between pre- and post-pubertal healthy girls. (XLSX 12 kb)
Additional file 2:
Most significant transcription factors targeting differentially methylated genes between pre- and post-pubertal healthy girls identified by GSEA. (DOCX 12 kb)
Additional file 3:
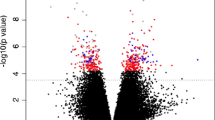
Volcano plot of differences in DNA methylation between CPP cases (n = 10) and pre-pubertal healthy girls (n = 15). Each point represents a CpG site (n = 443,042) with mean methylation difference on the x-axis and − log10 of FDR on the y-axis. Negative methylation differences indicate hypomethylation and positive differences hypermethylation in the CPP cases compared to the pre-pubertal controls. Green dots represent significantly differentially methylated CpGs (n = 417, FDR < 0.05, mean DNA methylation difference > 10%). (TIFF 168 kb)
Additional file 4:
Differentially methylated CpG sites between CPP and pre-pubertal control groups, with methylation differences > 10% and FDR < 0.05. (XLSX 50 kb)
Additional file 5:
Volcano plot of differences in DNA methylation between CPP cases (n = 10) and pubertal healthy girls (n = 18). Each point represents a CpG site (n = 443,042) with mean methylation difference on the x-axis and − log10 of FDR on the y-axis. Negative methylation differences indicate hypomethylation and positive differences hypermethylation in the CPP cases compared to the pubertal controls. Green dots represent significantly differentially methylated CpGs (n = 605, FDR < 0.05, mean DNA methylation difference > 10%). (TIFF 175 kb)
Additional file 6:
Differentially methylated CpG sites between CPP and pubertal control groups, with methylation differences > 10% and FDR < 0.05. (XLSX 71 kb)
Additional file 7:
Methylation index of the DLK1 and MKRN3 loci in healthy and CPP girls determined by Allele-Specific Methylated Multiplex Real-Time Quantitative Polymerase Chain Reaction. (DOCX 12 kb)
Additional file 8:
a) Expression of ZNFs selected from a group of ZNFs hypermethylated in CPP assessed in the MBH of female rhesus monkeys undergoing puberty. b) Lack of significant changes in MKRN3 and DLK1 expression between the early juvenile and peripubertal phases of monkey puberty. The mRNA levels obtained were expressed as fold-change with regard to the values observed in the EJ group. Bars represent mean ± s.e.m. (n = 4–7/group) (* = p < 0.05; vs EJ group; one-way ANOVA-SNK test). EJ, early juvenile; LJ, late juvenile; PUB, peripubertal. (TIFF 934 kb)
Additional file 9:
Normalization of signal intensities values from the Infinium I and II probes by the SWAN method, with beta value on the x-axis and density on the y-axis. (TIFF 151 kb)
Additional file 10:
Heatmap displaying the results of permutation tests performed for associations of the co-variables, as given by the RnBeads package. Pink boxes represent significant p-values (p < 0.01) and blue boxes represent non-significant p-values. (TIFF 265 kb)
Additional file 11:
Primers used to measure mRNA levels by qPCR in the hypothalamus of female rhesus monkeys. (DOCX 13 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bessa, D.S., Maschietto, M., Aylwin, C.F. et al. Methylome profiling of healthy and central precocious puberty girls. Clin Epigenet 10, 146 (2018). https://doi.org/10.1186/s13148-018-0581-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-018-0581-1